
Abstract
Purpose of Review
Gastroenteropancreatic NEN (GEP-NEN) are group of malignancies with significant clinical, anatomical and molecular heterogeneity. High-grade GEP-NEN in particular present unique management challenges.
Recent Findings
In the current era, multidisciplinary management with access to a combination of functional imaging and targeted molecular profiling can provide important disease characterisation, guide individualised management and improve patient outcome. Multiple treatment options are now available, and combination and novel therapies are being explored in clinical trials.
Summary
Precision medicine is highly relevant for a heterogenous disease like NEN. The integration of dual-tracer functional PET/CT imaging, molecular histopathology and genomic data has the potential to be used to gain a more comprehensive understanding of an individual patient’s disease biology for precision diagnosis, prognostication and optimal treatment allocation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neuroendocrine neoplasms (NENs) are an exemplar of the rational application of diagnostic modalities for characterisation and treatment selection in the age of precision medicine. NENs are a diverse group of malignancies arising from epithelial cells with neuronal differentiation and secretory capacity as part of the diffuse endocrine system. The term “neuroendocrine neoplasm” encompasses the well-differentiated neuroendocrine tumours (NETs) and the poorly differentiated neuroendocrine carcinomas (NECs), which have differing pathogenesis, behaviour and prognosis [1]. NEN is histopathologically divided into grade 1 NET (G1, Ki-67 < 3%), grade 2 NET (G2, Ki-67 3–20%), grade 3 NET (G3, Ki-67 > 20%), poorly differentiated NEC (small cell/large cell subtypes, Ki-67 usually > 55%) and mixed neuroendocrine-non-neuroendocrine neoplasm (MiNEN) [2••]. NEN most commonly arises from the gastrointestinal tract and pancreas (approximately 65%), collectively called gastroenteropancreatic NEN (GEP-NEN). The overall incidence of NEN is rising, currently, approximately 7.0–9.7 cases per 100,000 depending on geography [3,4,5]. NEN should thus be elevated from its historical “rare cancer” status to the “uncommon” cancer category, hence requiring increasing awareness.
Due to non-specific hormone secretory syndromes or symptomatology, NEN is often identified late: 60–85% of patients have incurable metastatic disease at diagnosis [2••, 6]. It is at this advanced stage that patients are typically referred for multidisciplinary assessment. Initial workup of NEN typically involves conventional radiology and histopathology assessments but these alone are inadequate to provide full characterisation for this complex heterogenous disease. This review will focus on the importance of precision evaluation and the need to improve and develop diagnostic paradigms to guide personalised therapeutic treatment of GEP-NEN. Access to molecular imaging and molecular testing can resolve diagnostic uncertainty, aid prognostication and guide therapeutic selection particularly for patients with higher-grade disease where disease heterogeneity is common. We will discuss the important role of molecular imaging with positron emission tomography (PET) using somatostatin receptor (SSTR) tracers, integrated with metabolic imaging using 2-[18F]fluoro-2-deoxy-d-glucose [18F]FDG (FDG) to non-invasively assess disease biology and heterogeneity. In addition, the development and integration of molecular testing with pathway-focussed histopathological analysis and both germline and tumour somatic mutational analysis can provide further important diagnostic insights, as well as treatment stratification for selected patients with GEP-NEN [2••].
Molecular Imaging: a Non-invasive Way to Understand Whole Body Disease Biology and Guide Treatment Selection
Radiology using computed tomography (CT) and magnetic resonance (MRI) remain the cornerstone of NEN imaging and are widely available for detecting and monitoring sites of disease. However, recognised limitations of CT include the inability to identify small malignant primary NEN lesions, lymph nodes or bone metastases which are prevalent for metastatic NEN [7••, 8, 9••]. The sensitivity and specificity for NEN detection or restaging may be reduced if serial scanning is performed using non-uniform protocols [10]. It is now well established that molecular PET/CT imaging using SSTR and FDG radiotracers play essential incremental roles in the staging, restaging and theranostics selection for patients with NEN, by characterising specific disease biology.
SSTR PET/CT Imaging
SSTR (particularly subtype 2) is commonly overexpressed on well-differentiated NEN and represents a useful molecular imaging and therapeutic target [11, 12•]. The initial approved modality [111In]In-DTPA-octreotide single-photon emission computed tomography (SPECT)/CT has become superseded by PET/CT imaging due to its superior imaging resolution, diagnostic performance and quantitation [13,14,15]. Even sub-centimetre lesions with high SSTR expression can be visualised with a high target-to-background ratio. Currently available FDA-approved SSTR-targeting PET radiotracers include [68 Ga]Ga-DOTATATE, [68 Ga]Ga-DOTATOC and [64Cu]Cu-DOTATATE. Existing guidelines from the European Neuroendocrine Tumor Society (ENETS) [7••], European Association of Nuclear Medicine (EANM) [8] and Society of Nuclear Medicine & multi-society workgroup for Molecular Imaging Appropriate Use Criteria [9••] support SSTR imaging for NEN diagnosis, initial staging after histologic diagnosis, pre-surgical assessment, treatment monitoring especially for NEN lesions seen predominantly on SSTR PET, and detection of recurrent disease and importantly for selection of patients for SSTR-targeted Peptide Receptor Radionuclide Therapy (PRRT).
FDG PET/CT Imaging
FDG is the most used oncological PET imaging agent. Uptake of this radiolabeled glucose analogue correlates with tissue metabolism and proliferation, where uptake is typically high in rapidly growing tumours or tumours with metabolic reprogramming favouring glycolysis. FDG is not a NEN-specific tracer, but FDG positivity is closely correlated with higher NEN tumour grade (typically G2 or G3 NET and NEC), poor differentiation and worse prognosis [16•, 17•, 18, 19]. Studies have established an inverse relationship between proliferation rate and SSTR positivity [20, 21]. A higher proliferation rate is expected for higher-grade disease in approximately 75% of G3 NET and around 90% of NEC cases [22, 23].
