
Abstract
Purpose of Review
This review presents an overview of the known neurocritical care complications of severe acute respiratory virus 2 (SARS-CoV-2). We present readers with a review of the literature of severe neurologic complications of SARS-CoV-2 and cases from our institution to illustrate these conditions.
Recent Findings
Neurologic manifestations are being increasingly recognized in the literature. Some patients can have severe neurologic manifestations, though the true prevalence is unknown.
Summary
Severe neurologic complications of COVID-19 include large vessel occlusion ischemic stroke, intracranial hemorrhage, encephalitis, myelitis, Guillain-Barre syndrome, status epilepticus, posterior reversible encephalopathy syndrome, and hypoxic-ischemic encephalopathy. These conditions can manifest in COVID-19 patients even in the absence of risk factors and must be promptly identified as they can have a high mortality if left untreated.
Similar content being viewed by others

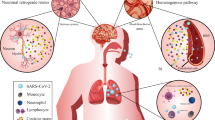
Introduction
In December 2019, patients in Wuhan, China, were diagnosed with a serious pneumonia subsequently found to be caused by the novel severe acute respiratory syndrome coronavirus (SARS-CoV-2). Between 36 and 69% patients with coronavirus experience neurologic manifestations, which can range from anosmia, headache, dizziness, and myopathy to encephalopathy, seizure, and acute cerebrovascular disease and are more common in patients who experience severe illness [1, 2]. Neurologic manifestations may be difficult to recognize in patients who are intubated and being treated with sedatives and paralytics. Several mechanisms of neuroinvasion have been suggested including transsynaptic spread across infected neurons such as the olfactory nerve and invasion across the vascular endothelium via leukocyte migration across the blood brain barrier. Secondary issues include evidence of hypercoagulability and thrombosis as well as consequences of a dramatic inflammatory response.
Large Vessel Occlusion Ischemic Stroke
Case 1: COVID-19 Presenting With Large Vessel Occlusion Ischemic Stroke
A 39-year-old man with a history of hypertension and hyperlipidemia presented with the sudden onset of left-sided weakness. The NIH stroke scale (NIHSS) was 18. Neurological examination was notable for right gaze preference, left face weakness, left-sided neglect, and left hemiplegia. CT angiogram of the head and neck showed right posterior cerebral artery occlusion and bilateral lung opacities. He underwent thrombectomy with TICI 2C recanalization complicated by intra-procedure atrial fibrillation which was controlled with amiodarone. Post-procedure his NIHSS improved to 4 with the improved left arm and leg strength and he was antigravity.
Chest X-ray on admission showed bilateral airspace opacities and nasopharyngeal PCR was positive for SARS-CoV-2 (Fig. 1a). MRI of the brain showed a large right posterior cerebral artery (PCA) territory stroke (Fig. 1b). Stroke work-up revealed a hemoglobin A1C of 11% and an ejection fraction of 33%. Inflammatory markers were elevated on admission with a ferritin of 1564 ng/mL, C-reactive protein (CRP) of 91.4 mg/L. He suffered acute hypoxic respiratory failure and required endotracheal intubation and transfer to the neurocritical care unit. A repeat CT scan a week later demonstrated the evolving hypodensity within the right PCA territory associated with sulcal effacement and mass effect on the temporal horn of the right lateral ventricle (Fig. 1c). His course was complicated by renal failure requiring initiation of continuous hemodialysis, and worsening hypoxemia and septic shock. He developed refractory metabolic acidosis and hyperkalemia and was unable to continue CVVHD. The patient expired on the eleventh day of his admission.

Acute ischemic stroke. a Chest X-ray on admission with bilateral airspace opacities; b Isotropic MRI sequence demonstrating a right posterior cerebral artery ischemic stroke; c CT head performed 1 week following MRI of the brain demonstrating the evolving hypodensity within the right PCA territory associated with sulcal effacement and mass effect on the temporal horn of the right lateral ventricle.
The relationship between COVID-19 and acute ischemic stroke (AIS) has been controversial, with some centers reporting higher rates of large vessel occlusion (LVO) during the COVID-19 outbreak in New York City while others reported no change in numbers [3,4,5]. Between 0.9 and 5.7% of patients with COVID-19 are diagnosed with AIS regardless of the presence of concurrent respiratory symptoms [1, 5,6,7,8,9]. Patients with AIS associated with COVID-19 have a median age in their 6th or 7th decade and have cardiovascular risk factors, although young healthy patients have also been diagnosed [3, 5, 6, 10]. AIS is usually diagnosed between 10 and 16 days after the onset of COVID-19 symptoms, and more than one-third of strokes occur in patients with severe infection and those requiring mechanical ventilation [3,4,5, 9]. Importantly, in young patients without cardiovascular risk factors and mild respiratory symptoms, the incidence of LVO is much higher than would be expected [3, 10].
Patients with coronavirus are at a high risk of thrombotic events despite the use of pharmacologic prophylaxis [1, 4]. Previous studies have shown that sepsis and viral infection are risk factors for ischemic and hemorrhagic stroke [11, 12]. Approximately 8.5% of stroke patients have had an episode of sepsis in the preceding year and the risk of a stroke remains 2.5 times higher the following year [11]. The risk appears to be even higher for COVID-19 patients who have at least 7.5 times higher risk for stroke compared to patients with influenza [4]. Several mechanisms for this hypercoagulable state have been proposed including the severe inflammatory state due to the release of activated neutrophils and proinflammatory cytokines, disseminated intravascular coagulation (DIC), or coagulopathy due to antiphospholipid antibodies (which can transiently rise in critical illness) [13,14,15,16]. Blood thromboelastography (TEG) studies suggest a state of hypercoagulability distinct from DIC [13].
