
Abstract
In inflammatory disorders such as rheumatoid arthritis, cytokines and danger signals are sensed by the central nervous system, which adapts behavior and physiologic responses during systemic stress. The central nervous system can also signal the periphery to modulate inflammation through efferent hormonal and neuronal pathways. The brain and spinal cord are involved in this bidirectional interaction. A variety of neuronal pathways that modulate synovial inflammation have been implicated, including the sympathetic and the parasympathetic branches of the autonomic system. Another mechanism, the dorsal root reflex, involves antidromic signaling along somatic afferent fibers that influences joint inflammation by releasing neuropeptides and other neuromediators in the periphery. Some of the neurotransmitters and neuroreceptors involved have been identified in preclinical models and represent novel targets for the treatment of rheumatic diseases.
Similar content being viewed by others

Introduction
The concept that the central nervous system (CNS) and the peripheral nervous system interact with the immune and innate mediators of host defense is well-established. Many mechanisms of this neuroimmune dialogue can influence inflammation and immunity on a systemic scale or at the regional and local levels. For instance, systemic control is under the influence of the hypothalamic-pituitary axis, which controls glucocorticoid release by the adrenal cortex (the HPA axis), sex hormone levels by the gonads, and the thyroid. The HPA axis provides an essential feedback mechanism on inflammation and is often blunted in a wide range of autoimmune and inflammatory diseases, including systemic lupus erythematosus and rheumatoid arthritis. A dynamic balance of glucocorticoid release is required to maintain the homeostasis between excess inflammation and response to stress. Pharmacologic or surgical interruption of the HPA axis can increase mortality resulting from septic shock, as seen in patients who require chronic glucocorticoid treatment.
The Autonomic Nervous System Link to Peripheral Inflammation
The systemic control exerted by the HPA axis takes place over hours, and other systems with similar feedback mechanisms between the periphery and the brain or spinal cord can control peripheral inflammation more rapidly and directly. The neurological control of the HPA axis and the autonomic system involves higher structures of the brain and brainstem. Modulation of inflammation is also under the control of somatic sensory fibers. Important steps in this neurological processing and feedback occur in the spinal cord. The spinal cord then signals back to the periphery by antidromic action potentials through afferent fibers [1]. The effect is mainly proinflammatory, and somatic denervation can actually protect from arthritis [2]. This antidromic dorsal root reflex can influence inflammation through the recruitment of inflammatory cells and by triggering vasoactive edema.
In addition to the systemic control exerted by the HPA axis and the local effects of the somatic fibers, the autonomic nervous system is considered essential for the control of the regional homeostasis at the level of individual organs. The autonomic nervous system comprises two main branches: the sympathetic and parasympathetic. They constantly control the metabolism and visceral output and exert both complementary and antagonizing functions. Branches of the vagal nerve and sympathetic fibers innervate immune organs, in which they can influence peripheral immune responses. Noradrenaline and adrenaline are the primary neurotransmitters of the sympathetic nervous system (SNS). Noradrenergic sympathetic nerve fibers originate from the spinal cord and innervate primary and secondary lymphoid organs. The SNS also directly innervates the joints and seems to plays a dual role in synovial inflammation, both proinflammatory and anti-inflammatory [3].
The vagus nerve that is formed by the 10th cranial nerves is the main branch of the parasympathetic nervous system. The immunoregulatory properties of the vagus nerve were initially characterized during evaluation of the inflammatory response in sepsis [4]. We describe how parasympathetic activation can suppress inflammation and how it relates to arthritis models. Similar to the HPA axis and the dorsal root reflex, a neuroimmune feedback mechanism modulates vagal activity. This mechanism has been termed the cholinergic reflex in reference to the central role of acetylcholine (ACh), the main neurotransmitter of the parasympathetic system [5]. Experimental evidence implicates the multifaceted effects of ACh in the central and peripheral control of the cholinergic reflex. In the periphery, the discovery that the anti-inflammatory effects of this pathway depend on the α7 nicotinic receptor initiated drug development efforts and preclinical testing of selective agonists of this receptor [6].
Thus, the study of these pathways has unraveled potential targets that are already validated in preclinical studies in inflammatory models. One hallmark of neuroimmune pathways it that the neurotransmitters involved and their receptors are often shared between the CNS and immune cells in the periphery. Indeed, lymphocytes, macrophages, and fibroblast-like synoviocytes (FLS) express an array of cell-surface receptors for neurotransmitters. To safely advance pharmacologic interventions to the clinical phase, it may be necessary to design strategies that spare the function of these receptors in the CNS. For instance, α7 agonists that penetrate the CNS are already in clinical development for treatment of psychiatric and degenerative diseases [7], while compounds without blood–brain barrier permeability are being developed for inflammatory diseases.