Dual-Tracer Imaging (SSTR and FDG Tracers)
This combined imaging approach can provide powerful complementary information to characterize NEN biology. It is well recognized that significant heterogeneity can exist within an individual patient, such that well-differentiated lesions (SSTR-expressing) can co-exist with higher-grade components (often FDG-avid) [24, 25]. SSTR imaging positivity is a marker of well-differentiated NEN. FDG positivity is a marker of disease metabolic activity and NEN aggressiveness. The use of dual-tracer imaging can assess the heterogeneity of disease biology within a patient, impacting on prognostication and management.
As a Prognostic Biomarker
Typically, patients with FDG positive/SSTR negative disease have a poor prognosis and shorter overall survival compared to patients with FDG positive/SSTR positive, or solely SSTR positive disease respectively (latter with best prognosis) [26•, 27]. Earlier institution of more aggressive treatments and frequent monitoring is warranted for patients with highly FDG-avid disease.
To Guide Biopsy Site
Tumour grading based on ease of access or location alone may not be representative of the true highest-grade disease given the potential disease heterogeneity. Dual-tracer imaging phenotype can guide the site for targeted biopsy. Typically, the lesion with the most intense FDG uptake is likely to represent a disease of the highest proliferative activity and grade [28].
To Guide Patient Management and Therapeutic Approach
Molecular imaging phenotype guides selection for PRRT and stratifies other systemic therapies. High SSTR expression at all disease sites is the main prerequisite for PRRT. PRRT can be effective even if lesions show FDG uptake provided that all these lesions also have high SSTR expression to allow therapeutic targeting [29, 30]. Spatially discordant (FDG positive/SSTR negative) disease cannot be targeted with PRRT alone, and in this case, other systemic or combination options should be considered [26•, 31]. Given the poorer prognosis, patients with highly FDG-avid disease (concordant or discordant) should be followed up more frequently following therapy.
The dual-tracer molecular imaging approach is therefore highly recommended for patients with (1) higher-grade disease including G2 and 3 NEN; (2) patients with presumed G1 disease but with non-SSTR-avid suspicious lesions on radiological imaging; (3) at the time of more rapid progression than expected for the grade (i.e. initial pathological sampling error or transformation to higher grade); (4) to assess heterogeneity and guide biopsy site; and (5) for theranostic selection and to guide therapeutic options [12•].
Whilst SSTR imaging is now widely considered the standard of care for NEN, the combined use with FDG PET/CT is yet to be universally applied due to geographical differences in resources and regulatory limitations. Its benefits warrant further prospective validation to enable integration in NEN management.
Histopathology: Defining Morphology and Protein Expression for Diagnosis and Prognostication
Histopathological evaluation of tumour morphology, proliferative index and immunohistochemical (IHC) biomarker expression is the foundation of NEN diagnosis and grading [1, 2••, 6]. As discussed, the use of molecular imaging phenotype will guide the biopsy site to ensure sampling representative of the highest-grade lesion. Guidelines specify a minimum requirement for structured reporting of morphology, immunostaining for expression of standard neuroendocrine differentiation markers (chromogranin A, synaptophysin and CD56 or INSM1), as well as proliferation markers (Ki-67/MIB1) (1). GEP-NENs are almost always pan-cytokeratin-positive, but CK7/CK20-negative. The use of morphology and proliferative index to stratify GEP-NENs into NETs (G1-3) or NECs has prognostic and therapeutic implications; however, the optimal parameters remain controversial, and predictors of treatment response are lacking [32]. Importantly, the assessment of the Ki-67 index may be limited by sample error due to inadequate sample size or scoring methodology and should be performed by pathologists with experience in NENs to ensure accuracy and reproducibility. Patients with GEP-NET G3 have better overall survival (OS) than patients with NEC at 43.6 vs 5.3 months [33]. Patients with NEC have been reported to have a better response to platinum-based chemotherapy than NET-G3, although overall survival remains lower [33]. It is important to recognise however that classification based on morphology alone may be challenging and molecular analysis is an essential adjunct.
IHC markers of neuroendocrine cell-of-origin and differentiation are essential to resolve the common diagnostic uncertainty around defining G3 NET versus NEC. Additional IHC markers of NET differentiation include somatostatin receptor type 2 (SSTR2), which can also be used to infer somatostatin analogue (SSA) sensitivity and utility of SSTR functional imaging and is reduced in poorly differentiated cancers [34, 35]. Nuclear staining for the neuroendocrine transcription factor Insulinoma-associated protein-1 (INSM1) has very high sensitivity and specificity (99 and 96% respectively) for GEP-NET, and 100% positive and negative predictive value for differentiating pancreatic NET from other pancreatic differentials including ductal adenocarcinoma, solid pseudopapillary neoplasm and acinar cell carcinoma [36,37,38]. Loss of immunostaining for alpha-thalassemia/mental retardation X-linked (ATRX) and Death Domain Associated Protein (DAXX, pancreatic NET) correlates with loss of function mutations and is associated with well-differentiated disease and may have prognostic value [34, 39••]. Retained expression of ATRX and DAXX, but the loss of expression of retinoblastoma 1 (RB1) and SMAD4, and altered p53 expression are typical of GEP-NECs [2••, 32, 34, 39••, 40, 41]. Glucose Transporter-1 (GLUT1) positivity is a marker of aggressive behaviour and poor prognosis in GEP-NET [42,43,44], and a potential surrogate for FDG PET/CT positivity. SSTR2, INSM1, ATRX, DAXX, RB1 and p53 IHC assessments are now more frequently available in anatomical pathology departments and should be incorporated as part of standard care for complex cases unable to be resolved by routine histological examination.
Genomics of GEP-NEN: a Nuanced Understanding of Individual Disease Biology Has the Potential to Inform Patient-Specific Treatment Strategies
Whilst the integration of molecular imaging and histopathology/IHC techniques have improved NEN characterisation and patient care, a precision medicine approach is needed to manage such complex heterogenous disease and improve individualised outcome. NETs and NECs have distinct genomic profiles and gene drivers (some can be inferred via IHC as in the previous section) such that the role of genomic analysis in GEP-NEN should extend beyond the consideration of germline testing for risk management alone. Rather, genomics can aid in diagnosis, prognosis, treatment selection and trial design.
