
Abstract
For bacterial cell surface display, the target protein needs to be linked to an anchoring motif, and it is essential to choose an appropriate anchoring motif for efficient and stable display of the protein on the cell surface. To isolate a potential anchoring motif that would allow a stable and enhanced display of target proteins on the surface of an Escherichia coli host, we analyzed the outer membrane proteome of E. coli. On the basis of this proteomic analysis, the outer membrane protein X (OmpX), which has a small, monomeric β-barrel structure and is highly expressed, was selected as a potential anchoring motif. The role of OmpX as an anchoring motif for cell surface display was demonstrated using three important industrial enzymes: endoxylanase, lipase, and alkaline phosphatase. Two different positions (Lys122, Val160) in the extracellular loops of OmpX were examined for C-terminal fusion, and the biological activities and localization of the displayed enzymes were analyzed. All three enzymes examined were efficiently displayed on the E. coli cell surface with high activity. These results reveal that the use of OmpX as an anchoring motif is an efficient method to display functional enzymes on the surface of an E. coli host.
Similar content being viewed by others


Introduction
Cell surface display, in which target proteins are expressed on the surface of host cells, is a powerful tool for biotechnological and industrial applications, including biocatalysis, bioremediation, and biosensors, and for high-throughput screening of protein libraries [1–3]. Anchoring to the cell allows the displayed proteins to become more stable than these would be in a free state and to also be freely accessible to substrate(s) or ligands without interference by a membrane barrier. Furthermore, a simple centrifugation following cell surface display of the proteins results in an assay reagent that can be used for various whole-cell applications. To date, many proteins ranging from small peptides to large, complex proteins have been successfully displayed on the surface of various hosts, including bacteria, yeast, spores, and others [2–5].
In surface display, target proteins are generally linked to an appropriate fusion partner (the so-called anchoring motif). Until now, various proteins such as outer membrane proteins (OMPs), lipoproteins, auto-transporters, surface appendage subunits, and S-layer proteins have been employed as anchoring motifs [2, 6–10]. In particular, OMPs, including OmpA, OmpC, FadL, LamB, and OprF, have been widely used because of advantages such as efficient transport signaling, strong anchoring structures (typical β-barrel structures), and resistance to proteolysis in the periplasm or media [2, 3, 11]. However, it is also well known that not all anchoring motifs permit the successful display of target proteins. One critical problem is that the overexpression of anchoring motifs on the cell surface often results in physiological effects on the rigidity of the cell membrane and can cause cell lysis [2, 3]. To reduce these deleterious effects of surface display, mild conditions (e.g., low temperatures and lower concentrations of the inducer) can be employed, but these mild conditions also result in reduced display of target proteins on the surface and consequently lead to reduced activity [12, 13].
One strategy to improve display efficiency is to use the abundant and stable OMPS of the host as anchoring motifs. It is known that Escherichia coli has approximately 150 proteins (4.6 % of the total proteins) in the outer membrane [14]. However, although E. coli has relatively fewer proteins in the outer membrane than in other compartments, it is not easy to find a suitable candidate for an anchoring motif as each protein in the outer membrane has unique characteristics (e.g., size and structure), as well as varying levels. To choose a potential anchoring motif among these proteins, it is necessary to investigate the OMP compositions of the host cells. Proteomic analysis is a powerful tool for this type of investigation. Recent developments in proteome and transcriptome analysis have given a better understanding regarding cell physiology [15, 16], and based on these analyses, new strategies have been developed to improve the physiology of the host cell and protein productivity under normal culture conditions [17].
To identify a potential anchoring motif allowing stable anchoring and enhanced display of target proteins, we first analyzed the outer membrane proteome of the E. coli XL10-Gold strain using two-dimensional gel electrophoresis (2-DE) and liquid chromatography–tandem mass spectrometry (LC–MS/MS). Based on the proteomic analysis, we chose the outer membrane protein X (OmpX) as a potential anchoring motif. We also demonstrated that three important industrial enzymes, endoxylanase from Bacillus sp., Pseudomonas fluorescens lipase, and alkaline phosphatase from E. coli, could be successfully displayed on the surface of the E. coli at high levels and with high activity using OmpX as an anchoring motif.
Materials and Methods
Bacterial Strains and Plasmids
All bacterial strains and plasmids used in this study are listed in Table 1. E. coli XL1-Blue was used as a host for general gene cloning, and E. coli XL10-Gold was used as the sole host for cell surface display. Polymerase chain reaction (PCR) was performed using a PCR Thermal Cycler MP (Takara Shuzo Co., LTD., Shiga, Japan) with the Expand High Fidelity PCR System (Roche Molecular Biochemicals, Mannheim, Germany). The primers used for the PCR reactions are listed in Table 2. The truncated ompX genes encoding 122 and 160 amino acids from the N-terminus were amplified by PCR using each primer set. Each PCR product was digested with EcoRI and XbaI restriction enzymes and then cloned into the pTrc99A vector to yield pTrcOmpXK122 and pTrcOmpXV160, respectively. For the expression of the ompX gene without fusion as a negative control, the complete ompX gene was amplified and cloned into the EcoRI and XbaI sites of pTrc99A to generate pTrcOmpX. The Bacillus sp. endoxylanase gene [18] was amplified by PCR, and the PCR products were cloned into the XbaI and HindIII sites of pTrcOmpXK122 and pTrcOmpXV160 to yield pTrcOmpXK122EX and pTrcOmpXV160EX. A P. fluorescens SIK W1 lipase gene [19] was amplified by PCR and cloned into the same sites of pTrcOmpXK122 and pTrcOmpXV160 to yield pTrcOmpXK122PL and pTrcOmpXV160PL. In addition, the mature truncated form of E. coli W3110 alkaline phosphatase gene (phoA) was amplified by PCR, and the PCR product was cloned into the same site of pTrcOmpXK122 to yield pTrcOmpXK122AP. In all constructs used for the expression of endoxylanase, lipase, and alkaline phosphatase, a FLAG tag (DYKDDDDK) was fused to the C-terminus of each target protein for simple detection of expression and localization. All DNA manipulations, including restriction digestion, ligation, and agarose gel electrophoresis, were carried out using standard methods [20].