Spinal Control of Synovial Inflammation
Acute peripheral inflammation leads to a series of activation events in the spinal cord. During the development of experimental arthritis, somatosensory neurons become hyperexcitable in response to innocuous stimuli (allodynia) [8]. This hyperexcitability is caused by C-fiber activation, and afferent action potentials trigger the release of excitatory amino acid glutamate and aspartate within the spinal cord [9]. The resulting hyperalgesia is dependent on activation of glutamate receptors of the N-methyl-D-aspartate (NMDA) subtype and can be blocked by intrathecal administration of NMDA receptor antagonists [10].
In return, the spinal cord can signal back to the periphery via several neuronal routes to control different parameters of local and regional inflammation. The first inflammatory events that were shown to be provoked by neurogenic inflammation were arteriolar vasodilation, plasma extravasation, and hyperalgesia. These events are caused by the release of inflammatory mediators such as substance P, calcitonin gene-related peptide, or vasoactive intestinal peptide by primary afferent fibers [11]. Antidromic action potentials originate in the spinal cord toward the periphery and travel through primary afferent neurons to mediate the release of these neuromediators, mostly through small afferent fibers such as C fibers.
These acute events do not usually lead to protracted inflammation by themselves, unless tissue injury and cell death occur. In synovitis, the transition to the chronic phase usually involves the recruitment and inappropriate activation of cells of the innate immune system. For instance, activated neutrophils and other resident sentinel cells of the joint release destructive enzymes. The endogenous danger signals released by the damaged tissue start and amplify inflammation and can also increase input to the spinal cord via the somatic afferents.
The ability of the spinal cord to regulate neutrophil recruitment was first investigated in sterile inflammation. Intrathecal administration of adenosine agonists decreased neutrophil accumulation in a model of acute peripheral inflammation provoked by the intradermal injection of carrageenan [12]. The mechanism involves A1 receptor stimulation in the spinal cord, which in turn inhibits activation of NMDA receptors by excitatory amino acids such as glutamate (Fig. 1). Thus, the same excitatory mediators that activate spinal neurons during local inflammation also control an efferent mechanism involved in neutrophil recruitment. Strikingly, the proinflammatory effect of NMDA receptor activation in the spinal cord, which can be antagonized by intrathecal adenosine, is caused by a decrease in adenosine levels in the periphery.

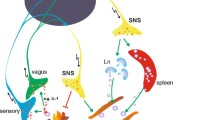
Schematic description of the main neuronal pathways involved in the regulation of synovial inflammation. Vagal nerve firing leads to acetylcholine (ACh) release in the periphery. ACh is an agonist for the α7 nicotinic receptor on macrophages and fibroblast-like synoviocytes (FLS) and decreases the release of cytokines and chemokines. Vagal fibers are detected in peripheral organs such as the bronchi (dashed lines), but not in the synovium. In organs not directly innervated by the vagus, such as the spleen or the joints, vagal stimulation is postulated to stimulate autonomic fibers to release norepinephrine (NE), which in turn triggers ACh release via a non-neuronal cholinergic system. The peripheral increase in ACh is sensed by the nicotinic α7 receptor, which downregulates the release of proinflammatory cytokines. Central and intrathecal administration of p38 inhibitors stimulates the vagal nerve and decreases synovial inflammation. In rheumatoid arthritis, loss of noradrenergic fibers inhibits the anti-inflammatory action of the sympathetic system (β2 receptors) and the vagal system. Stimulation of peripheral C-afferent fibers releases excitatory amino acid (eg, glutamate) in the spinal cord, which then binds to N-methyl-D-aspartate (NMDA) receptors to decrease adenosine release by afferent fibers. Adenosine has an anti-inflammatory effect in the periphery by binding to A2 receptors on neutrophils and FLS and on central A1 receptors that inhibit NMDA receptor activation. AG autonomic ganglion; IL interleukin; Mφ macrophage; PMN polymorphonuclear leukocyte; Pp38 phospho-p38 mitogen-activated protein kinase; SB i. t. intrathecal SB203580 (p38 inhibitor); TNF tumor necrosis factor
The anti-inflammatory effect of adenosine in the periphery is mediated by the A2 receptor expressed by neutrophils rather than the A1 receptor that mediates NMDA suppression in the spinal cord [12]. The efferent neuronal route is dependent on sensory nerves because the effect of central A1 agonists was abolished after rhizotomy, a surgical procedure that selectively interrupts somatic sensory fibers, but not after sympathectomy [13]. Notably, other studies also implicate additional glutamate receptors, such as the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor, in reflex anti-inflammatory mechanisms.
These experiments demonstrate that the action of spinal mechanisms on inflammation is not limited to acute vasoactive events. The control exerted on the innate immune system suggested that spinal cord events might play a role during arthritis. The rapidity of the proinflammatory response evoked by antidromic signaling helps control acute insults. As it is triggered by pain and inflammation that are also cardinal symptoms of arthritis, the possibility existed that it could aggravate joint disease.