Germline Testing
Germline testing is currently only recommended for GEP-NET patients with features of clinical endocrine tumour syndromes [45,46,47,48]. It has long been known that approximately 10% of GEP-NEN is associated with germline mutations driving the classical syndromes of multiple endocrine neoplasia type 1 (MEN1, encoding the histone modifying Menin 1 protein), as well as neurofibromatosis 1 (NF1), von Hippel Lindau (VHL) and tuberous sclerosis (TSC1/TSC2). To challenge this paradigm, the seminal International Cancer Genome Consortium study involving whole genome sequencing of 98 apparently sporadic pancreatic NETs revealed previously unknown germline alterations in up to 17% of patients including homologous recombination DNA repair genes (BRCA2 and CHEK2) as well as the base-excision DNA repair gene MUTYH [49••, 50]. For patients with small intestinal GEP-NET (SI-NET), long been considered a sporadic disease notorious for a paucity of recurrent driver genes (with the exception of somatic CDKN1B in a minor fraction), germline mutations in IMPK, OGG1 and DNA repair-associated genes including CHEK2, RAD51C, ATM and MUTYH have recently also been identified [50,51,52]. The pathogenicity and clinical significance of these defects in SI-NET are at present unclear [53, 54••].
Recognising the cohort of patients with GEP-NEN who harbour DNA repair defects and have SSTR-expressing disease on molecular imaging could inform the rational allocation to combination PRRT and drugs that inhibit alternative/rescue DNA repair pathways, such as Poly-ADP Ribose (PARP) inhibitors to maximise radiosensitivity. Such a therapeutic strategy is under active investigation in the PARLuNET trial (NCT05053854), and NCT04086485. Patients with tumours driven by DNA repair defects might also plausibly benefit from a combination of radionuclide therapy and DNA-damaging agents used in the treatment of advanced NET including the antimetabolite capecitabine and the alkylating agent temozolomide [31, 55].
Somatic Profiling
Genomic profiling of NEN reveals recurrent features and has a clear diagnostic application. NETs typically have few driver mutations [56••, 57••]. Sporadic NETs frequently harbour somatic mutations in MEN1 but also VHL and TSC2 [58, 59]. Loss of function mutations in chromatin-modifying genes ATRX/DAXX corresponds to alternative lengthening of telomeres (ALT), chromosomal instability and recurrent genome-wide patterns of chromosomal loss [49••, 60, 61, 62••, 63••, 64••, 65]. Mutations in histone modifiers (e.g. SETD2, KMT2C) and chromatin remodelling genes (e.g. SWI/SNF subunits ARID1A, SMARCA4) and the PI3K/AKT/mTOR pathway (e.g. PIK3CA, PTEN, DEPDC5) are recurrent in NETs [49••, 60, 66]. YY1 mutations are enriched in insulinomas [62••, 67]. Some novel gene fusions including EWSR1-BEND2 and NET1-AKR1C3/4 have been reported in GEP-NETs [49••, 63••, 64••). The vast majority of NECs (small and large cell type) harbour mutations in TP53 plus either RB1 or CCNE1 and MYC amplifications [63••]. TP53 mutations are also common in G3NET [63••]. NEC can have tissue of origin mutation patterns, including mutations in KRAS (pancreatic NEC), APC and BRAF (colorectal NEC) [63••], while NOTCH1/2/3 inactivating mutations are enriched in non-pancreatic GI and lung NECs [63••, 68].
Somatic testing can potentially lead to targeted treatment or trial allocation in NEN, and comprehensive genomic profiling is endorsed at clinical discretion in NEN NCCN guidelines [45]. The NCI-MATCH study found that 10% of patients with unspecified subtypes of “neuroendocrine cancer” who underwent tumour panel gene testing were allocated to trials [69]. Studies of somatic mutational testing in cohorts of patients with NEN observed that, depending on the NEN subtype, more than 20% of tumours tested harbour at least one potentially actionable mutation for on-label or off-label therapies as per clinical genomic databases [64••, 70]. Commonly implicated targetable pathways include DNA repair (e.g. BRCA2, ATM, RAD51C); activation of PI3K/Akt1/mTOR signalling and inhibition of the negative PI3K/mTOR pathway regulator PTEN; and amplification of growth factor receptor signalling including EGFR, ERBB2 and FGFR [54••, 63••, 64••, 70, 71]. A small proportion of NEN harbour actionable gene fusions including NTRK fusions (multiple NEN subtypes) and ALK fusions (lung NEN) with case reports of treatment response to entrectanib and alectanib, respectively [72,73,74]. MGMT inactivation via methylation has been demonstrated to occur broadly in NEN; however, the most appropriate MGMT promotor methylation assay thresholds for NEN and their use to predict disease response to temozolomide have not yet been clearly established [75,76,77]. Somatic testing for high tumour mutational burden (TMB; TMB-high > 10 mutations/Mb) can identify patients in whom immune checkpoint inhibitor (ICI) therapy may be effective, though this has been found in only approximately 5–6% of NEN [45, 78•]. TMB-high NEN have been found to harbour defects in DNA repair (MSI, MUTYH-deficiency) or to have smoking-associated (lung NEN) or treatment-associated (alkylating agent) mutational signatures [66].
Liquid Biopsy
The detection and analysis of circulating tumour DNA (ctDNA) from blood sampling are a non-invasive method to overcome procedural risks and the issues of undersampling of disease heterogeneity inherent in tissue biopsy. Given the limitations in sensitivity and specificity of current markers such as chromogranin A for diagnosis/prognostication in NEN, novel non-invasive biomarkers are sorely needed. Feasibility has been demonstrated by Zakka et al. who undertook ctDNA analysis using Guardant360® assay (73 gene panel) of 320 patients with NEN, finding molecular alterations in 87.5% of patients [79••]. Other novel ctDNA biomarkers under investigation in NEN include copy number change and methylation pattern [80, 81••]. Another approach, the NETest™, is a 51-gene panel detecting circulating tumour RNA, the levels of which are extrapolated to reflect “-omic” biological pathway perturbations reported as a “disease activity score” between 0 and 100% [82]. The NETest™ is not in widespread use due to limited independent validation and assay complexity.