A recent case series suggests that alteplase (tPA) was not associated with symptomatic complications and most patients have clinical neurologic improvement [17]. However, some centers have reported that COVID-19 patients with LVO have a high mortality despite treatment with tPA and mechanical thrombectomy, though their mortality may be driven primarily by the severity of their COVID-19 disease [5]. The hypercapnia and hypoxia associated with acute respiratory distress syndrome (ARDS) can cause cerebral vasodilation and may accelerate the development of malignant cerebral edema. Early reports indicate that COVID-19 patients with malignant cerebral edema secondary to a large hemispheric AIS can have positive outcomes with decompressive hemicraniectomy [18]. Based on the hypercoagulable profile of patients with coronavirus, hospitals are creating individual anticoagulation protocols to use treatment dose anticoagulation for the prevention of thrombotic events in these patients [19, 20]. Low molecular weight heparin (LMWH) (40–60 mg enoxaparin/day) for 7 days was associated with a better 28-day mortality in patients with severe COVID-19 coagulopathy [8].
Intracranial Hemorrhage
Case 2: Massive Intracranial Hemorrhage in a Patient With COVID-19
A 77-year-old woman with a history of diabetes and hypertension presented to the emergency department with 5 days of productive cough and shortness of breath. Her initial oxygen saturation was 76%. Chest X-ray showed bilateral infiltrates. Admission serum studies showed WBC 15.7 ×10E3/uL, LDH 651 u/L, CRP 272 mg/L, ESR 73 mm/HR, d-dimer >20 μg/mL, troponin of 2.3 ng/mL, and creatinine of 5 mg/dL. EKG did not show ischemic changes. A nasopharyngeal swab confirmed COVID-19 infection. She was started on a heparin infusion per hospital protocol for empiric anticoagulation due to elevated d-dimer.
On the third day of her admission, she was found unresponsive with a right gaze preference and sluggish pupils. Heparin drip was immediately stopped. A CT scan revealed a large right frontal acute intraparenchymal hemorrhage with surrounding edema and 14.2 mm midline shift. There was right uncal herniation and reduced mammillary pontine distance consistent with downward central herniation. She was given protamine for reversal of heparin infusion. She was taken for emergent decompressive hemicraniectomy but post-operative exam was consistent with brain death. Post-operative head CT showed global cerebral edema and diffuse ischemic injury and she was declared dead by neurologic criteria.
Compared to ischemic stroke, intracranial hemorrhage (ICH) is a rare complication of COVID-19. In a case series from Wuhan, China, only 1 out of 88 COVID-19 patients had an ICH, though no patients were on anticoagulation [21]. In an Italian case series where anticoagulation status was not commented upon, 5 out of 26 patients with COVID-19 and neurological manifestations had an ICH [22]. From the onset of COVID symptoms, the average time to ICH is 32 days [23].
There are several hypotheses as to why COVID-19 patients can develop ICH even in the absence of vascular abnormalities, including mechanisms related to the infection itself. One hypothesis is reduction in ACE receptor availability. Viral particles enter hosts via the angiotensin-converting enzyme 2 (ACE-2) receptor in the human cell surface, which has reduced expression in patients with hypertension [24, 25]. As SARS-CoV-2 occupies human ACE-2 receptors, their expression is further reduced, which has been suggested to elevate blood pressure and increase the risk of ICH [24, 25]. Intraparenchymal and subdural hemorrhage has also been found in a patient with COVID-19 related meningoencephalitis whose cerebrospinal fluid (CSF) PCR was positive for SARS-CoV-2, suggesting direct infection may cause parenchymal hemorrhage [26].
Another proposed mechanism is the coagulopathy associated with the disease. Patients with COVID-19 can develop coagulopathy with a pattern distinct from DIC with prolonged prothrombin time (PT), increased d-dimer, and decreased platelet count [27,28,29]. COVID-19 infection may also result in thrombocytopenia through the formation of autoantibodies, immune complexes or direct infection of hematopoietic stem cells, megakaryocytes, and platelets [30]. Immune thrombocytopenia purpura may also be triggered by COVID-19 [31].
Empiric anticoagulation has been shown to lower the 28-day mortality among patients with severe COVID-19 or associated coagulopathy [8]. Thus, medical centers have created anticoagulation protocols most often based on elevated d-dimers [19, 20, 32]. However, therapeutic anticoagulation places patients at an increased risk of ICH [33]. A recent report of 33 COVID-19 positive patients with ICH found that 66.7% of them were receiving therapeutic dose anticoagulation and 9% were receiving prophylactic dosing. Of these patients, 25% had punctate hemorrhages, 60.7% had small-moderate size hemorrhages, 14.3% had a large hemorrhage without evidence of herniation, and 15.2% had parenchymal hemorrhages with mass effect and herniation [34]. While most of the hemorrhages without mass effect were thought to be likely a result of hemorrhagic transformation of ischemic stroke, the hemorrhages with evidence of mass effect and herniation were thought to be likely primary hemorrhages. In a small case series, the patients whose hemorrhage caused mass effect had 100% mortality [34]. In addition, patients undergoing treatment with extracorporeal membrane oxygenation for COVID-19-associated severe acute respiratory distress syndrome (ARDS) with refractory hypoxia are at increased risk of ICH as therapeutic anticoagulation is required to maintain therapy [35]. Thus, while therapeutic anticoagulation may benefit some patients, there should be a low threshold to acquire head imaging in these patients.
Guillain-Barre Syndrome
Case 3: Autoimmune Demyelinating Polyneuropathy Following COVID-19 Infection
A 67-year-old woman presented to the emergency department with back pain, anorexia, nausea, and dry cough. At that time, she had a COVID-19 nasopharyngeal swab done which was positive. She was discharged home with a lidocaine patch for back pain but developed bilateral lower extremity weakness and numbness as well saddle anesthesia, followed later by involvement of the upper extremities, prompting a return to the emergency room.