Indeed, intrathecal administration of a selective A1 adenosine agonist (cyclohexyladenosine [CHA]) inhibited inflammation and joint destruction in the rat model of adjuvant arthritis [14]. The chondroprotective effects were linked to a decreased activation of activator protein-1 in the joints, which is a transcription factor involved in the production of metalloproteinases.
The expression of the c-fos gene in the spinal cord, a marker of neuronal activation, was increased during adjuvant arthritis [14]. This is caused by painful and inflammatory stimuli in the periphery that cause excitatory amino acid release in the spinal cord. However, c-fos expression is only transiently decreased after indirect blockade of NMDA receptors by CHA, suggesting the existence of additional spinal mechanisms [13].
New findings from neuropathic pain studies helped us discover a previously unsuspected player in the processing of pain and inflammatory signals by the spinal cord—the microglial cells. Although glial cells were originally considered passive supporters of neurons in the CNS, they actually contribute to the chronicity of neuropathic pain. It is mainly the spinal microglial cells that contribute to neuronal activation in these models. Strikingly, cytokines and signaling pathways that mediate peripheral inflammation are also expressed in the CNS during microglial activation: the mitogen-activated protein kinases (MAPKs) and cytokines interleukin (IL)-1 and tumor necrosis factor (TNF) [15].
The MAPK system, and the p38 MAPK in particular play a central role in microglia activation in chronic pain models [16–18]. After nerve injury, p38 is activated by several stimuli, including osmotic stress and cytokines such as IL-1 and TNF. Activated p38 augments the production of the same inflammatory mediators, resulting in a self-sustaining activation loop.
The role of p38 activation in the spinal cord was investigated in adjuvant arthritis in the rat. In this model, administration of a very small dose of a p38 inhibitor (0.1% of the dose required for systemic administration) in the intrathecal space not only controlled pain but also reduced inflammation and joint destruction [19]. TNF levels were increased in the cerebrospinal fluid of arthritic rats, and administration of a subclinical dose of etanercept intrathecally also suppressed arthritis and spinal p38 activation. Because p38 inhibition in the CNS also suppressed intrathecal TNF, the most likely mechanism is that TNF both induces and is induced by p38 activation. Recently, another group demonstrated that intrathecal administration of etanercept is also effective in the antigen-induced arthritis model [20••].
The Vagal Cholinergic Pathway
The efferent mechanisms by which p38 inhibition in the CNS downregulates arthritis are not completely understood. Downstream of p38, an efferent neural route that ultimately leads to vagus nerve activation appears to contribute. Borovikova and colleagues [4] demonstrated that the vagus nerve and cholinergic stimulation play an important role in limiting the inflammatory response. The pathway is composed of the efferent vagus nerve, the release of ACh in the periphery by terminal fibers of the vagus, and nicotinic receptors composed of the α7 subunit [6].
The vagus nerve is the major parasympathetic division of the autonomic nervous system. It provides constant and rapid regulation of organ function, including heart rate and gut motility. The cholinergic vagal anti-inflammatory system was first studied in acute models of inflammation. Initial work focused on sepsis and endotoxemia models in which electric stimulation of the vagus reduced cytokine production, including TNF, and prevented shock [4]. An opposite effect was obtained in vagotomized animals in similar models [21]. The latter observation is explained by the existence of a reflex mechanism by which cytokines released at the inflammatory site activate afferent fibers of the vagus nerve. The afferent arm of the cholinergic reflex routes through to the dorsal motor nucleus of the vagus to stimulate the release of ACh, the main neurotransmitter released by terminal vagal fibers.
Interruption of this homeostatic mechanism in vagotomized animals aggravates inflammation. In animals with an intact vagal nerve, this reflex operates to reduce the release of proinflammatory cytokines in the periphery, thereby contributing to the homeostasis of the system by reducing inflammation.
Modulation of the Cholinergic Anti-inflammatory Reflex by the Central Nervous System
The heart is innervated by the two divisions of the autonomic nervous system. Their physiologic function is to adapt and anticipate heart rate and output (eg, during a fight or flight response). The cardiac autonomic tone can be studied with noninvasive electrocardiogram recordings. Mathematical analysis of the electrocardiogram can provide quantitative measurements of sympathetic and parasympathetic activities [22]. Vagal stimulation leads to ACh release, which has a negative chronotropic effect on heart rate. ACh was identified as the first neurotransmitter with this historic observation. The cardiac vagal tone correlates with the cholinergic anti-inflammatory reflex, at least in the acute setting [23]. This characteristic can be exploited to measure vagal activity by noninvasive methods in inflammatory models.