Treatment Selection for GEP NEN: Current Approach and Future Perspectives for Precision Therapy
The selection of therapy for NENs is currently primarily based on histology (grade), primary site, structural/functional imaging, IHC and clinical behaviour. As described, molecular characterisation (e.g. TMB status) may have a role in future treatment decision-making.
Grade 1 and 2 NETs—More Indolent Disease
First-line Therapy
Somatostatin analogues (SSAs, either depot octreotide or Lanreotide) have demonstrated antisecretory and antiproliferative effects in terms of disease progression, but without significant overall survival benefit [83,84,85]. The most favourable effect was observed in patients with low hepatic tumour load [83] and in Ki-67 < 10% [85].
Second Line and Beyond
PRRT, molecular targeted agents (MTAs: everolimus and sunitinib) and chemotherapy. These are utilised in patients not suitable for SSA, if there is rapid disease progression, or poor prognostic features (high burden, high grade or FDG-avid disease).
For PRRT, patient selection is based upon functional imaging demonstrating high tumour SSTR expression without discordant FDG-avid disease (where performed). Its approval was based on phase III NETTER-1 trial, which demonstrated a 20-month PFS rate benefit in midgut NET in favour of 177Lu-DotaTate PRRT (65.2%) versus high dose Octreotide LAR alone (10.8%) [86]. The lack of OS advantage can be explained by high cross over into the PRRT group [87••]. Several series have demonstrated the benefit of PRRT in other primary sites, especially pancreatic NETs [88]. A meta-analysis compared 177Lu DOTATATE PRRT with everolimus and observed that the ORR and PFS were greater for PRRT: 47% vs 12% and 25.7 vs 14.7 months, respectively (P < 0.001) [89••]. A randomised phase II trial in patients with pancreatic NEN also confirmed the superiority of PRRT versus sunitinib [90••]. The completed COMPETE phase III study has compared 177Lu DOTANOC PRRT to everolimus (NCT03049189). Current trials are evaluating PRRT combined with PARP inhibitors (NCT05053854) and capecitabine (NCT02736448). Retreatment of patients with PRRT is feasible, with a recent meta-analysis demonstrating a median PFS of 12.5 months and OS of 26.8 months, with a similar safety profile as initial therapy [91••].
Regarding MTAs, everolimus has demonstrated increased PFS relative to placebo in pancreatic NETs (HR = 035, P < 0.001) [92], non-functional pulmonary NETs and GEP NETs (HR = 0.48, P < 0.00001) [93]. Sunitinib has a PFS advantage relative to placebo for pancreatic NETs: HR = 0.42, P < 0.001 [94].
In terms of chemotherapy, modern phase III trials are lacking. Patients selected are those with progression post-SSA, PRRT or MTAs, if unsuitable for PRRT, or those with large volume or rapidly progressive disease. The integration of dual-tracer molecular imaging plays an important role in identifying patients with these poor prognostic features. Chemotherapy is more active in patients with pancreatic NETs, with ORR from 31 to 70% and OS exceeding 40 months [95•]. Regimens include capecitabine plus temozolomide (CapTem), temzolomide, FOLFOX, capecitabine-oxaliplatin (CapOx) and streptozotocin-5FU. The activity of CapTem was confirmed by the randomised phase II E211 trial [96••].
The optimal therapy sequencing of the available options, however, has not been validated. The SEQTOR study (GETNE 1206) randomised patients with progressive pancreatic NET to everolimus followed by streptozotocin-5FU upon progression (arm A), or the reverse sequence (arm B). On initial analysis, both sequential strategies showed similar efficacy and PFS [97••].
Grade 3 NETs and NECs
The treatment approach for patients with G3 NETs and NECs differs substantially given their histopathology, imaging characteristics and genomics (see Table 1). Given the more aggressive nature of the disease, early institution of therapy is important to optimise patient outcome: the integration of molecular imaging and molecular profiling could play an important role for these patients.
First-line Therapy
The clinical behaviour of NECs is similar to extensive-stage small cell lung cancer (SCLC) [22]. Treatment is platinum-based (cisplatin/carboplatin) plus etoposide (EP), with median survival ranging from 9.5 to 19 months [98••, 99, 100, 101•, 102, 103] ad a short median PFS from 4 to 6 months with an ORR of 30–50% [98••, 100, 104]. Irinotecan plus cisplatin, based on Japanese randomised trials, showed similar or superior response rates relative to EP [105, 106••]. Differentiation status in G3 disease [102, 107] and a Ki-67 ≤ 60% predict less benefit from platin-based chemotherapy [100]. In G3 NET, the ORR to platin-based regimens is < 5%, with PFS < 3 months, but prolonged OS [18, 100, 108, 109].
Hence, patients with G3 NETs benefit from similar therapies used in G2 NETs [18, 22]. Several heterogeneous retrospective series have indicated activity for CapTem in G3 NETs: ORR varies from 30 to 51%, median PFS of 9 to 15.3 months and OS from 19 to 29.3 months [110,111,112,113,114]. The optimal threshold for higher ORR is a Ki-67 from 10 to 40% [115]. Data for other therapies in G3 NET is limited. The pivotal SSA phase III trials had not included G3NET [83, 116] and so their use should be limited to patients with confirmed SSTR expression, no FDG discordance (this should be closely monitored), or for management of secretory syndromes [117]. The data on MTAs in G3 NET is sparse. Everolimus has been evaluated in patients with G3NETs (Ki-67 20–55%) in the first/second-line setting (N = 15), with a median PFS of 6 months, and OS of 28 months [118]. A completed German study (EVINEC) has evaluated everolimus as a second-line treatment for G3 NET and G3 NEC (NCT02113800). Sunitinib was evaluated in 31 patients with pancreatic grade 3 NET/NECs: with partial response in 4 and stable disease seen in 14 patients [119]. A completed Nordic phase II study has evaluated temozolomide and everolimus as first-line treatment in metastatic G3NET (Ki-67 21–55%) (NCT02248012).