Her physical examination revealed weakness in all extremities, depressed biceps, triceps, and brachioradialis reflexes, and areflexia of the patellar and Achilles tendons. She reported pins and needles sensation from the knee down bilaterally. Lumbar puncture (LP) revealed cytoalbuminologic dissociation with WBC 0/uL and protein of 222 mg/dL. She was hyponatremic on admission to 119 mEq/L. Electromyography and nerve condiction studies (EMG and NCS) were deferred to reduce clinician exposure. She was transferred to the neurocritical care unit for initiation of plasma exchange (PLEX) for progression of weakness now involving the left side of the face. PLEX was chosen over intravenous immunoglobulin (IVIG) given the potential for thrombotic complications with both COVID-19 infection and IVIG.
Her course was complicated by PEA arrest in the setting of hypoxia with return of spontaneous circulation after 4 min. Tracheostomy was placed due to inability to wean from the ventilator. Her neurological exam improved after 5 sessions of PLEX. She was transferred to a rapid weaning unit where she was weaned off the ventilator and her tracheostomy was decannulated. Her neurologic exam improved, but she had residual lower extremity weakness and was discharged to acute rehabilitation [36].
Guillain-Barre syndrome (GBS) is a monophasic immune-related polyradiculopathy usually provoked by an antecedent infection. The infection triggers an immune response which cross-reacts with proteins of the Schwann cell surface membrane or myelin, resulting in demyelination. In the acute motor axonal neuropathy variant, the membranes of the axon are the primary target for injury. The etiology of GBS in COVID-19 is unknown, but is hypothesized to occur as a consequence of molecular mimicry due to similarities between the ACE-2 receptor and various glycoproteins and gangliosides on cell surfaces [37]. Another theory is that nerve damage occurs due to T-cell activation and release of inflammatory mediators due to the heightened immune response.
The exact incidence of acute sensorimotor polyneuropathy in COVID-19 is unknown, but several cases have been observed in patients with SARS-CoV-2. Although GBS has been reported in patients who only experienced anosmia and ageusia, most cases are reported in patients with fever and respiratory or gastrointestinal symptoms [38, 39]. The onset of GBS symptoms has been about 5–10 days after COVID-19 symptoms onset [38, 40,41,42,43,44]. The most common subtype is AIDP, followed by AMSAN, MFS, and AMAN [45].
Diagnostic studies are similar to those of GBS not associated to COVID-19. About 75% of patients have cytoalbuminologic dissociation in the CSF and antiganglioside antibodies and SARS-CoV-2 have not been detected [39,40,41,42,43,44,45,46]. EMG and NCS most commonly show demyelination, but there has been evidence of axonal damage in some cases [38,39,40,41,42,43,44, 46].
In most cases, patients improved after one course of IVIG. However, some patients have required a second course of IVIG or PLEX [39,40,41,42,43,44, 47]. The additional risk of thrombotic complications associated with IVIG must be considered, though no IVIG-related thrombotic complications have been reported in COVID-19 patients.
Encephalitis and Myelitis
There is evidence that coronaviruses can invade the central nervous system [48]. Direct invasion has been suspected in numerous cases, but only proven by histopathology, imaging, or CSF in a small fraction of cases.
The pathophysiology of COVID-19 encephalitis is unknown, but it may be a consequence of direct invasion of the virus into the neural tissue, as a consequence of the hyperinflammatory state, or molecular mimicry [49,50,51]. Brain autopsies of patients with SARS-CoV revealed tissue edema, neuronal degeneration, and SARS-CoV viral particles [52]. Similarly, low levels of SARS-CoV-2 have been found in brain autopsies of COVID-19 patients [53]. The hyperinflammatory state and massive cytokine release induced by SARS-CoV-2 could alter the permeability of the blood-brain barrier, which could result in activation of the neuroinflammatory cascade [54]. SARS-CoV-2 may also induce production of antibodies against neural or glial cells in a mechanism similar to that demonstrated in HSV-1, Ebstein-Barr virus, and Japanese encephalitis [50, 55].
Presenting symptoms of COVID-19 encephalitis vary and can include disorientation, dizziness, dysarthria, refractory seizures, and psychosis [51, 56,57,58,59]. First symptoms of encephalitis are reported to be between 3 and 11 days after onset of respiratory symptoms, which in many cases have been mild [51, 57,58,59,60]. Work-up should include a LP which can show a viral inflammatory pattern with elevated opening pressure, lymphocytic pleocytosis, and elevated protein [51, 59,60,61]. SARS-CoV-2 PCR has been tested in the CSF with inconsistent results, hypothesized to be because the viral particles are mainly cell-bound or because viral concentration in the CSF is below the level of detection [51, 58, 60, 62]. Other serum inflammatory markers such as IL-6, IL-8, TNF-a, and B2 microglobulin have been detected at increased levels which normalize as the course of the disease improves [51]. The most common MRI findings are cortical signal abnormalities on FLAIR images, cortical diffusion restriction, and leptomeningeal enhancement [63]. Acute necrotizing encephalitis has also been reported [57]. Most reported cases respond to intravenous steroids and IVIG [51, 58].
Status Epilepticus
At least 0.5% of patients have seizures as a neurological manifestation of COVID-19 infection [21]. Critical illness, acute neurologic insult, hypoxia, metabolic derangements, or organ failure can provoke seizures in patients with COVID-19 [64,65,66,67]. Seizures may also occur as a result of cortical irritation due to the interaction of SARS-CoV-2 with endothelial receptors and glial tissue receptors [68]. Seizures and status epilepticus have been reported in patients without prior history of seizures and with varying degrees of respiratory symptoms [65, 66, 69,70,71].