The quantitative measure of heart rate variability to monitor vagal nerve activity has been used in preclinical models to explore vagal outflow. We used computerized frequency domain analysis, a standardized method of quantifying vagal activity. Intrathecal administration of a p38 inhibitor at the same dose that suppressed arthritis increased vagal activity, suggesting a link between spinal mechanisms and the cholinergic anti-inflammatory pathway [24•]. This experimental protocol targets the delivery of the p38 inhibitor to the lumbar spine. Because vagal activity is controlled by the dorsal motor nucleus located in the floor of the fourth ventricle, the increase in vagal activity seen after p38 blockade in the lumbar spine suggests the existence of an ascending neuronal tract to the dorsal motor nucleus. Another p38 inhibitor, CNI-1493, can activate the vagus when administered via intracerebroventricular injection, confirming the role of the p38 MAPK on vagal tone [25]. Even though the anatomic route of administration is different, this protocol was shown to decrease acute peripheral paw inflammation in the carrageenan model [26]. The anti-inflammatory effects on the paws were abolished by vagotomy and mimicked by direct electrical stimulation of the vagus.
The link between TNF-mediated spinal activation and the autonomic system was also studied in a monoarticular model of knee arthritis [20••]. Administration of etanercept by the same intrathecal route at the lumbar spine level reduced inflammation [19] and pain behavior, consistent with the role of TNF in activation of p38 in spinal microglia [17]. Electrocardiogram recordings showed that intrathecal TNF prevented the increase in heart rate seen in arthritic rats. This is compatible with an increased sympathetic drive or a decreased vagal tone. Cardiac sympathetic drive was increased in that study, but direct vagal measures were not performed. Thus, spinal mechanisms also influence the sympathetic system. The role of sympathetic nerves in joint inflammation is discussed further below.
In addition to these spinal mechanisms, central cholinergic muscarinic receptors modulate the vagal tone [23]. Different subtypes of ACh receptors in the brain regulate vagal output. The two main classes of ACh receptors are classified by their response to agonists as muscarinic or nicotinic. The control of vagal tone by brain structures is mediated by muscarinic receptors of the M1 and M2 subtypes [23]. Activation of brain M1 receptors or inhibition of presynaptic M2 receptors activates efferent vagus activity and reduces peripheral TNF levels.
Increasing central ACh levels by targeting endogenous mechanisms could provide a more physiologic approach to re-establish the autonomic balance. Indeed, rheumatoid arthritis patients have a decreased vagal output and enhanced sympathetic tone [27]. The decrease in vagal tone was inversely correlated with the level of the proinflammatory factor high mobility group box-1 [28]. α7 Agonists also could be used because they were shown to retain their efficacy in vitro on primary cells from rheumatoid arthritis patients [29, 30, 31•].
Recently, an endogenous mechanism that regulates acetylcholinesterase levels was discovered. In the steady state, ACh levels are tightly and rapidly regulated by cholinesterases. During inflammation, serum acetylcholinesterase levels decrease, which suppresses inflammation by boosting ACh concentration. This effect is mediated by the degradation of the mRNA encoding for acetylcholinesterase by a microRNA (miR-132) [32]. miRNAs bind to mRNA targets and usually prevent the production of the corresponding protein. As miR-132 is expressed in the brain and the periphery, its anti-inflammatory effect is expected to target central and peripheral cholinergic mechanisms of the vagal reflex.
The regulation of the vagal anti-inflammatory pathway by brain acetylcholinesterases is confirmed by pharmacologic studies. In the rodent model of carrageenan-induced paw inflammation, central stimulation of the vagus by cholinesterase inhibitors significantly decreases paw swelling [24•]. The high-frequency component of heart rate variability, a measure of vagal activity that correlates with its anti-inflammatory effects, is increased by the same drug [24•]. Finally, administration of a centrally acting muscarinic receptor antagonist decreased the anti-inflammatory effect of the cholinesterase inhibitor galantamine [33].
Peripheral Mediators of the Cholinergic Anti-inflammatory Pathway
The peripheral anti-inflammatory effects of ACh released by terminal vagus fibers are mediated by the α7 subtype of nicotinic receptors. Nicotinic receptors are ligand-gated ion channels formed by the hetero- or homocombination of 17 different subunits. The α7 receptor was first identified in neurons as a homopentamer with high calcium permeability. Several lines of evidence indicate that the α7 receptor mediates most of the anti-inflammatory effect of the vagal cholinergic pathway. α7 Knockout mice have no overt basal phenotype but suffer from increased inflammatory responses in various models [21]. Vagal stimulation does not suppress inflammation in these knockout mice. In vitro, application of α7 agonists reduces proinflammatory cytokine production by various cell types, including macrophages and FLS [30, 31•]. α7 Agonists reduce inflammation in a broad variety of animal models, including in collagen-induced arthritis [34].