Second Line and Beyond
Patients with NECs/G3 NETs may benefit from subsequent chemotherapy [100]. Options for G3 NETs include chemotherapy (subject to prior exposure), MTAs (as above) and PRRT. In the case of NEC, patients that have progressed in ≥ 3 months post platinum-based treatment may still be platinum-sensitive (100). Other regimens include FOLFIRI, FOLFOX and CapTem. In terms of Irinotecan-5FU-based regimens, the ORR ranges from 17 to 40%, PFS 4–5.8 months and OS 5–11 months [120,121,122]. For Oxaliplatin-5FU, PR ranges from 23 to 29%, PFS 4.5 months and OS 9.9 months [123,124,125]. CapeTem has also demonstrated activity in this setting [113]. However, patients with Ki-67 > 55% have worse outcomes [126]. The SEcond-line therapy in NEuroendocrine CArcinomas (SENECA) phase II study is evaluating FOLFIRI or CAPTEM post failure of first-line chemotherapy in patients with lung and NEC [127].
PRRT is an option, as G3NETs have the greater propensity for SSTR expression relative to NECs: its utility here has been reported by several small studies [31, 128,129,130, 131••]. In the largest series reported (N = 69 G3NET/NEC), the median PFS was 9.6 months, and the median OS was 19.9 months; for patients with Ki-67 ≤ 55% (n = 53), the median PFS was 11 months and OS 22 months, for those with Ki-67 > 55% (n = 11), 4 months and 7 months, respectively [31]. An analysis of 4 studies where PRRT was used in the second/third line setting: overall PFS was 19 months in G3NET, 11 months for NEC (Ki-67 ≤ 55%) and 4 months for NEC (Ki-67 > 55%) [131••]. Thus, PRRT may be considered for patients in G3 NENs with Ki-67 < 55% [31, 131••]. Current trials include the phase III COMPOSE study of 177Lu-DOTANOC versus systemic therapy (NCT04919226) and the NETTER-2 phase III trial randomising patients to PRRT versus high dose SSA (NCT03972488). PRRT is being combined with Nivolumab (NCT04525638).
ICI is also promising in progressive high-grade NET and NEC, based on their higher TMB; the latter is greater in NECs and with microsatellite instability noted in 14% of NECs [132]. A meta-analysis of 10 heterogenous, single-arm studies of ICI in NEN (N = 464) found a pooled ORR of 15.5% [133). The response was based on primary site: with thoracic NEN being more likely to respond than GEP-NEN (ORR 24.7% vs 9.5% respectively) and well-differentiated tumours having a lower response rate than NECs (ORR 10.4% vs 22.7% respectively) [133]. Very limited activity has been observed with single-agent immunotherapy [134, 135], relative to combined PD1 and CTL4 blockade. From the CA209-538 study, 29 patients with heavily pre-treated NETs were treated with a combination of ipilimumab and nivolumab. Overall, in the 13 (45%) with high-grade disease, the ORR was 24% and a DCR of 72% [136••]. The SWOG S1609 DART trial reported the results of the high-grade G3 NET/NEC cohort (N = 19) with a median Ki-67 value of 80%. The ORR was 26% and the clinical benefit rate (stable disease for ≥ 6 months plus PR and CR) was 32% [137••]. Other trials are yet to be reported, including a phase II trial of PDR001 (PD-L1 inhibitor) (NCT02955069), Nivolumab combined with EP (NCT03980925) and toripalimab in pancreatic NEN (NCT03043664, NCT02939651 and NCT03147404). Even within TMB-high NENs, however, there is a heterogeneous response to ICI highlighting the need for further biomarkers for stratification.
Perspectives for Precision Therapy Utilizing Multidisciplinary Diagnostic Approaches
NEN is a challenging, heterogenous disease with different clinical, imaging, pathological and genomic complexities to consider in each patient. Multiple treatment options are now available, and combination and novel therapies are being explored in clinical trials. However, clinical treatment selection and sequencing are still mainly based on disease grade, primary site, agent availability and local protocols, without personalisation. Precision medicine is highly relevant for a heterogenous disease like NEN. In the current era, the integration of molecular imaging (SSTR and FDG PET/CT) and molecular profiling (IHC profile and genomic analyses) can provide important disease characterisation, to guide precision management and individualised treatment selection/sequencing (see Fig. 1). This is particularly crucial for patients with advanced high-grade NENs and to resolve G3 NET vs NEC disease biology, as clinical behaviour and treatment options can differ significantly. It is also imperative to focus on incorporating prospective serial translational genomic analysis of tissue and blood, to develop novel liquid biopsy and tumour testing methodologies to understand NEN pathogenesis, discover predictive and prognostic biomarkers to explain the differential response to therapy and subsequently guide future trial design for rational treatment allocation. Using multidisciplinary diagnostic approaches should be the focus of future development to improve individualised therapy and patient outcomes.

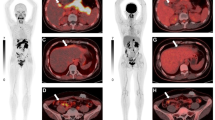
A case example of a 54-year-old female, with a previous history of treated localised breast cancer, and previously resected grade 1 (Ki-67 2%) pancreatic NET. She presented with new, multiple hepatic (A) and mesenteric nodal metastases (B). A Ga-68 DOTATATE PET/CT (C) showed metastatic disease in the liver, nodes and bones with high SSTR expression. FDG PET/CT (D) showed some lesions with concordant FDG avidity. The lesion with the highest metabolic activity (mesenteric node, E) was targeted for biopsy and diagnosis. Histopathology (F) showed monotonous cuboidal cells with granular eosinophilic cytoplasm, ovoid nuclei and fine chromatin. By IHC Ki-67 labelling index was 25% (G) and DAXX expression was lost (H). Other IHC (not shown) demonstrated expression of SSTR2 and synaptophysin, retained ATRX and Rb, a p53 wild-type pattern, and no staining for chromogranin or multiple breast markers. Overall, the features were supportive of a G3 NET and not breast carcinoma or NEC. Genomic sequencing confirmed DAXX mutation and MEN1 mutation, typical for NET. The patient proceeded to receive PRRT treatment for metastatic G3 NET
Conclusion
We are in an exciting era for the biological interrogation of neuroendocrine neoplasms to guide precision management by incorporating molecular imaging assessment with clinically relevant molecular pathology pathway and genomic evaluation. Our technological capability for precision diagnosis needs to be developed in parallel with therapy advancements in patients with advanced-stage higher-grade NEN and globally is only a reality for patients who have geographical or financial access to major NEN referral centres [138]. It is therefore imperative not only to place molecular imaging and genomics at the centre of NEN patient management but to also show the symptomatic, survival and health economic benefits of doing so through high-quality research such that these technologies are widely supported by guidelines and imbursed by regulatory bodies.