Detection of clinical seizures in COVID-19 patients may be difficult due to the use of paralytic agents and isolation measures. 89.3% of seizures in critically ill patients have no clinical signs [72]. Patients with COVID-19 can have rhythmic movements, acute encephalopathy, or slow awakening after sedation which can be concerning for seizures or nonconvulsive status epilepticus (NCSE). Some patients may experience periods of impaired consciousness, while others may have focal motor seizures that progress to NCSE [69, 70].
The most common indications for EEG in critically-ill patients with COVID-19 are encephalopathy, motor seizure-like events, poor recovery of consciousness after discontinuation of sedation, and gaze deviation. Use of EEG, however, is limited by the need to minimize contact to protect healthcare workers, prone-positioning, and contamination risk to equipment [66]. Thirty-eight to 40.9% of COVID-19 positive patients who had an EEG for concern of encephalopathy or seizure-like event had epileptiform discharges with frontal spikes as the predominant pattern [66, 67]. The presence of epileptiform discharges was not associated with the presence of renal or hepatic failure [66].
When available, some institutions opt for a rapid response 8-channel EEG instead of routing or continuous EEG to reduce the time of patient contact [73]. As one considers anti-epileptic therapy in these patients, special consideration must be given to concomitant circumstances that may affect antiseizure medication pharmacokinetics, such as renal or hepatic failure or extra corporeal membrane oxygenation [67]. While there currently is no data on the optimal duration of anti-epileptic therapy in COVID-19 patients with new onset seizures or status epilepticus, one may consider continuing anti-epileptic therapy throughout the course of the hospitalization with possible discontinuation as an outpatient depending on the clinical course.
Posterior Reversible Encephalopathy Syndrome
Posterior reversible encephalopathy syndrome (PRES) can develop in COVID-19 patients even without dramatic fluctuations in blood pressure [74,75,76,77,78]. The pathophysiology of PRES remains controversial, but it is hypothesized to occur due to impairments in cerebral autoregulation or endothelial dysfunction [79]. Some studies suggest that SARS-CoV-2 and its related systemic inflammatory response can induce endothelial dysfunction, disrupting the integrity of the blood brain barrier making it highly permeable and susceptible to the development of cerebral edema [80]. In addition, hypoxia in patients with severe respiratory symptoms may increase anaerobic metabolism in the brain which can lead to cerebral vasodilation and development of interstitial edema [81].
The presenting symptoms can vary and include altered mental status, agitation, blurry vision, cortical blindness, seizures refractory to multiple antiseizure medications, and status epilepticus [74, 75, 77, 78, 82, 83]. PRES has been reported both on presentation and in the hospital when infectious symptoms have begun to improve [74,75,76,77,78]. MRI shows the typical PRES findings of symmetric vasogenic edema with parieto-occipital predominance and can at times show scattered foci of hemorrhage [74,75,76,77,78, 82]. EEG is advised if there is any concern for concurrent seizures. In most cases, the symptoms and imaging findings normalize after strict blood pressure control [74, 76,77,78, 82, 83].
Hypoxic-Ischemic Encephalopathy
Case 4: Hypoxic-Ischemic Encephalopathy in a COVID-19 Patient
A 59-year-old woman with a history of prediabetes presented to the emergency department with respiratory distress and acute hypoxic respiratory failure. Her oxygen saturation on presentation was 40% which improved to 65% with noninvasive supplemental oxygen. She was emergently intubated and transferred to the critical care unit. On hospital day 4, she was noted to have a persistent upward gaze deviation which did not resolve with benzodiazepines. Rapid response 8-channel EEG showed occasional left temporal lateralized periodic discharges (LPDs) (Fig. 2a). She was started on levetiracetam for presumed seizure. Despite this, mental status continued to be poor with the patient only opening her eyes, but without tracking or purposeful movements.

Hypoxic-ischemic encephalopathy. a Rapid response limited 8-channel EEG showing LPDs most prominent in the T5-O1 channel (4–5); b and c MRI with confluent areas of predominantly subcortical FLAIR hyperintensity (b) and restricted diffusion (c).
MRI of the brain showed multiple patchy scattered areas of FLAIR hyperintensity and restricted diffusion involving the cortical gray matter of the cerebral hemispheres, as well as areas of confluent involvement of the cerebral white matter including centrum semiovale and corona radiata, bilaterally. The areas of restricted diffusion did not correspond to any particular vascular territory and also involved the caudate heads and putamen bilaterally. There was corresponding mild gyral expansion and sulcal effacement in the involved areas of cortex. LP revealed RBC 1280/uL WBC 0/uL, and protein and glucose within the reference range. An encephalitis biofire panel was negative.
She completed a course of PLEX due to initial concern for autoimmune encephalitis without improvement in mental status. Repeat MRI showed extensive brain parenchyma symmetric signal changes bilaterally which appeared more confluent compared to the prior study as well as small new subarachnoid hemorrhage in the high convexities (Fig. 2b and c). She did not have any further improvement in her mental status and she was transferred to a skilled nursing facility, as many as 74% of hospitalized COVID-19 patients have encephalopathy at some point during their admission [84]. Given the hypoxia and severe respiratory symptoms often associated with COVID-19 infection, hypoxia is an important cause of encephalopathy along with sepsis, metabolic derangements, organ failure, and prolonged use of paralytics and sedatives. Shortly after the hypoxic insult, patients can have agitation, confusion, disorientation, altered awareness, and myoclonus [2]. Imaging can often show diffuse edema, loss of gray-white matter differentiation, and decreased basal ganglia attenuation. EEG often shows non-specific changes and bifrontal slowing [2, 85].