In acute inflammation, α7 expression by splenic macrophages is required for the anti-inflammatory effect of vagal stimulation. The anatomic route is most likely indirect because vagal cholinergic fibers are not detectable in the spleen. Rather, recent findings suggest that a splenic non-neuronal cholinergic system leads to ACh release during the vagal anti-inflammatory reflex [35••]. This non-neuronal cholinergic system may be controlled by catecholaminergic endings of the splenic nerve found in the vicinity of splenic macrophages.
Vagotomy very clearly antagonizes the anti-inflammatory effect of electrical and pharmacologic activation of the vagus in acute inflammation of the paws [26]. In the murine collagen-induced arthritis model, vagotomy can exacerbate arthritis, supporting a role for the vagus in chronic synovitis [34]. However, the effect of denervation in chronic models must be interpreted with caution because undesired side effects of the procedures on metabolic functions may confound the results. An effect on the spleen could modulate the recirculation of macrophages and lymphocytes to the synovium [35••]. Alternatively, the synovium contains a non-neuronal cholinergic system and catecholaminergic fibers [36]. The influence of the vagus on the synovium could communicate through this system, similar to the model proposed in the spleen [35••]. α7 Receptors are also highly expressed in the synovial intimal lining and by cultured FLS. In vitro, α7 agonists suppress the cytokines and chemokines produced by stimulated FLS [30, 31•].
The mechanism by which α7 regulates inflammatory responses depends on the model studied. α7 Receptor stimulation antagonizes the nuclear factor-κB pathway in macrophages in vitro [37]. Activation of a Janus kinase 2–signal transducer and activator of transcription 3 anti-inflammatory pathway was described in a murine model of surgical intestinal inflammation [38]. In synoviocytes, instability of IL-6 mRNA decreases IL-6 protein levels [30]. In addition, it is not yet clear whether calcium fluxes are required for the anti-inflammatory action of the α7 receptor, or if other nonionic signals are involved. Because of the diversity of the mechanisms proposed, the development of α7 agonists is still mainly based on the calcium-dependent neuronal model, which may not be fully relevant to their application to inflammatory diseases such as rheumatoid arthritis.
The Sympathetic Nervous System
The SNS also exerts complex control on joint inflammation [3]. Along with the dorsal root reflex, it is a critical proinflammatory component of neurogenic inflammation. Sympathetic outflow can participate in the chronic phase of arthritis and at this stage can exhibit both pro- and anti-inflammatory actions. The duality of this system can be explained in part by the fact that the SNS innervates the joints and the secondary lymphoid organs.
For instance, systemic sympathectomy protects from arthritis in experimental models, but only if performed before immunization with the inducing protocol [39]. In the late stages of experimental arthritis, the SNS markedly decreases disease activity, and sympathectomy exacerbates joint inflammation [40]. The sympathetic neurotransmitter norepinephrine directly influences the T-cell–dependent stage of experimental arthritis by activating CD4+ T cells [41]. Thus, in experimental models, the proinflammatory influence of the SNS occurs before the onset of synovitis. In the later phases of experimental arthritis, the net effect of the SNS is anti-inflammatory and can be enhanced by the β-adrenergic agonist salbutamol [42].
Generalized loss of sympathetic fibers occurs in some inflammatory diseases, potentially altering disease activity. In rheumatoid arthritis, the decreased synovial sympathetic innervation decreases norepinephrine levels. At low concentrations, this neurotransmitter predominantly binds to the higher-affinity α-adrenoreceptors that are proinflammatory [3].
Rational therapeutic approaches that target the SNS will have to reconcile the detrimental influence of the SNS on adaptive immunity and its anti-inflammatory function in the joints. Sympathetic activity is globally increased in patients with rheumatoid arthritis despite loss of fibers specifically to the joint [27] and can potentially contribute to autoimmunity. However, systemic sympathetic blockade could increase the local inflammation in the joints. Cortisol has a permissive effect on β-adrenergic signaling in synovial tissue, and local administration of a β-agonist combined with a corticoid has been suggested [43]. Sympathetic fiber loss in the rheumatoid arthritis synovium can also contribute to the loss of synergy with endogenous or therapeutic corticoids [43]. Counteracting the mechanisms that lead to the depletion of sympathetic terminals in the synovium could represent an elegant approach. Some of the factors involved are described. For instance, increased concentrations of the soluble nerve factor neuropilin-2 synergize with the repellent semaphoring 3F to deplete sympathetic fibers in rheumatoid arthritis [44•].
Therapeutic Interventions
Among the different neuroimmune mechanisms that have been described (Table 1), much of the current translational research effort is focused on the vagal cholinergic pathway. The elegant and detailed map of this pathway suggests several possible strategies for treating inflammatory disorders. Indeed, a recent Cochrane review confirmed that nicotine itself is an effective agent in inflammatory bowel disease [45].