References
Papers of particular interest, published recently, have been highlighted as: • of importance •• of major importance
Perren A, Couvelard A, Scoazec JY, Costa F, Borbath I, DelleFave G, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: pathology: diagnosis and prognostic stratification. Neuroendocrinology. 2017;105(3):196–200.
•• Nagtegaal ID, Odze RD, Klimstra D, Paradis V, Rugge M, Schirmacher P, et al. The 2019 WHO classification of tumours of the digestive system. Histopathology. 2020;76(2):182–8. WHO guidelines for GEP-NEN classification.
Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, et al. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017;3(10):1335–42.
Michael M, Thursfield V, Te Marvelde L, Kong G, Hicks RJ. Incidence, prevalence, and survival trends for neuroendocrine neoplasms in Victoria, Australia, from 1982 to 2019: based on site, grade, and region. Asia Pac J Clin Oncol 2021;18(5):e187–e528
Takayanagi D, Cho H, Machida E, Kawamura A, Takashima A, Wada S, et al. Update on epidemiology, diagnosis, and bio markers in gastroenteropancreatic neuroendocrine neoplasms. Cancers (Basel). 2022;14(5):1119
Garcia-Carbonero R, Sorbye H, Baudin E, Raymond E, Wiedenmann B, Niederle B, et al. ENETS consensus guidelines for high-grade gastroenteropancreatic neuroendocrine tumors and neuroendocrine carcinomas. Neuroendocrinology. 2016;103(2):186–94.
•• Sundin A, Arnold R, Baudin E, Cwikla JB, Eriksson B, Fanti S, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: radiological, nuclear medicine & hybrid imaging. Neuroendocrinology. 2017;105(3):212–44. Comprehensive review of radiology and nuclear medicine functional imaging for NEN.
Bozkurt MF, Virgolini I, Balogova S, Beheshti M, Rubello D, Decristoforo C, et al. Guideline for PET/CT imaging of neuroendocrine neoplasms with (68)Ga-DOTA-conjugated somatostatin receptor targeting peptides and (18)F-DOPA. Eur J Nucl Med Mol Imaging. 2017;44(9):1588–601.
•• Hope TA, Bergsland EK, Bozkurt MF, Graham M, Heaney AP, Herrmann K, et al. Appropriate use criteria for somatostatin receptor PET imaging in neuroendocrine tumors. J Nucl Med : Off Publ, Soc Nucl Med. 2018;59(1):66–74. SNMMI guidelines for SSTR PET/CT imaging in NEN.
Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9(1):61–72.
Reubi JC, Waser B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging. 2003;30(5):781–93.
• Kong G, Hicks RJ. Peptide receptor radiotherapy: current approaches and future directions. Curr Treat Options Oncol. 2019;20(10):77. Current outline of functional imaging and dual tracer imaging approach for NEN.
Deppen SA, Blume J, Bobbey AJ, Shah C, Graham MM, Lee P, et al. 68Ga-DOTATATE compared with 111In-DTPA-octreotide and conventional imaging for pulmonary and gastroenteropancreatic neuroendocrine tumors: a systematic review and meta-analysis. J Nucl Med : Off Publ, Soc Nucl Med. 2016;57(6):872–8.
Sadowski SM, Neychev V, Millo C, Shih J, Nilubol N, Herscovitch P, et al. Prospective study of 68Ga-DOTATATE positron emission tomography/computed tomography for detecting gastro-entero-pancreatic neuroendocrine tumors and unknown primary sites. J Clin Oncol. 2016;34(6):588–96.
Skoura E, Michopoulou S, Mohmaduvesh M, Panagiotidis E, Harbi MA, Toumpanakis C, et al. The impact of 68Ga-DOTATATE PET/CT imaging on management of patients with neuroendocrine tumors: experience from a national referral center in the United Kingdom. J Nucl Med. 2016;57(1):34–40.
• Binderup T, Knigge U, Loft A, Federspiel B, Kjaer A. 18F-fluorodeoxyglucose positron emission tomography predicts survival of patients with neuroendocrine tumors. Clin Cancer Res. 2010;16(3):978–85. Important study showing the prognostic role of FDG PET/CT in NEN.
• Garin E, Le Jeune F, Devillers A, Cuggia M, de Lajarte-Thirouard AS, Bouriel C, et al. Predictive value of 18F-FDG PET and somatostatin receptor scintigraphy in patients with metastatic endocrine tumors. J Nucl Med : Off Publ, Soc Nucl Med. 2009;50(6):858–64. Important study showing the prognostic role of FDG PET/CT in NEN.
Heetfeld M, Chougnet CN, Olsen IH, Rinke A, Borbath I, Crespo G, et al. Characteristics and treatment of patients with G3 gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2015;22(4):657–64.
Zhang P, Yu J, Li J, Shen L, Li N, Zhu H, et al. Clinical and prognostic value of PET/CT imaging with combination of (68)Ga-DOTATATE and (18)F-FDG in gastroenteropancreatic neuroendocrine neoplasms. Contrast Media Mol Imaging. 2018;2018:2340389.
Ezziddin S, Logvinski T, Yong-Hing C, Ahmadzadehfar H, Fischer HP, Palmedo H, et al. Factors predicting tracer uptake in somatostatin receptor and MIBG scintigraphy of metastatic gastroenteropancreatic neuroendocrine tumors. J Nucl Med : Off Publ, Soc Nucl Med. 2006;47(2):223–33.
Kayani I, Bomanji JB, Groves A, Conway G, Gacinovic S, Win T, et al. Functional imaging of neuroendocrine tumors with combined PET/CT using 68Ga-DOTATATE (DOTA-DPhe1, Tyr3-octreotate) and 18F-FDG. Cancer. 2008;112(11):2447–55.