Culture Conditions
All cells were cultivated in Luria–Bertani (LB) medium (10 g/L of tryptone, 5 g/L of yeast extract, 5 g/L of NaCl, and 50 μg/L of ampicillin) at 37 °C and 200 rpm. At an OD600 value of 0.5, cells were induced with 1 mM isopropyl-β-d-thiogalactopyranoside for the production of OmpX-fused proteins and surface display. After induction, the cells displaying endoxylanase and lipase were further cultured for 3 h at 25°C, and the cells displaying alkaline phosphatase were further cultured for 3 h at 30°C. Cells were then harvested by centrifugation at 6,000 rpm at 4°C for 10 min for further analysis.
Fractionation of Outer Membrane Proteins
For proteomic analysis, the OMPs were prepared as previously described [21] and then resolubilized in sample rehydration buffer (8 M urea, 2 M thiourea, 4 % (w/v) CHAPS, 20 mM DTT, 1 % (v/v) protease inhibitor cocktail (Roche Diagnostics GmbH, Germany), and 0.8 % (v/v) IPG buffer (GE Healthcare Life Sciences, Giles, UK)). Insoluble materials were removed by centrifugation at 12,000 rpm for 10 min. The supernatant was collected, and the protein concentration was determined by the Bradford method. The resulting solution was stored at −80 °C for 2-DE analysis. For confirmation of the localization of OmpX-fused proteins, the OMPs were prepared by sodium lauryl sarcosinate enrichment as described previously [22].
Proteome Analysis Using 2-DE and LC–MS/MS
2-DE experiments were performed using the IPGphor IEF system (GE Healthcare) and Protean II xi Cell (Bio-Rad, Hercules, CA, USA) as previously described [21]. After gel electrophoresis, the protein spots were visualized using a silver staining kit (GE Healthcare), and the stained gels were scanned using a UMAX PowerLook 2100XL Scanner (UMAX Technologies, Taipei, Taiwan). PDQuest 2-D Analysis Software (Bio-Rad) was used to automate the process of finding protein spots within the image and quantifying the spot densities as a percentage of the volume.
Samples for the MS/MS analysis were prepared as previously described [21]. Tryptic peptides (10-μL aliquots) were analyzed using a nano-LC/MS system consisting of an Ultimate HPLC system (LC Packings, Amsterdam, Netherlands) and a quadrupole-time-of-flight MS (Micromass, Manchester, UK) equipped with a nano-ESI source. The acquired MS/MS spectra were processed using MassLynx v.3.5 (Waters, Milford, MA, USA). The MASCOT search server (version 2.0, Matrix Science, London, UK) was used for the identification of protein spots by querying the sequences of the tryptic peptide fragments. The reference database used for the identification of target proteins was NCBI (http://www.ncbi.nlm.nih.gov).
Proteinase K Treatment
After cultivation, cells were harvested by centrifugation at 6,000 rpm for 5 min at 4°C. The cells were washed twice with phosphate-buffered saline (PBS; 135 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, and 1.4 mM KH2PO4, pH 7.2) and resuspended in proteinase K (Qiagen, Valencia, CA, USA) diluted in PBS (1:100). The cell suspension was incubated for 1 h at room temperature, and the proteinase K reaction was stopped by adding phenylmethylsulfonyl fluoride to a final concentration of 10 μM.
Confocal Immunofluorescence Microscopy
After cultivation, cells were harvested by centrifugation at 6,000 rpm for 5 min at 4 °C and washed with PBS. The cells were incubated with ANTI-FLAG M2 antibody conjugated with fluorescein isothiocyanate (FITC) (Sigma-Aldrich, Co., St. Louis, MO, USA) diluted (1:500) in PBS for 2 h at 25 °C. Prior to microscopic observation, the cells were washed five times with PBS to remove unbound antibody probes. The cells were mounted on poly-l-lysine-coated microscopic glass slides and examined by confocal microscopy (Carl Zeiss, Jena, Germany). Photographs were taken with a Carl Zeiss LSM 410. The samples were excited at 488 nm, and the images were filtered by a long-pass 505-nm filter.