Patients treated with cholinesterase inhibitors have decreased levels of inflammatory cytokines compared with controls [46]. Similar to the experimental setting, this decrease could be caused by central activation of the vagus and peripheral increase in ACh, the main anti-inflammatory neurotransmitter released by parasympathetic fibers. Other strategies could be developed to boost the anti-inflammatory effect of the vagal output, such as targeting the central regulatory pathways with specific muscarinic agonists, or by decreasing endogenous cholinesterase levels with therapeutic microRNAs based on the effect of miR-132 [32]. Electric vagal nerve stimulators that are already approved for the treatment of neurological disorders also could be envisioned [47]. The limitation of these approaches for the treatment of severe synovitis is their lack of specificity. Indeed, cholinergic side effects are responsible for the narrow therapeutic range of cholinesterase inhibitors.
Selective α7 agonists are currently considered a safer and more specific alternative for future therapeutic trials in inflammatory diseases. Expression of the α7 receptor has been detected on lymphocytes, macrophages, and FLS. Several compounds are effective in preclinical in vivo and in vitro models, including GTS-21, AR-R17779, and PNU-282987. They differ by their selectivity toward other classes of nicotinic receptors, with GTS-21 being less selective. GTS-21 has already been tested in clinical trials for the treatment of cognitive disorders and schizophrenia, with no adverse events reported. AR-R17779 reduced arthritis in the collagen arthritic model [34], whereas PNU-282987 decreased IL-6 and chemokine release by FLS in vitro [30].
Nicotinic receptor activation by α7 agonists causes a rapid and long-lasting desensitization, which may impair the therapeutic potential of these compounds [48]. Allosteric modulators such as PNU-120596 can convert desensitized receptors back to the open configuration. In neurons, this increases the calcium signals. Most of the studies on the properties of the α7 receptor were performed using patch clamp experiments in overexpression systems. It is not yet known whether these results also apply to inflammatory cell types that express lower endogenous levels of the receptor. Therefore, it is difficult to identify which type of agonists should be used to treat inflammation.
Conclusions
Distinct neuroimmune mechanisms can influence the outcome of generalized or localized inflammation in inflammatory arthritis. The presence of a feedback loop is characteristic of most neuroimmune homeostatic mechanisms, including the HPA axis and the vagal cholinergic reflex [5]. Increased glucocorticoid release is essential for the metabolic response to stress, and cytokines such as IL-1, IL-6, and TNF can influence the hypothalamus. The same signals are believed to be involved in the activation of the vagal anti-inflammatory reflex. Inflammatory cytokines are also released in the spinal cord during pain and inflammation. They participate in the activation of neuronal and microglial cells and modify neuropathic pain responses and neuronal feedback to the inflamed tissue [13, 14, 19].
Spatially restricted neural mechanisms can modulate synovial inflammation. Three main neurological routes are demonstrated to have an effect on synovitis in the experimental setting: antidromic signaling through somatic afferents and the two branches of the autonomic nervous system, the parasympathetic and the sympathetic system. Autonomic dysregulation is a hallmark of systemic autoimmune disease, including rheumatoid arthritis, providing indirect evidence for a role of the sympathetic system and the cholinergic anti-inflammatory pathway in human disease [26].
The role of the cholinergic anti-inflammatory pathway has been evaluated in a wide range of experimental models of chronic and acute inflammation. In the periphery, most of its beneficial effects are linked to the stimulation of the α7 nicotinic receptors. This subunit is expressed in the synovial tissue of rheumatoid arthritis patients and by primary FLS [30, 31•], supporting a link between the cholinergic vagal pathway and synovial inflammation.
The α7 receptor is also expressed in the CNS and is considered as a therapeutic target for the treatment of schizophrenia and Alzheimer’s disease. Some α7-specific compounds developed for the treatment of CNS diseases also display anti-inflammatory activities in preclinical models of arthritis [34]. Preclinical evaluation of these compounds is still required to determine whether α7 agonists that do not penetrate into the CNS will have a better therapeutic index.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Rees H, Sluka KA, Westlund KN, Willis WD: Do dorsal root reflexes augment peripheral inflammation? Neuroreport 1994, 5:821–824.
Kane D, Lockhart JC, Balint PV, et al.: Protective effect of sensory denervation in inflammatory arthritis (evidence of regulatory neuroimmune pathways in the arthritic joint). Ann Rheum Dis 2005, 64:325–327.
Straub RH, Harle P: Sympathetic neurotransmitters in joint inflammation. Rheum Dis Clin North Am 2005, 31:43–59, viii.
Borovikova LV, Ivanova S, Zhang M, et al.: Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405:458–462.
Tracey KJ: The inflammatory reflex. Nature 2002, 420:853–859.
Wang H, Yu M, Ochani M, et al.: Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421:384–388.
Olincy A, Harris JG, Johnson LL, et al.: Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry 2006, 63:630–638.