Patients can also have prolonged or delayed encephalopathy even after respiratory symptoms have abated or after prolonged intubation. MRI may show confluent T2 hyperintensities in the periventricular, deep, and subcortical white matter and restricted diffusion [86,87,88]. In some cases, punctate microbleeds in the juxtacortical and callosal white matter, internal capsules, brainstem, and cerebellar peduncles may also be present [86, 87]. This pattern is similar to what has been described in delayed posthypoxic leukoencephalopathy (DPHL). The mechanism behind DPHL is thought to be necrosis of oligodendrocytes in arterial border zones due to hypoxia with subsequent failure of myelin turnover. This can result in widespread demyelination with axonal sparing [89].
Conclusion
As neurologic manifestations of COVID-19 infection are increasingly identified, neurointensivists must recognize severe neurologic manifestations. Hospitals should work to implement frequent neurological checks in order to promptly identify these severe neurologic manifestations as these can happen unexpectedly in patients without risk factors. In addition, clinicians should attempt obtain head imaging and neurology consultation when there is a neurological change in the patient. Since the use of sedation or paralytics can mask neurological deterioration, head imaging and neurology consultation should also be obtained in patients with delayed recovery from sedation. Consideration to bedside tests can be made if the patient is too unstable to move for imaging; for example, bedside EEG, transcranial Dopplers, and optic nerve sheath diameters may help diagnose and manage an unstable patient. Patients with severe neurological complications should be monitored closely in a neurocritical care unit when possible, as yet there are no COVID-19-specific treatments available, and treatment should follow the standard of care for other etiologies of the disease. However, further study should include the development of risk stratification instruments to identify the patients with COVID-19 who are most likely to benefit from therapeutic anticoagulation.
References and Recommended Reading
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–7. https://doi.org/10.1016/j.thromres.2020.04.013.
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. 2020;382(23):2268–70. https://doi.org/10.1056/NEJMc2008597.
Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, et al. Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med. 2020;382(20):e60. https://doi.org/10.1056/NEJMc2009787.
Merkler AE, Parikh NS, Mir S, Gupta A, Kamel H, Lin E, et al. Risk of ischemic stroke in patients with coronavirus disease 2019 (COVID-19) vs patients with influenza. JAMA Neurol. 2020;77:1366. https://doi.org/10.1001/jamaneurol.2020.2730.
Yaghi S, Ishida K, Torres J, Grory BM, Raz E, Humbert K, et al. SARS-CoV-2 and Stroke in a New York Healthcare System. Stroke. 2020;51(7):2002–11. https://doi.org/10.1161/STROKEAHA.120.030335.
Li Y, Li M, Wang M, Zhou Y, Chang J, Xian Y et al. Acute cerebrovascular disease following COVID-19: a single center, retrospective, observational study. Stroke Vasc Neurol. 2020:svn-2020-000431. https://doi.org/10.1136/svn-2020-000431.
Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, et al. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun. 2020;87:18–22. https://doi.org/10.1016/j.bbi.2020.03.031.
Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18(5):1094–9. https://doi.org/10.1111/jth.14817.
Hess DC, Eldahshan W, Rutkowski E. COVID-19-related stroke. Transl Stroke Res. 2020;11(3):322–5. https://doi.org/10.1007/s12975-020-00818-9.
Ashrafi F, Zali A, Ommi D, Salari M, Fatemi A, Arab-Ahmadi M, et al. COVID-19-related strokes in adults below 55 years of age: a case series. Neurol Sci. 2020;41(8):1985–9. https://doi.org/10.1007/s10072-020-04521-3.
Boehme AK, Ranawat P, Luna J, Kamel H, Elkind MSV. Risk of acute stroke after hospitalization for sepsis. Stroke. 2017;48(3):574–80. https://doi.org/10.1161/STROKEAHA.116.016162.
Grau AJ, Buggle F, Becher H, Zimmermann E, Spiel M, Fent T, et al. Recent bacterial and viral infection is a risk factor for cerebrovascular ischemia. Neurology. 1998;50(1):196–203. https://doi.org/10.1212/WNL.50.1.196.
Panigada M, Bottino N, Tagliabue P, Grasselli G, Novembrino C, Chantarangkul V, et al. Hypercoagulability of COVID-19 patients in intensive care unit: a report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost. 2020;18(7):1738–42. https://doi.org/10.1111/jth.14850.
Manousakis G, Jensen MB, Chacon MR, Sattin JA, Levine RL. The interface between stroke and infectious disease: infectious diseases leading to stroke and infections complicating stroke. Curr Neurol Neurosci Rep. 2009;9(1):28–34. https://doi.org/10.1007/s11910-009-0005-x.
Vazquez-Garza E, Jerjes-Sanchez C, Navarrete A, Joya-Harrison J, Rodriguez D. Venous thromboembolism: thrombosis, inflammation, and immunothrombosis for clinicians. J Thromb Thrombolysis. 2017;44(3):377–85. https://doi.org/10.1007/s11239-017-1528-7.
Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med. 2020;382(17):e38. https://doi.org/10.1056/NEJMc2007575.
Carneiro T, Dashkoff J, Leung LY, Nobleza COHS, Marulanda-Londono E, Hathidara M, et al. Intravenous tPA for acute ischemic stroke in patients with COVID-19. J Stroke Cerebrovasc Dis. 2020;29(11):105201. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.105201.
Liang JW, Reynolds AS, Reilly K, Lay C, Kellner CP, Shigematsu T et al. COVID-19 and decompressive hemicraniectomy for acute ischemic stroke. Stroke. STROKEAHA.120.030804. https://doi.org/10.1161/STROKEAHA.120.030804.
BMC COVID Anticoagulation Algorithm Update.