Measurement of Enzyme Activity
The activity of Bacillus sp. endoxylanase on E. coli cell surface was assayed by the 3,5-dinitrosalicylic acid (DNS) method [18]. After cell cultivation, cells were harvested by centrifugation at 6,000 rpm for 5 min at 4°C. The cells were washed twice with 1× PBS and mixed with a substrate solution of 2 % (w/v) beechwood xylan (Sigma-Aldrich) in 100 mM sodium phosphate buffer (pH 6.3). The reaction mixture was incubated at 37°C in a shaking incubator for 8 h. An aliquot of the reaction mixture was centrifuged at 12,000 × g for 10 min to remove the remaining cells and xylan. The supernatant of the reaction mixture was boiled for 5 min in a 3× volume of DNS solution containing (per liter) 7.5 g DNS, 14 g NaOH, 216.1 g Rochelle salt, 5.4 mL phenol, and 5.9 g Na2S2O5. The reactants were cooled to room temperature for 5 min, and the absorbance was detected by a spectrophotometer (Optizen pop, Mecasys Co., Ltd., Yuseong-gu, Daejeon, South Korea) at 550 nm.
The activity of P. fluorescens lipase on E. coli surface was assayed by a spectrophotometric method using p-nitrophenyl decanoate as described previously but with slight modifications [22]. After cell cultivation, cells were harvested by centrifugation at 6,000 rpm for 5 min at 4°C. The cells were washed twice with PBS, and then cells were added to 3 mL of a substrate solution consisting of 10 mM p-nitrophenyl decanoate (Sigma-Aldrich) in acetonitrile–ethanol–Tris–HCl (with a volume ratio of 1:4:95). The reaction mixture was incubated at 37°C for approximately 15 min until a yellow reaction product was visible, and the reaction was stopped by adding 2 μL of 0.5 M EDTA. The lipase activity was determined using the following formula: [optical density at 410 nm/(minutes of reaction) (optical density at 600 nm)] [23].
The alkaline phosphatase activity was assayed by a spectrophotometric method using p-nitrophenyl phosphate as described previously [23]. After cell cultivation, cells were harvested by centrifugation at 6,000 rpm at 4°C for 5 min. The cells were washed twice with PBS and resuspended in 0.1 mL of 1 M Tris–HCl (pH 8.0). The cell suspension was added to 1 mL of alkaline phosphatase substrate solution consisting of 2 mM p-nitrophenyl phosphate (Sigma-Aldrich) in 1 M Tris–HCl (pH 8.0). The reaction mixture was incubated at 37°C for approximately 5 min until a yellow reaction product was visible, and 0.1 mL of 1 M K2HPO4 was added to stop the reaction. The alkaline phosphatase activity was determined by the following formula: [optical density at 410 nm/(minutes of reaction) (optical density at 600 nm)] [23].
Other Analytical Methods
Protein samples were analyzed by electrophoresis on 12 % (w/v) SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel. For immunodetection of the FLAG-tag fused protein, a monoclonal ANTI-FLAG M2 antibody (Sigma-Aldrich) and goat anti-mouse immunoglobulin G (IgG)-horseradish peroxidase conjugate (Invitrogen Corp., Carlsbad, CA, USA) were used. An ECL kit (Amersham ECL Prime Western Blotting Detection Reagent, GE Healthcare) was used for signal detection.
Results
Proteome Analysis of Outer Membrane Fractions
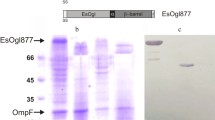
To identify a potential anchoring motif for surface display, the outer membrane proteome of E. coli XL10-Gold was first analyzed. During the exponential growth phase, cells were harvested and the OMPs were fractionated and analyzed. On 2-D gels, approximately 100 protein spots were reproducibly observed, and 41 unique proteins were identified by nano-LC–MS/MS analysis (Fig. 1) (detailed information for all identified proteins is provided in the supplementary data). On the 2-D gels, as expected, the most dominant protein was OmpA, which is the major protein in the outer membrane channels, and other well-known proteins, including AcrA, GatZ, AtpF, OmpW, FtsZ, OmpT, TolC, and FadL, were also detected at high levels (Fig. 1). In choosing a potential anchoring motif from the proteomic analysis, we considered the following criteria. First, the size of the anchoring motif was considered. For cell surface display, the fusion protein should cross two membranes (inner and outer membranes), and generally the translocation efficiency is highly dependent on protein sizes and we need to consider the total size of fusion protein (anchoring motif + passenger proteins). In this aspect, it is suggested that the smaller anchoring motifs are more profitable for surface display than the big-sized anchoring motif. Of course, big-sized anchoring motifs have also been successfully used for cell surface display [24], but in many cases, their use for cell surface display has been demonstrated with relatively small-sized proteins [2, 25, 26]. Second, we considered the levels of the proteins in the outer membrane fraction. A higher level of a protein indicates its stable localization in the outer membrane, and thus the use of that protein as an anchoring motif may ensure the stable and efficient display of target proteins on the cell surface. Among the 41 identified proteins, two proteins, OmpW and OmpX, fulfilled the cited criteria, i.e., they had relatively low molecular weight (22.9 and 18.6 kDa, respectively) and high levels on proteomic analysis (Fig. 1). Among the two candidates, OmpW may be a better candidate than OmpX because OmpW content was much higher than that of OmpX in the membrane fraction (Fig. 1). However, when we examined the cell surface display of P. fluorescens SIK W1 lipase with OmpW, all E. coli XL10-Gold cells producing OmpW-linked lipase showed severe cell lysis (data not shown). Therefore, OmpX was chosen as a final candidate for cell surface display, and we focused on OmpX in all further studies.