Sorbye H, Baudin E, Perren A. The problem of high-grade gastroenteropancreatic neuroendocrine neoplasms: well-differentiated neuroendocrine tumors, neuroendocrine carcinomas, and beyond. Endocrinol Metab Clin North Am. 2018;47(3):683–98.
Walter T, Tougeron D, Baudin E, Le Malicot K, Lecomte T, Malka D, et al. Poorly differentiated gastro-entero-pancreatic neuroendocrine carcinomas: are they really heterogeneous? Insights from the FFCD-GTE national cohort. Eur J Cancer. 2017;79:158–65.
Tang LH, Untch BR, Reidy DL, O’Reilly E, Dhall D, Jih L, et al. Well-differentiated neuroendocrine tumors with a morphologically apparent high-grade component: a pathway distinct from poorly differentiated neuroendocrine carcinomas. Clin Cancer Res. 2016;22(4):1011–7.
Shi C, Gonzalez RS, Zhao Z, Koyama T, Cornish TC, Hande KR, et al. Liver metastases of small intestine neuroendocrine tumors: Ki-67 heterogeneity and World Health Organization grade discordance with primary tumors. Am J Clin Pathol. 2015;143(3):398–404.
• Chan DL, Pavlakis N, Schembri GP, Bernard EJ, Hsiao E, Hayes A, et al. Dual somatostatin receptor/FDG PET/CT imaging in metastatic neuroendocrine tumours: proposal for a novel grading scheme with prognostic significance. Theranostics. 2017;7(5):1149–58. One of the first studies highlighting the role of dual tracer imaging in patients with NEN.
Carideo L, Prosperi D, Panzuto F, Magi L, Pratesi MS, Rinziv illo M, et al. Role of combined [(68)Ga]Ga-DOTA-SST Ana logues and [(18)F]FDG PET/CT in the management of GEP NENs: a systematic review. J Clin Med. 2019;8(7):1032
de Mestier L, Armani M, Cros J, Hentic O, Rebours V, Cadiot G, et al. Lesion-by-lesion correlation between uptake at FDG PET and the Ki67 proliferation index in resected pancreatic neuroendocrine tumors. Dig Liver Dis. 2019;51(12):1720–24
Kashyap R, Hofman MS, Michael M, Kong G, Akhurst T, Eu P, et al. Favourable outcomes of (177)Lu-octreotate peptide receptor chemoradionuclide therapy in patients with FDG-avid neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2015;42(2):176–85.
Ambrosini V, Kunikowska J, Baudin E, Bodei L, Bouvier C, Capdevila J, et al. Consensus on molecular imaging and theranostics in neuroendocrine neoplasms. Eur J Cancer. 2021;146:56–73.
Thang SP, Lung MS, Kong G, Hofman MS, Callahan J, Michael M, et al. Peptide receptor radionuclide therapy (PRRT) in European Neuroendocrine Tumour Society (ENETS) grade 3 (G3) neuroendocrine neoplasia (NEN) - a single-institution retrospective analysis. Eur J Nucl Med Mol Imaging. 2018;45(2):262–77.
Hayes AR, Furnace M, Shah R, Rundell C, Muller G, Dehbi HM, et al. High-grade gastroenteropancreatic neuroendocrine neoplasms and improved prognostic stratification with the new world health organization 2019 classification: a validation study from a single-institution retrospective analysis. Pancreas. 2021;50(4):516–23.
Milione M, Maisonneuve P, Spada F, Pellegrinelli A, Spaggiari P, Albarello L, et al. The clinicopathologic heterogeneity of grade 3 gastroenteropancreatic neuroendocrine neoplasms: morphological differentiation and proliferation identify different prognostic categories. Neuroendocrinology. 2017;104(1):85–93.
Konukiewitz B, Schlitter AM, Jesinghaus M, Pfister D, Steiger K, Segler A, et al. Somatostatin receptor expression related to TP53 and RB1 alterations in pancreatic and extrapancreatic neuroendocrine neoplasms with a Ki67-index above 20. Mod Pathol. 2017;30(4):587–98.
Kaemmerer D, Träger T, Hoffmeister M, Sipos B, Hommann M, Sänger J, et al. Inverse expression of somatostatin and CXCR4 chemokine receptors in gastroenteropancreatic neuroendocrine neoplasms of different malignancy. Oncotarget. 2015;6(29):27566–79.
Tanigawa M, Nakayama M, Taira T, Hattori S, Mihara Y, Kondo R, et al. Insulinoma-associated protein 1 (INSM1) is a useful marker for pancreatic neuroendocrine tumor. Med Mol Morphol. 2018;51(1):32–40.
Zhang Q, Huang J, He Y, Cao R, Shu J. Insulinoma-associated protein 1(INSM1) is a superior marker for the diagnosis of gastroenteropancreatic neuroendoerine neoplasms: a meta-analysis. Endocrine. 2021;74(1):61–71.
González I, Lu HC, Sninsky J, Yang C, Bishnupuri K, Dieckgraefe B, et al. Insulinoma-associated protein 1 expression in primary and metastatic neuroendocrine neoplasms of the gastrointestinal and pancreaticobiliary tracts. Histopathology. 2019;75(4):568–77.
•• Mafficini A, Scarpa A. Genetics and epigenetics of gastroenteropancreatic neuroendocrine neoplasms. Endocr Rev. 2019;40(2):506–36. Comprehensive review highlighting different biology of GEP-NET and GEP-NEC and therapeutically relevant genomic pathway alterations.
Metovic J, La Salvia A, Rapa I, Napoli F, Birocco N, Pia Bizzi M, et al. Molecular subtypes of extra-pulmonary neuroendocrine carcinomas identified by the expression of neuroendocrine lineage-specific transcription factors. Endocr Pathol. 2022;33(3):388–99.
Busico A, Maisonneuve P, Prinzi N, Pusceddu S, Centonze G, Garzone G, et al. Gastroenteropancreatic high-grade neuroendocrine neoplasms: histology and molecular analysis, two sides of the same coin. Neuroendocrinology. 2020;110(7–8):616–29.