Draisci G, Iadarola MJ: Temporal analysis of increases in c-fos, preprodynorphin and preproenkephalin mRNAs in rat spinal cord. Brain Res Mol Brain Res 1989, 6:31–37.
Sluka KA, Westlund KN: An experimental arthritis in rats: dorsal horn aspartate and glutamate increases. Neurosci Lett 1992, 145:141–144.
Schaible HG, Grubb BD, Neugebauer V, Oppmann M: The effects of NMDA antagonists on neuronal activity in cat spinal cord evoked by acute inflammation in the knee joint. Eur J Neurosci 1991, 3:981–991.
Lin Q, Wu J, Willis WD: Dorsal root reflexes and cutaneous neurogenic inflammation after intradermal injection of capsaicin in rats. J Neurophysiol 1999, 82:2602–2611.
Bong GW, Rosengren S, Firestein GS: Spinal cord adenosine receptor stimulation in rats inhibits peripheral neutrophil accumulation. The role of N-methyl-D-aspartate receptors. J Clin Invest 1996, 98:2779–2785.
Sorkin LS, Moore J, Boyle DL, et al.: Regulation of peripheral inflammation by spinal adenosine: role of somatic afferent fibers. Exp Neurol 2003, 184:162–168.
Boyle DL, Moore J, Yang L, et al.: Spinal adenosine receptor activation inhibits inflammation and joint destruction in rat adjuvant-induced arthritis. Arthritis Rheum 2002, 46:3076–3082.
Raghavendra V, Tanga FY, DeLeo JA: Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci 2004, 20:467–473.
Kim SY, Bae JC, Kim JY, et al.: Activation of p38 MAP kinase in the rat dorsal root ganglia and spinal cord following peripheral inflammation and nerve injury. Neuroreport 2002, 13:2483–2486.
Svensson CI, Marsala M, Westerlund A, et al.: Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem 2003, 86:1534–1544.
Ji RR, Suter MR: p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain 2007, 3:33.
Boyle DL, Jones TL, Hammaker D, et al.: Regulation of peripheral inflammation by spinal p38 MAP kinase in rats. PLoS Med 2006, 3:e338.
•• Boettger MK, Weber K, Grossmann D, et al.: Spinal TNF-alpha neutralization reduces peripheral inflammation and hyperalgesia and suppresses autonomic responses in experimental arthritis. Arthritis Rheum 2010, 62:1308–1318. This study and the study by Boyle et al. [19] demonstrate that spinal activation by the proinflammatory cytokine TNF increases peripheral arthritis. TNF activates the p38 MAPK in spinal microglial cells. Local administration in the cerebrospinal fluid of a p38 MAPK inhibitor or the anti-TNF agent etanercept attenuates synovial inflammation.
Rosas-Ballina M, Tracey KJ: Cholinergic control of inflammation. J Intern Med 2009, 265:663–679.
Heart rate variability: standards of measurement, physiological interpretation and clinical use. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Circulation 1996, 93:1043–1065.
Pavlov VA, Ochani M, Gallowitsch-Puerta M, et al.: Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc Natl Acad Sci U S A 2006, 103:5219–5223.
• Waldburger JM, Boyle DL, Edgar M, et al.: Spinal p38 MAP kinase regulates peripheral cholinergic outflow. Arthritis Rheum 2008, 58:2919–2921. This study and the study by Bernik et al. [25] show that inhibition of the p38 MAPK pathway in the CNS increases the vagal anti-inflammatory pathway, suggesting that some of the effect of p38 inhibitors is mediated by this central mechanism.
Bernik TR, Friedman SG, Ochani M, et al.: Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med 2002, 195:781–788.
Borovikova LV, Ivanova S, Nardi D, et al.: Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton Neurosci 2000, 85:141–147.
Evrengul H, Dursunoglu D, Cobankara V, et al.: Heart rate variability in patients with rheumatoid arthritis. Rheumatol Int 2004, 24:198–202.
Goldstein RS, Bruchfeld A, Yang L, et al.: Cholinergic anti-inflammatory pathway activity and High Mobility Group Box-1 (HMGB1) serum levels in patients with rheumatoid arthritis. Mol Med 2007, 13:210–215.
Bruchfeld A, Goldstein RS, Chavan S, et al.: Whole blood cytokine attenuation by cholinergic agonists ex vivo and relationship to vagus nerve activity in rheumatoid arthritis. J Intern Med 2010 Feb 18 (Epub ahead of print).
Waldburger JM, Boyle DL, Pavlov VA, et al.: Acetylcholine regulation of synoviocyte cytokine expression by the alpha7 nicotinic receptor. Arthritis Rheum 2008, 58:3439–3449.