The representative outer membrane proteome map of E. coli XL10-Gold grown in LB medium. The positions of the molecular mass standards are indicated in kilodaltons (kDa) on the vertical axes, and the approximate pI scales are indicated on the horizontal axes
Construction of the Cell Surface Display System
An OmpX protein possesses four extracellular loops that form a β-sheet protruding from the cell surface [27], and these loops can be used for fusion with target proteins. In this work, a C-terminal truncation strategy was employed for protein display, and two different positions in the extracellular loops of OmpX were examined for fusions: the Lys122 position in the third extracellular loop (loop 3) and the Val160 position in the fourth extracellular loop (loop 4) (Fig. 2a). As model proteins for display, three important industrial enzymes with different molecular weight, namely, Bacillus sp. endoxylanase (23.3 kDa), P. fluorescens SIK W1 lipase (49.9 kDa), and E. coli alkaline phosphatase (49.4 kDa), were examined. Each coding gene fused with a FLAG tag was amplified by PCR and cloned into the C-terminus of the OmpX truncated forms (Fig. 2b). After transformation of each construct into E. coli XL10-Gold, the display efficiency and biological activities of displayed proteins were analyzed.

Construction of OmpX-fused display system. a The structure of OmpX of E. coli. Each arrow indicates the fusion position (Lys122 in the third extracellular loop and Val160 in the fourth extracellular loop) of OmpX with endoxylanase, lipase, and alkaline phosphatase. b Schematic diagram of the expression system for OmpX-fused target genes. POI protein of interest, E restriction enzymes EcoRI, X XbaI, H HindIII
Display of Bacillus sp. Endoxylanase on E. coli Cell Surface
After cultivation of E. coli cells harboring pTrcOmpXK122EX and pTrcOmpXV160EX, the presence of endoxylanase on the surface was first examined by SDS-PAGE and western blotting with whole-cell lysates and the outer membrane fraction. Both fusion constructs showed high expression levels, as well as highly efficient localization of fusion proteins on the outer membrane (Fig. 3). The localization of endoxylanase on the cell surface was further confirmed by proteinase K treatment. Whole cells displaying endoxylanase on the surface were treated with proteinase K, and the membrane fraction proteins were analyzed by western blotting. When cells were treated with proteinase K, displayed endoxylanase for both could not be detected by western blotting (Fig. 4a). Next, proteinase K-treated and untreated cells were labeled with a FITC-conjugated probe, and fluorescence signals were analyzed by a confocal microscope. When the cells were not treated with proteinase K, each cell showed a high fluorescent signal (Fig. 4b, i and iii). In contrast, none of the proteinase K-treated cells exhibited fluorescence, which indicates removal of endoxylanase on the cell surface by proteinase K (Fig. 4b, ii and iv). These results clearly indicate the successful display of endoxylanase on the cell surface.

Display of endoxylanase on E. coli cell surface. SDS-PAGE and western blotting analysis for total and membrane fractions of E. coli harboring a pTrcOmpXK122EX and b pTrcOmpXV160EX. T total proteins, M outer membrane protein fractions. Arrows indicate the OmpX-fused endoxylanases

Confirmation of localization of OmpX-fused endoxylanase by proteinase K treatment. a Western blot analysis with outer membrane fractions of E. coli harboring pTrcOmpXK122EX (lanes 1 and 3) and pTrcOmpXV160EX (lanes 2 and 4). Lanes 1 and 2, proteinase K-untreated samples; lanes 3 and 4, proteinase K-treated samples. The solid arrow and dashed arrow indicate the endoxylanase fused to Lys122 and Val160, respectively. b Confocal immunofluorescence microscope image of E. coli harboring pTrcOmpXK122EX (i, ii) and pTrcOmpXV160EX (iii, iv). i and iii proteinase K-untreated cells, ii and iv proteinase K-treated cells. Each scale bar represents 5 μm
The endoxylanase activity of whole cells was assayed by the DNS method using xylan as a substrate. The relative amount of converted reducing sugars from xylan by endoxylanase was measured by a spectrophotometer at 550 nm. Immediately after being mixed with xylan, E. coli cells displaying both OmpX-fused endoxylanase showed significant increases in reducing sugars (Fig. 5). With the two fused forms, the fusion at Lys122 of OmpX (pTrcOmpXK122EX) showed a slightly higher activity (approximately 1.4-fold) than that of the fusion at Val160 (pTrcOmpXV160EX). When cells were treated with proteinase K, all cells showed a significant decrease (more than twofold) in activities, which coincided well with previous results using confocal immunofluorescence microscopy and western blotting.