Mount Sinai COVID-19 Anticoagulation Algorithm. Version 1.1. April 2020.
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77(6):683–90. https://doi.org/10.1001/jamaneurol.2020.1127.
Md Noh MSF. COVID-19 and cerebral hemorrhage: proposed mechanisms. J Neuroradiol. 2020. https://doi.org/10.1016/j.neurad.2020.05.007.
Benger M, Williams O, Siddiqui J, Sztriha L. Intracerebral haemorrhage and COVID-19: clinical characteristics from a case series. Brain Behav Immun. 2020;88:940–4. https://doi.org/10.1016/j.bbi.2020.06.005.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–80.e8. https://doi.org/10.1016/j.cell.2020.02.052.
Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5(4):562–9. https://doi.org/10.1038/s41564-020-0688-y.
Al-Olama M, Rashid A, Garozzo D. COVID-19-associated meningoencephalitis complicated with intracranial hemorrhage: a case report. Acta Neurochir. 2020;162(7):1495–9. https://doi.org/10.1007/s00701-020-04402-w.
Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135(23):2033–40. https://doi.org/10.1182/blood.2020006000.
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–20. https://doi.org/10.1056/NEJMoa2002032.
Yang X, Yang Q, Wang Y, Wu Y, Xu J, Yu Y, et al. Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost. 2020;18(6):1469–72. https://doi.org/10.1111/jth.14848.
Xu P, Zhou Q, Xu J. Mechanism of thrombocytopenia in COVID-19 patients. Ann Hematol. 2020;99(6):1205–8. https://doi.org/10.1007/s00277-020-04019-0.
Magdi M, Rahil A. Severe immune thrombocytopenia complicated by intracerebral haemorrhage associated with coronavirus infection: a case report and literature review. Eur J Case Rep Intern Med. 2019;6(7):001155. https://doi.org/10.12890/2019_001155.
Tassiopoulos AK RJ, Rutigliano D, Jawa RS, Talamini MA. Stony brook anticoagulation protocol for COVID-19 PATIENTS. Department of surgery, Stony Brook University Medical Center.
Kvernland A, Kumar A, Yaghi S, Raz E, Frontera J, Lewis A, Czeisler B, Kahn DE, Zhou T, Ishida K, Torres J, Riina HA, Shapiro M, Nossek E, Nelson PK, Tanweer O, Gordon D, Jain R, Dehkharghani S, Henninger N, de Havenon A, Grory BM, Lord A, Melmed K Anticoagulation use and hemorrhagic stroke in SARS-CoV-2 patients treated at a New York Healthcare System. Neurocrit Care 2020. doi:https://doi.org/10.1007/s12028-020-01077-0.
Dogra S, Jain R, Cao M, Bilaloglu S, Zagzag D, Hochman S, et al. Hemorrhagic stroke and anticoagulation in COVID-19. J Stroke Cerebrovasc Dis. 2020;29(8):104984. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.104984.
Heman-Ackah SM, Su YS, Spadola M, Petrov D, Chen HI, Schuster J, et al. Neurologically devastating intraparenchymal hemorrhage in COVID-19 patients on extracorporeal membrane oxygenation: a case series. Neurosurgery. 2020;87(2):E147–E51. https://doi.org/10.1093/neuros/nyaa198.
Abrams RMC, Kim BD, Markantone DM, Reilly K, Paniz-Mondolfi AE, Gitman MR, et al. Severe rapidly progressive Guillain-Barré syndrome in the setting of acute COVID-19 disease. J Neurovirol. 2020;26:1–3. https://doi.org/10.1007/s13365-020-00884-7.
Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin–angiotensin–aldosterone system inhibitors in patients with Covid-19. N Engl J Med. 2020;382(17):1653–9. https://doi.org/10.1056/NEJMsr2005760.
Dalakas MC. Guillain-Barré syndrome: the first documented COVID-19–triggered autoimmune neurologic disease. Neurol Neuroimmunol Neuroinflamm. 2020;7(5):e781. https://doi.org/10.1212/NXI.0000000000000781.
Scheidl E, Canseco DD, Hadji-Naumov A, Bereznai B. Guillain-Barré syndrome during SARS-CoV-2 pandemic: a case report and review of recent literature. J Peripher Nerv Syst. 2020;25(2):204–7. https://doi.org/10.1111/jns.12382.
Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, Franciotta D, Baldanti F, Daturi R, Postorino P, Cavallini A, Micieli G Guillain–Barré syndrome associated with SARS-CoV-2. N Engl J Med 2020;382(26):2574–2576. doi:https://doi.org/10.1056/NEJMc2009191.
Alberti P, Beretta S, Piatti M, Karantzoulis A, Piatti ML, Santoro P, et al. Guillain-Barré syndrome related to COVID-19 infection. Neurol Neuroimmunol Neuroinflamm. 2020;7(4):e741. https://doi.org/10.1212/NXI.0000000000000741.
Coen M, Jeanson G, Culebras Almeida LA, Hübers A, Stierlin F, Najjar I, et al. Guillain-Barré syndrome as a complication of SARS-CoV-2 infection. Brain Behav Immun. 2020;87:111–2. https://doi.org/10.1016/j.bbi.2020.04.074.
Arnaud S, Budowski C, Wing N, Tin S, Degos B. Post SARS-CoV-2 Guillain-Barré syndrome. Clin Neurophysiol. 2020;131(7):1652–4. https://doi.org/10.1016/j.clinph.2020.05.003.
Ottaviani D, Boso F, Tranquillini E, Gapeni I, Pedrotti G, Cozzio S, et al. Early Guillain-Barré syndrome in coronavirus disease 2019 (COVID-19): a case report from an Italian COVID-hospital. Neurol Sci. 2020;41(6):1351–4. https://doi.org/10.1007/s10072-020-04449-8.