• van Maanen MA, Stoof SP, van der Zanden EP, et al.: The alpha7 nicotinic acetylcholine receptor on fibroblast-like synoviocytes and in synovial tissue from rheumatoid arthritis patients: a possible role for a key neurotransmitter in synovial inflammation. Arthritis Rheum 2009, 60:1272–1281. This study and the studies by Bruchfeld et al. [29] and Waldburger et al. [30] show that the α7 nicotinic receptor is expressed in the joints of rheumatoid arthritis patients. α7 Agonists suppress cytokine and chemokine release by peripheral blood mononuclear cells [28] and FLS [29, 30•], providing a rationale for their clinical development.
Shaked I, Meerson A, Wolf Y, et al.: MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity 2009, 31:965–973.
Pavlov VA, Parrish WR, Rosas-Ballina M, et al.: Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun 2009, 23:41–45.
van Maanen MA, Lebre MC, van der Poll T, et al.: Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum 2009, 60:114–122.
•• Rosas-Ballina M, Ochani M, Parrish WR, et al.: Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A 2008, 105:11008–11013. This study shows that the anti-inflammatory effects of the cholinergic pathway in organs not directly innervated by the vagus could be mediated by an alternative neuroanatomic route involving catecholaminergic fibers.
Grimsholm O, Rantapaa-Dahlqvist S, Dalen T, Forsgren S: Unexpected finding of a marked non-neuronal cholinergic system in human knee joint synovial tissue. Neurosci Lett 2008, 442:128–133.
Wang H, Liao H, Ochani M, et al.: Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004, 10:1216–1221.
de Jonge WJ, van der Zanden EP, The FO, et al.: Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005, 6:844–851.
Lorton D, Lubahn C, Klein N, et al.: Dual role for noradrenergic innervation of lymphoid tissue and arthritic joints in adjuvant-induced arthritis. Brain Behav Immun 1999, 13:315–334.
Straub RH, Rauch L, Fassold A, et al.: Neuronally released sympathetic neurotransmitters stimulate splenic interferon-gamma secretion from T cells in early type II collagen-induced arthritis. Arthritis Rheum 2008, 58:3450–3460.
Harle P, Pongratz G, Albrecht J, et al.: An early sympathetic nervous system influence exacerbates collagen-induced arthritis via CD4 + CD25+ cells. Arthritis Rheum 2008, 58:2347–2355.
Malfait AM, Malik AS, Marinova-Mutafchieva L, et al.: The beta2-adrenergic agonist salbutamol is a potent suppressor of established collagen-induced arthritis: mechanisms of action. J Immunol 1999, 162:6278–6283.
Straub RH, Gunzler C, Miller LE, et al.: Anti-inflammatory cooperativity of corticosteroids and norepinephrine in rheumatoid arthritis synovial tissue in vivo and in vitro. FASEB J 2002, 16:993–1000.
• Fassold A, Falk W, Anders S, et al.: Soluble neuropilin-2, a nerve repellent receptor, is increased in rheumatoid arthritis synovium and aggravates sympathetic fiber repulsion and arthritis. Arthritis Rheum 2009, 60:2892–2901. This paper describes the surprising synergistic nerve repellent effect between semaphoring 3F, a nerve repellent factor, and the soluble form of its receptor neuropilin-2. The measured increase in the concentration of soluble neuropilin-2 in rheumatoid arthritis synovial fluid thus could contribute to the loss of sympathetic fibers in the synovium rather than neutralizing the repellent semaphoring 3F.
McGrath J, McDonald JW, Macdonald JK: Transdermal nicotine for induction of remission in ulcerative colitis. Cochrane Database Syst Rev 2004, 4:CD004722.
Reale M, Iarlori C, Gambi F, et al.: Acetylcholinesterase inhibitors effects on oncostatin-M, interleukin-1 beta and interleukin-6 release from lymphocytes of Alzheimer’s disease patients. Exp Gerontol 2005, 40:165–171.
Schachter SC: Vagus nerve stimulation: where are we? Curr Opin Neurol 2002, 15:201–206.
Briggs CA, Gronlien JH, Curzon P, et al.: Role of channel activation in cognitive enhancement mediated by alpha7 nicotinic acetylcholine receptors. Br J Pharmacol 2009, 158:1486–1494.
Acknowledgments
This work was supported by an American College of Rheumatology Research and Education Foundation Within Our Reach grant, by the Swiss National Science Foundation, and by the Swiss Society of Rheumatology/Essex grant.
Disclosure
Dr. Firestein has served as a consultant for Merck & Co., Pfizer, and Takeda Pharmaceutical Co., has received research grants from Intellikine, and had expenses covered for attendance at an advisory board meeting by Pfizer. No other potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Waldburger, JM., Firestein, G.S. Regulation of Peripheral Inflammation by the Central Nervous System. Curr Rheumatol Rep 12, 370–378 (2010). https://doi.org/10.1007/s11926-010-0124-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11926-010-0124-z