Comparison of xylan hydrolysis with E. coli XL10-Gold harboring pTrcOmpXK122EX, pTrcOmpXV160EX, and pTrcOmpX. In each clone, the gray bar represents the proteinase K-untreated samples and the black bar represents the proteinase K-treated samples
Display of P. fluorescens Lipase on the E. coli Cell Surface
To demonstrate the general use of OmpX as an anchoring motif for cell surface display, we examined the P. fluorescens lipase as another model protein that catalyzes the hydrolysis of oils and transesterification of esters and is of a much larger size (49.9 kDa) than the endoxylanase (23.3 kDa). After cultivation of cells harboring pTrcOmpXK122PL and pTrcOmpXV160PL, lipase display on the E. coli cell surface was confirmed by SDS-PAGE and western blotting. In both constructs, a high level of gene expression was obtained, and localization of lipase on the outer membrane was successfully confirmed though the display efficiencies were lower than those of endoxylanase (Fig. 6). Lipase display on the cell surface was also confirmed by proteinase K treatment. As shown in Fig. 7, when cells were treated with proteinase K, the lipase band was not detected by western blotting, and the cells did not show any fluorescent signal under the confocal microscope, indicating the localization of lipase on E. coli cell surface. The activity of lipase on the cell surface was assayed by using p-nitrophenyl decanoate as a substrate. E. coli cells displaying lipase at two fusion positions (Lys122 and Val160) of OmpX showed a high activity, while other negative controls such as E. coli host (no plasmid) or E. coli cells harboring pTrcOmpX did not show any enzymatic activity (Fig. 8). Similar to that of endoxylanase, the fusion at Lys122 of OmpX (pTrcOmpXK122PL) showed a slightly higher activity (approximately 1.3-fold) compared to the fusion at Val160 (pTrcOmpXV160PL). As expected, all proteinase K-treated cells showed relatively lower enzymatic activities, which coincided with the results of the confocal immunofluorescence microscopy. After lyophilization of cells displaying lipase, the specific activity of the lipase was also determined using the same substrate (p-nitrophenyl decanoate). E. coli producing OmpXK122-lipase showed a fourfold higher activity (3,070 ± 57 U/mg lyophilized E. coli cells) than OmpXV160-lipase (supplementary materials, Fig. S1). The lipase activity obtained with the OmpXK122 display system was also higher than that obtained with other display systems, including OprF [12] and FadL [22] display systems, by more than tenfold; the specific lipase activity was 378 ± 35 and 248 ± 33 U/mg of lyophilized cells when using OprF and FadL as anchoring motifs, respectively. These results suggest that OmpX anchoring motif (particularly C-terminal fusion at K122) provides the efficient platform for displaying functional lipase on cell surface.

Display of P. fluorescens lipase on E. coli cell surface. SDS-PAGE and western blotting analysis for total and membrane fractions of E. coli harboring a pTrcOmpXK122PL and b pTrcOmpXV160PL. Arrows indicate the OmpX-fused lipase. Lane T total proteins, lane M outer membrane protein fractions

Confirmation of localization of lipase by proteinase K treatment. a Western blot analysis with outer membrane fractions of E. coli harboring pTrcOmpXK122PL (lanes 1 and 3) and pTrcOmpXV160PL (lanes 2 and 4). Lanes 1 and 2 proteinase K-untreated samples, lanes 3 and 4 proteinase K-treated samples. The solid arrow and dashed arrow indicate the lipase fused to Lys122 and Val160, respectively. b Confocal immunofluorescence microscope image of E. coli harboring pTrcOmpXK122PL (i, ii) and pTrcOmpXV160PL (iii, iv). i and iii proteinase K-untreated cells, ii and iv proteinase K-treated cells. Each scale bar represents 5 μm

Comparison of lipase activity with E. coli XL10-Gold harboring pTrcOmpXK122PL, pTrcOmpXV160PL, pTrcOmpX, and no plasmid. In each clone, gray bar represents the proteinase K-untreated samples and black bar represents the proteinase K-treated samples
Display of Alkaline Phosphatase on the E. coli Cell Surface
Alkaline phosphatase from E. coli was also tested as an additional model protein for surface display using OmpX. Alkaline phosphatase is a hydrolase enzyme which removes phosphate groups from molecules and also has a high molecular weight (49.4 kDa). Alkaline phosphatase was fused only to the extracellular loop 3 of OmpX, as the extracellular loop 3 showed a more efficient display of passenger proteins than loop 4 in both previous experiments (endoxylanase and lipase). In this fusion format, the expression level and display of alkaline phosphatase was confirmed by SDS-PAGE and western blotting with whole-cell lysates and outer membrane fraction proteins of E. coli harboring pTrcOmpXK122AP. Alkaline phosphatase was expressed highly, and a more efficient display was shown than that of the former examples (Fig. 9a). In addition, the surface localization of the alkaline phosphatase was examined by proteinase K treatment, and all experiments clearly confirmed the successful display of alkaline phosphatase on the surface of host cells (Fig. 9b, c). The activity of the surface-displayed alkaline phosphatase was assayed using p-nitrophenyl phosphate as a substrate. Alkaline phosphatase displayed on E. coli cell surface showed a much higher activity than the other negative control cells, including E. coli XL10-Gold (no plasmid) and E. coli harboring pTrcOmpX (Fig. 10). When the cells were treated with proteinase K, much lower activity (approximately 50 % of the untreated cell) could be obtained. From those results, we could conclude that alkaline phosphatase was indeed highly active on the surface of host cells.