Caress JB, Castoro RJ, Simmons Z, Scelsa SN, Lewis RA, Ahlawat A et al. COVID-19–associated Guillain-Barré syndrome: the early pandemic experience. Muscle Nerve. 2020. https://doi.org/10.1002/mus.27024.
Zhao H, Shen D, Zhou H, Liu J, Chen S. Guillain-Barré syndrome associated with SARS-CoV-2 infection: causality or coincidence? Lancet Neurol. 2020;19(5):383–4. https://doi.org/10.1016/S1474-4422(20)30109-5.
Virani A, Rabold E, Hanson T, Haag A, Elrufay R, Cheema T, et al. Guillain-Barré syndrome associated with SARS-CoV-2 infection. IDCases. 2020;20:e00771. https://doi.org/10.1016/j.idcr.2020.e00771.
Morgello S. Coronaviruses and the central nervous system. J NeuroVirol. 2020;26:459–73. https://doi.org/10.1007/s13365-020-00868-7.
Li YC, Bai WZ, Hashikawa T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol. 2020;92(6):552–5. https://doi.org/10.1002/jmv.25728.
Armangue T, Leypoldt F, Málaga I, Raspall-Chaure M, Marti I, Nichter C, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol. 2014;75(2):317–23. https://doi.org/10.1002/ana.24083.
Pilotto A, Odolini S, Masciocchi S, Comelli A, Volonghi I, Gazzina S, et al. Steroid-responsive encephalitis in coronavirus disease 2019. Ann Neurol. 2020;88(2):423–7. https://doi.org/10.1002/ana.25783.
Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202(3):415–24. https://doi.org/10.1084/jem.20050828.
Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N, et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology. 2020;77(2):198–209. https://doi.org/10.1111/his.14134.
Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood–brain barrier: structural components and function under physiologic and pathologic conditions. J NeuroImmune Pharmacol. 2006;1(3):223–36. https://doi.org/10.1007/s11481-006-9025-3.
Smatti MK, Cyprian FS, Nasrallah GK, Al Thani AA, Almishal RO, Yassine HM. Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses. 2019;11(8):762. https://doi.org/10.3390/v11080762.
Efe IE, Aydin OU, Alabulut A, Celik O, Aydin K. COVID-19−associated encephalitis mimicking glial tumor. World Neurosurg. 2020;140:46–8. https://doi.org/10.1016/j.wneu.2020.05.194.
Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B. COVID-19-associated acute hemorrhagic necrotizing encephalopathy: imaging features. Radiology. 2020;296(2):E119–e20. https://doi.org/10.1148/radiol.2020201187.
Zhang T, Rodricks MB, Hirsh E. COVID-19-associated acute disseminated encephalomyelitis: a case report. medRxiv. 2020:2020.04.16.20068148. https://doi.org/10.1101/2020.04.16.20068148.
Bernard-Valnet R, Pizzarotti B, Anichini A, Demars Y, Russo E, Schmidhauser M et al. Two patients with acute meningoencephalitis concomitant with SARS-CoV-2 infection. Eur J Neurol. https://doi.org/10.1111/ene.14298.
Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis. 2020;94:55–8. https://doi.org/10.1016/j.ijid.2020.03.062.
Paniz-Mondolfi A, Bryce C, Grimes Z, Gordon RE, Reidy J, Lednicky J, et al. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J Med Virol. 2020;92(7):699–702. https://doi.org/10.1002/jmv.25915.
Huang YH, Jiang D, Huang JT. SARS-CoV-2 detected in cerebrospinal fluid by PCR in a Case of COVID-19 encephalitis. Brain Behav Immun. 2020;87:149. https://doi.org/10.1016/j.bbi.2020.05.012.
Kandemirli SG, Dogan L, Sarikaya ZT, Kara S, Akinci C, Kaya D, et al. Brain MRI findings in patients in the intensive care unit with COVID-19 infection. Radiology. 2020;201697:E232–5. https://doi.org/10.1148/radiol.2020201697.
Lu L, Xiong W, Liu D, Liu J, Yang D, Li N, et al. New onset acute symptomatic seizure and risk factors in coronavirus disease 2019: a retrospective multicenter study. Epilepsia. 2020;61(6):e49–53. https://doi.org/10.1111/epi.16524.
Vollono C, Rollo E, Romozzi M, Frisullo G, Servidei S, Borghetti A, et al. Focal status epilepticus as unique clinical feature of COVID-19: a case report. Seizure. 2020;78:109–12. https://doi.org/10.1016/j.seizure.2020.04.009.
Galanopoulou AS, Ferastraoaru V, Correa DJ, Cherian K, Duberstein S, Gursky J, et al. EEG findings in acutely ill patients investigated for SARS-CoV-2/COVID-19: a small case series preliminary report. Epilepsia Open. 2020;5(2):314–24. https://doi.org/10.1002/epi4.12399.
Ayub N, Cohen J, Jing J, Jain A, Tesh R, Mukerji SS et al. Clinical electroencephalography findings and considerations in hospitalized patients with coronavirus SARS-CoV-2. medRxiv. 2020. https://doi.org/10.1101/2020.07.13.20152207.
Baig AM, Khaleeq A, Ali U, Syeda H. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. 2020;11(7):995–8. https://doi.org/10.1021/acschemneuro.0c00122.
Balloy G, Leclair-Visonneau L, Péréon Y, Magot A, Peyre A, Mahé P-J, et al. Non-lesional status epilepticus in a patient with coronavirus disease 2019. Clin Neurophysiol. 2020;131(8):2059–61. https://doi.org/10.1016/j.clinph.2020.05.005.