Display of alkaline phosphatase on E. coli cell surface. a SDS-PAGE and western blotting analysis for total (T) and membrane (M) fractions of E. coli harboring pTrcOmpXK122AP. bWestern blotting analysis with the membrane fraction of E. coli harboring pTrcOmpXK122AP without proteinase K treatment (lane 1) or with proteinase K treatment (lane 2). c Confocal immunofluorescence microscope image of E. coli harboring pTrcOmpXK122AP. i proteinase K-untreated, ii proteinase K-treated. Each scale bar represents 5 μm. Arrows indicate the OmpX-fused alkaline phosphatase

Comparison of alkaline phosphatase activity with E. coli XL10-Gold harboring pTrcOmpXK122AP, pTrcOmpX, and no plasmid. In each clone, gray bar represents the proteinase K-untreated samples and black bar represents the proteinase K-treated samples
Discussion
In E. coli, cell surface display entails the translocation of passenger proteins fused to carrier proteins (anchoring motifs) and the presentation of recombinant proteins or peptides on the host surface. For efficient translocation and stable immobilization on the surface, the choice of anchoring motif is the most important issue in the development of a cell surface display system [3]. Here, we developed a new cell surface display system using the E. coli outer membrane protein, OmpX, as an anchoring motif. To select this potential anchoring motif for cell surface display, we performed a proteome analysis of outer membrane fractions. Among the 41 identified proteins, two proteins, OmpX and OmpW, were chosen initially, both of which have relatively low molecular weight and high level in the proteome, and this may allow the stable anchoring of passenger proteins with high levels. In addition, both proteins are channel proteins with β-barrel domains [27, 28], and the exposed loops of the β-barrel structure can be utilized as suitable anchoring sites to display recombinant proteins. However, fusion with OmpW resulted in severe lysis of cells and thus could not be used for cell surface display. Even though OmpW is more abundant than OmpX on the membrane, most OmpW is localized to the poles of rod-shaped E. coli cells and is not randomly distributed on the entire surface, in contrast to OmpX and other OMPs (OmpA, OmpC, etc.) [29–31]. Targeting of OmpW fused with passenger proteins may have decreased the integrity of the outer membrane, which can subsequently cause cell lysis. Therefore, OmpX was chosen as an anchoring motif for cell surface display, and the usefulness of OmpX for cell surface display was successfully demonstrated with three different enzymes.
The use of OmpX as an anchoring motif for cell surface display was previously demonstrated. The Daugherty group (University of San Diego, USA) developed a bacterial display system using circularly permuted OmpX (CPX), which presents both N- and C-termini on the external cell surface [17]. In addition, the CPX system was also used for high-throughput library screening, in which high-affinity peptide ligands were successfully isolated [17, 32]. However, in those experiments, only small peptides (approximately 0.8–1.6 kDa) were used for display, but the display of much larger proteins has not yet been demonstrated. In the present work, we successfully demonstrated the surface display of three different enzymes, of which the molecular weights were relatively high (20–50 kDa) using C-terminal fusion to OmpX; to the best of our knowledge, this is the first report on the use of OmpX for the surface display of large proteins. Although there are several reports about the display of much larger proteins such as P450 (117 kDa) and alpha-amylase (70 kDa) using various anchoring motifs [33, 34], the most current display systems are primarily restricted to display of peptides and small proteins [3, 35]. As demonstrated here and previously [17, 32], an OmpX-fused display system can accommodate various proteins with a broader range of sizes (from a small peptide to a large protein) with high efficiency.
The efficiency and stability of the displayed protein with an anchoring motif are closely related to the loops of the protein fusion [12, 36]; therefore, the proper choice of loops for the protein fusion is an important aspect in protein display. OmpX has four extracellular loops, and in this work two different positions (Lys122 and Val160 position) in the third and fourth extracellular loops of OmpX were examined for display. In both examples (endoxylanase and lipase), fusion to loop 3 (Lys122) showed slightly higher activities of displayed enzymes than fusion to loop 4 (Val160) did, indicating a more efficient exposure of enzymes to the cell surface with fusion to loop 3. These results demonstrate that a relatively short anchoring motif (truncation in loop 3) is enough to anchor to the outer membrane with high stability and may also be more suitable for the efficient translocation of fusion proteins to the cell surface. Regarding the amino acid residues at the fusion position, two residues (Lys122 and Val160) were used for fusion, and other residues were not examined in the present work. Previously, Lee et al. [12] reported that, using the Pseudomonas aeruginosa OprF anchoring motif, four different positions (Lys164, Ala196, Val188, and Arg213) in the same fifth extracellular loop of OprF were tested for protein display. They showed that Ala196 and Arg213 were not suitable for the display of large proteins, possibly due to membrane instability and cell growth inhibition. In contrast, Lys164 and Val188 residue allowed for the efficient display of large proteins with effective cell growth and suggested this as a potential fusion point in the OprF anchoring motif. In the present work, no severe growth limitations were observed with both fusion formats. It is not certain that both Lys122 and Val160 residues are the optimal positions for protein fusion, but we can say that both positions are suitable for protein fusion in an OmpX display system.
To improve the display efficiency of OmpX-fused proteins on cell surface, several strategies can be considered. First, the display efficiency of protein is highly dependent on the gene expression level and the choice of promoter is one of the important factors in gene expression levels. The use of strong promoters (T7, arabinose promoter, etc.) may increase the gene expression levels, but it can cause a high accumulation of insoluble aggregates in the cytoplasm. In contrast, the use of a weak promoter can increase protein solubility and display efficiency although the expression level is not high. In this work, trc promoter, which is known to have a moderate activity, was used for all expressions, but other tunable or mild constitutive promoters can be a good choice. In this aspect, we examined OmpX own promoter which has a weak and constitutive activity. However, compared to trc promoter, the expression level under the OmpX own promoter was too low, and any detectable amount of OmpX-fused protein (endoxylanase) was not obtained (Supporting material, Fig. S2), but we think that it is worth to examine other promoters and it needs to be optimized. Second, the use of ompX gene knock-out mutant strain can be considered to enhance the efficiency of protein display. The protein amounts of which a cell can accommodate on the surface are highly limited, and so the deletion of ompX gene in the host strain may prevent the possible competition between endogenous OmpX and OmpX-fused proteins in the localization on the cell surface; hence, the display efficiency may be improved. OmpX is also a non-lethal protein, so a knock-out mutant may have no problem in cell growth and higher gene expression can be achieved.
In conclusion, we isolated a potential anchoring motif (OmpX) based on a proteomic analysis of the OMP fraction. Furthermore, the potential usefulness of the anchoring motif for cell surface display was also successfully demonstrated with three important industrial enzymes (endoxylanase, lipase, and alkaline phosphatase). As demonstrated with lipase, C-terminal fusion strategy using the OmpX as an anchoring motif (particularly fusion at Lys122) provides a more stable and efficient platform for displaying functional lipase on the cell surface than other anchoring motifs. Due to high selectivity, stability, and easy purification/separation, the cited enzymes displayed on the cell surface can be used as a cost-effective system in various bioconversion processes, and the present system based on OmpX can be also used for other applications such as protein engineering screening and vaccine development. Last decade, there was big progress in OMICs study, and their applications to the development of new biological systems increase more and more [37]. To the best of our knowledge, this is the first report on the application of proteome analysis for cell surface display system, and we believe that our strategy of isolating a potential anchoring motif based on a proteomic analysis of host cells should be useful for the development of new display systems in other bacterial systems.
References
Jose, J. (2006). Autodisplay: efficient bacterial surface display of recombinant proteins. Applied Microbiology and Biotechnology, 69(6), 607–614.
Lee, S. Y., Choi, J. H., & Xu, Z. (2003). Microbial cell-surface display. Trends in Biotechnology, 21(1), 45–52.
Bloois, E. V., Winter, R. T., Kolmar, H., & Fraaije, M. W. (2011). Decorating microbes: surface display of proteins on Escherichia coli. Trends in Biotechnology, 29(2), 79–86.
Boder, E. T., & Wittrup, K. D. (1997). Yeast surface display for screening combinatorial polypeptide libraries. Nature Biotechnology, 15(6), 553–557.
Knecht, L. D., Pasini, P., & Daunert, S. (2011). Bacterial spores as platforms for bioanalytical and biomedical applications. Anaytical and Bioanalytical Chemistry, 400(4), 977–989.
Binder, U., Matschiner, G., Theobald, I., & Skerra, A. (2010). High-throughput sorting of an Anticalin library via EspP-mediated functional display on the Escherichia coli cell surface. Journal of Molecular Biology, 400(4), 783–802.
Samuelson, P., Gunneriusson, E., Nygren, P. A., & Stahl, S. (2002). Display of proteins on bacteria. Journal of Bacteriology, 96(2), 129–154.
Silhavy, T. J., Kahne, D., & Walker, S. (2010). The bacterial cell envelope. Cold Spring Harbor Perspectives in Biology, 2(5), a000414.
Valls, M., Gonzalez-Duarte, R., Atrian, S., & Lorenzo, V. D. (1998). Bioaccumulation of heavy metals with protein fusions of metallothionein to bacterial OMPs. Biochimie, 80(10), 855–861.
Park, T. J., Lee, S. Y., Park, J. P., & Lee, S. J. (2011). Quantitative studies of carbohydrate–protein interaction using functionalized bacterial spores in solution and on chips. Biotechnology and Bioprocess Engineering, 16(1), 190–195.
Fairman, J. W., Noinaj, N., & Buchanan, S. K. (2011). The structural biology of beta-barrel membrane proteins: a summary of recent reports. Current Opinion in Structural Biology, 21(4), 523–531.
Lee, S. H., Choi, J. I., Han, M. J., Choi, J. H., & Lee, S. Y. (2005). Display of lipase on the cell surface of Escherichia coli using OprF as an anchor and its application to enantioselective resolution in organic solvent. Biotechnology and Bioengineering, 90(2), 223–230.
Yang, Z., Liu, Q., Wang, Q., & Zhang, Y. (2008). Novel bacterial surface display systems based on outer membrane anchoring elements from the marine bacterium Vibrio anguillarum. Applied and Environmental Microbiology, 74(14), 4359–4365.
Molloy, M. P., Herbert, B. R., Slade, M. B., Rabilloud, T., Nouwens, A. S., Williams, K. L., & Gooley, A. A. (2000). Proteomic analysis of the Escherichia coli outer membrane. European Journal of Biochemistry, 267(10), 2871–2881.
Han, M. J., Lee, S. Y., Koh, S. T., Noh, S. G., & Han, W. H. (2010). Biotechnological applications of microbial proteomes. Journal of Biotechnology, 145(4), 341–349.
Chao, T. C., & Hansmeier, N. (2012). The current state of microbial proteomics: where we are and where we want to go. Proteomics, 12(4–5), 638–650.
Rice, J. J., Schohn, A., Bessette, P. H., Boulware, K. T., & Daugherty, P. S. (2006). Bacterial display using circularly permuted outer membrane protein OmpX yields high affinity peptide ligands. Protein Science, 15(4), 825–836.
Jeong, K. J., Lee, P. C., Park, I. Y., Kim, M. S., & Kim, S. C. (1998). Molecular cloning and characterization of an endoxylanase gene of Bacillus sp. in Escherichia coli. Enzyme and Microbial Technology, 22(7), 599–605.