Hepburn M, Mullaguri N, George P, Hantus S, Punia V, Bhimraj A, et al. Acute symptomatic seizures in critically ill patients with COVID-19: is there an association? Neurocrit Care. 2020. https://doi.org/10.1007/s12028-020-01006-1.
Somani S, Pati S, Gaston T, Chitlangia A, Agnihotri S. De novo status epilepticus in patients with COVID-19. Ann Clin Transl Neurol. 2020;7(7):1240–4. https://doi.org/10.1002/acn3.51071.
Newey CR, Kinzy TG, Punia V, Hantus S. Continuous electroencephalography in the critically Ill: clinical and continuous electroencephalography markers for targeted monitoring. J Clin Neurophysiol. 2018;35(4).
Yazbeck M, Sra P, Parvizi J. Rapid response electroencephalography for urgent evaluation of patients in community hospital intensive care practice. J Neurosci Nurs. 2019;51(6):308–12. https://doi.org/10.1097/jnn.0000000000000476.
Princiotta Cariddi L, Tabaee Damavandi P, Carimati F, Banfi P, Clemenzi A, Marelli M, et al. Reversible encephalopathy syndrome (PRES) in a COVID-19 patient. J Neurol. 2020;267:1–4. https://doi.org/10.1007/s00415-020-10001-7.
Franceschi AM, Ahmed O, Giliberto L, Castillo M. Hemorrhagic posterior reversible encephalopathy syndrome as a manifestation of COVID-19 infection. AJNR Am J Neuroradiol. 2020;41(7):1173–6. https://doi.org/10.3174/ajnr.A6595.
Gómez-Enjuto S, Hernando-Requejo V, Lapeña-Motilva J, Ogando-Durán G, Fouz-Ruiz D, Domingo-García J, et al. Verapamil as treatment for refractory status epilepticus secondary to PRES syndrome on a SARS-Cov-2 infected patient. Seizure. 2020;80:157–8. https://doi.org/10.1016/j.seizure.2020.06.008.
Kaya Y, Kara S, Akinci C, Kocaman AS. Transient cortical blindness in COVID-19 pneumonia; a PRES-like syndrome: case report. J Neurol Sci. 2020;413:116858. https://doi.org/10.1016/j.jns.2020.116858.
Doo FX, Kassim G, Lefton DR, Patterson S, Pham H, Belani P. Rare presentations of COVID-19: PRES-like leukoencephalopathy and carotid thrombosis. Clin Imaging. 2020;69:94–101. https://doi.org/10.1016/j.clinimag.2020.07.007.
Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. Am J Neuroradiol. 2008;29(6):1043–9. https://doi.org/10.3174/ajnr.A0929.
Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect. 2020;53(3):368–70. https://doi.org/10.1016/j.jmii.2020.03.005.
Wilson MH, Edsell ME, Davagnanam I, Hirani SP, Martin DS, Levett DZ, et al. Cerebral artery dilatation maintains cerebral oxygenation at extreme altitude and in acute hypoxia--an ultrasound and MRI study. J Cereb Blood Flow Metab. 2011;31(10):2019–29. https://doi.org/10.1038/jcbfm.2011.81.
Kishfy L, Casasola M, Banankhah P, Parvez A, Jan YJ, Shenoy AM, et al. Posterior reversible encephalopathy syndrome (PRES) as a neurological association in severe Covid-19. J Neurol Sci. 2020;414:116943. https://doi.org/10.1016/j.jns.2020.116943.
Parauda SC, Gao V, Gewirtz AN, Parikh NS, Merkler AE, Lantos J, et al. Posterior reversible encephalopathy syndrome in patients with COVID-19. J Neurol Sci. 2020;416:117019. https://doi.org/10.1016/j.jns.2020.117019.
Garg RK, Paliwal VK, Gupta A. Encephalopathy in patients with COVID-19: a review. J Med Virol. 2020;93:206–22. https://doi.org/10.1002/jmv.26207.
Scullen T, Keen J, Mathkour M, Dumont AS, Kahn L. Coronavirus 2019 (COVID-19)–associated encephalopathies and cerebrovascular disease: the New Orleans experience. World Neurosurg 2020. doi: https://doi.org/10.1016/j.wneu.2020.05.192, 141, e437, e446.
Radmanesh A, Derman A, Lui YW, Raz E, Loh JP, Hagiwara M, et al. COVID-19 -associated diffuse leukoencephalopathy and microhemorrhages. Radiology. 2020;202040:E223–7. https://doi.org/10.1148/radiol.2020202040.
Breit H, Jhaveri M, John S. Concomitant delayed posthypoxic leukoencephalopathy and critical illness microbleeds. Neurol Clin Pract. 2018;8(5):e31–e3. https://doi.org/10.1212/CPJ.0000000000000513.
Zamora CA, Nauen D, Hynecek R, Ilica AT, Izbudak I, Sair HI, et al. Delayed posthypoxic leukoencephalopathy: a case series and review of the literature. Brain Behav. 2015;5(8):e00364-e. https://doi.org/10.1002/brb3.364.
King F, Morris NA, Schmahmann JD. Delayed Posthypoxic Leukoencephalopathy: improvement with antioxidant therapy. Case Rep Neurol. 2015;7(3):242–6. https://doi.org/10.1159/000441892.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Daniella C. Sisniega declares that she has no conflict of interest. Alexandra S. Reynolds declares that she has no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Critical Care Neurology
Rights and permissions
About this article

Cite this article
Sisniega, D.C., Reynolds, A.S. Severe Neurologic Complications of SARS-CoV-2. Curr Treat Options Neurol 23, 14 (2021). https://doi.org/10.1007/s11940-021-00669-1
Accepted:
Published:
DOI: https://doi.org/10.1007/s11940-021-00669-1