Ahn, J. H., Pan, J. G., & Rhee, J. S. (1999). Identification of the tliDEF ABC transporter specific for lipase in Pseudomonas fluorescens SIK W1. Journal of Bacteriology, 181(6), 1847–1852.
Sambrook, J., & Russell, D. (2001). Molecular cloning: a laboratory manual (3rd ed.). Cold Spring Harbor: Cold Spring Harbor Laboratory.
Han, M. J., Lee, J. W., & Lee, S. Y. (2005). Enhanced proteome profiling by inhibiting proteolysis with small heat shock proteins. Journal of Proteome Research, 4(6), 2429–2434.
Lee, S. H., Choi, J. I., Park, S. J., Lee, S. Y., & Park, B. C. (2004). Display of bacterial lipase on the Escherichia coli cell surface by using FadL as an anchoring motif and use of the enzyme in enantioselective biocatalysis. Applied and Environmental Microbiology, 70(9), 5074–5080.
Kriakov, J., Lee, S. H., & Jacobs, W. R. (2003). Identification of a regulated alkaline phosphatase, a cell surface-associated lipoprotein, in Mycobacterium smegmatis. Journal of Bacteriology, 185(16), 4983–4991.
Wentzel, A., Christmann, A., Adams, T., & Kolmar, H. (2001). Display of passenger proteins on the surface of Escherichia coli K-12 by the enterohemorrhagic E. coli intimin EaeA. Journal of Bacteriology, 183(24), 7273–7284.
Sousa, C., Kotrba, P., Ruml, T., Cebolla, A., & Lorenzo, V. D. (1998). Metalloadsorption by Escherichia coli cells displaying yeast and mammalian metallothioneins anchored to the outer membrane protein LamB. Journal of Bacteriology, 180(9), 2280–2284.
Nicolay, T., Lemoine, L., Lievens, E., Balzarini, S., Vanderleyden, J., & Spaepen, S. (2012). Probing the applicability of autotransporter based surface display with the EstA autotransporter of Pseudomonas stutzeri A15. Microbial Cell Factory, 11, 158.
Vogt, J., & Schulz, G. E. (1999). The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence. Structure, 7(10), 1301–1309.
Hong, H., Patel, D. R., Tamm, L. K., & Berg, B. V. D. (2006). The outer membrane protein OmpW forms an eight-stranded beta-barrel with a hydrophobic channel. Journal of Biological Chemistry, 281(11), 7568–7577.
Lai, E. M., Nair, U., Phadke, N. D., & Maddock, J. R. (2004). Proteomic screening and identification of differentially distributed membrane proteins in Escherichia coli. Molecular Microbiology, 52(4), 1029–1044.
Verhoeven, G. S., Alexeeva, S., Dogterom, M., & Blaauwen, T. D. (2009). Differential bacterial surface display of peptides by the transmembrane domain of OmpA. PLoS One, 4(8), e6739.
Xu, Z., & Lee, S. Y. (1999). Display of polyhistidine peptides on the Escherichia coli cell surface by using outer membrane protein C as an anchoring motif. Applied and Environmental Microbiology, 65(11), 5142–5147.
Rice, J. J., & Daugherty, P. S. (2008). Directed evolution of a biterminal bacterial display scaffold enhances the display of diverse peptides. Protein Engineering, Design & Selection, 21(7), 435–442.
Yim, S. K., Kim, D. H., Jung, H. C., Pan, J. G., Kang, H. S., Ahn, T., & Yun, C. H. (2010). Surface display of heme- and diflavin-containing cytochrome P450 BM3 in Escherichia coli: a whole cell biocatalyst for oxidation. Journal of Microbiology and Biotechnology, 20(4), 712–717.
Narita, J., Okano, K., Tateno, T., Tanino, T., Sewaki, T., Sung, M. H., Fukuda, H., & Kondo, A. (2006). Display of active enzymes on the cell surface of Escherichia coli using PgsA anchor protein and their application to bioconversion. Applied Microbiology and Biotechnology, 70(5), 564–572.
Rutherford, N., & Mourez, M. (2006). Surface display of proteins by Gram-negative bacterial autotransporters. Microbial Cell Factories, 5, 22.
Lang, H., Maki, M., Rantakari, A., & Korhonen, T. K. (2000). Characterization of adhesive epitopes with the OmpS display system. European Journal of Biochemistry, 267(1), 163–170.
Baek, J. H., Han, M. J., Lee, S. H., & Lee, S. Y. (2010). Enhanced display of lipase on the Escherichia coli cell surface based on transcriptome analysis. Applied and Environmental Microbiology, 76(3), 971–973.
Acknowledgments
This study was supported by the Advanced Biomass R and D Center of Global Frontier Project (Grant No. ABC-2011-0031363) and Converging Research Center Program (Grant Nos. 2012 K001386 and 2009–0093652) through the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology, and by Brain Korea 21 project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Sung Sun Yim and Seul Ji An contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 290 kb)
Rights and permissions
About this article
Cite this article
Yim, S.S., An, S.J., Han, MJ. et al. Isolation of a Potential Anchoring Motif Based on Proteome Analysis of Escherichia coli and Its Use for Cell Surface Display. Appl Biochem Biotechnol 170, 787–804 (2013). https://doi.org/10.1007/s12010-013-0236-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-013-0236-9