Fujino M, Aishima S, Shindo K, Oda Y, Morimatsu K, Tsutsumi K, et al. Expression of glucose transporter-1 is correlated with hypoxia-inducible factor 1α and malignant potential in pancreatic neuroendocrine tumors. Oncol Lett. 2016;12(5):3337–43.
Binderup T, Knigge UP, Federspiel B, Sommer P, Hasselby JP, Loft A, et al. Gene expression of glucose transporter 1 (GLUT1), hexokinase 1 and hexokinase 2 in gastroenteropancreatic neuroendocrine tumors: correlation with F-18-fluorodeoxyglucose positron emission tomography and cellular proliferation. Diagnostics (Basel). 2013;3(4):372–84.
Sampedro-Núñez M, Bouthelier A, Serrano-Somavilla A, Mar- tínez-Hernández R, Adrados M, Martín-Pérez E, et al. LAT-1 and GLUT-1 carrier expression and its prognostic value in gas- troenteropancreatic neuroendocrine tumors. Cancers (Basel). 2020;12(10):2968
Shah MH, Goldner WS, Benson AB, Bergsland E, Blaszkowsky LS, Brock P, et al. Neuroendocrine and adrenal tumors, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2021;19(7):839–68.
Falconi M, Eriksson B, Kaltsas G, Bartsch DK, Capdevila J, Caplin M, et al. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology. 2016;103(2):153–71.
Pavel M, Öberg K, Falconi M, Krenning EP, Sundin A, Perren A, et al. Gastroenteropancreatic neuroendocrine neoplasms: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(7):844–60.
Halfdanarson TR, Strosberg JR, Tang L, Bellizzi AM, Bergsland EK, O’Dorisio TM, et al. The North American neuroendocrine tumor society consensus guidelines for surveillance and medical management of pancreatic neuroendocrine tumors. Pancreas. 2020;49(7):863–81.
•• Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543(7643):65–71. Seminal International Cancer Genome Consortium paper describing novel germaline mutations (e.g. BRCA2, CHEK2) and the somatic mutations landscape of pancreatic NET.
Dumanski JP, Rasi C, Björklund P, Davies H, Ali AS, Grönberg M, et al. A MUTYH germline mutation is associated with small intestinal neuroendocrine tumors. Endocr Relat Cancer. 2017;24(8):427–43.
Perez K, Kulke MH, Chittenden A, Ukaegbu C, Astone K, Alexander H, et al. Clinical implications of pathogenic germline variants in small intestine neuroendocrine tumors (SI-NETs). JCO Precis Oncol. 2021;5:808–16.
Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet. 2013;45(12):1483–6.
Helderman NC, Elsayed FA, van Wezel T, Terlouw D, Langers AMJ, van Egmond D, et al. Mismatch repair deficiency and MUTYH variants in small intestine-neuroendocrine tumors. Hum Pathol. 2022;125:11–7.
•• Scarpa A. The landscape of molecular alterations in pancreatic and small intestinal neuroendocrine tumours. Ann Endocrinol (Paris). 2019;80(3):153–8. Very good review of GEP-NET genomics.
Pavlakis N, Ransom DT, Wyld D, Sjoquist KM, Wilson K, Gebski V, et al. Australasian gastrointestinal trials group (AGITG) CONTROL NET Study: 177Lu-DOTATATE peptide receptor radionuclide therapy (PRRT) and capecitabine plus temozolomide (CAPTEM) for pancreas and midgut neuroendocrine tumours (pNETS, mNETS)—Final results. J Clin Oncol. 2022;40(16_suppl):4122-.
Pan-cancer analysis of whole genomes. Nature. 2020;578(7793):82-93. International Cancer Genome Consortium (ICGC)/The Cancer Genome Atlas (TCGA) Pan-Cancer Analysis of Whole Genomes describes the paucity of driver mutations in pancreatic NEN, suggesting other mechanisms for carcinogenesis.
•• Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature. 2019;575(7781):210–6. Seminal study of mutational landscape of metastatic cancer. Further highlights the dearth of driver mutations in NEN (especially SI-NEN).
Corbo V, Dalai I, Scardoni M, Barbi S, Beghelli S, Bersani S, et al. MEN1 in pancreatic endocrine tumors: analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr Relat Cancer. 2010;17(3):771–83.
Görtz B, Roth J, Krähenmann A, de Krijger RR, Muletta-Feurer S, Rütimann K, et al. Mutations and allelic deletions of the MEN1 gene are associated with a subset of sporadic endocrine pancreatic and neuroendocrine tumors and not restricted to foregut neoplasms. Am J Pathol. 1999;154(2):429–36.
Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331(6021):1199–203.
Marinoni I, Kurrer AS, Vassella E, Dettmer M, Rudolph T, Banz V, et al. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology. 2014;146(2):453-60.e5.
•• Hong X, Qiao S, Li F, Wang W, Jiang R, Wu H, et al. Whole-genome sequencing reveals distinct genetic bases for insulinomas and non-functional pancreatic neuroendocrine tumours: leading to a new classification system. Gut. 2020;69(5):877–87. Evidence for whole genome sequencing of pancreatic NET for prognostically relevant subtyping by copy number, ATRX/DAXX. Describes genetic difference between insulinoma and non-functioning NET.
•• Yachida S, Totoki Y, Noe M, Nakatani Y, Horie M, Kawasaki K, et al. Comprehensive genomic profiling of neuroendocrine carcinomas of the gastrointestinal system. Cancer Discov. 2022;12(3):692–711. Recent comprehensive somatic analysis of GEP-NEN (focus on NEC) describing molecular subtypes and novel epigenetic, viral drivers for NEN development.
•• van Riet J, van de Werken HJG, Cuppen E, Eskens F, Tesselaar M, van Veenendaal LM, et al. The genomic landscape of 85 advanced neuroendocrine neoplasms reveals subtype-heterogeneity and potential therapeutic targets. Nat Commun. 2021;12(1):4612. Study involving comprehensive genomic analysis of advanced NEN revealing genomic subtypes relating to grade and location as well as highlighting discovery of clinically actionable targets.
Lawrence B, Blenkiron C, Parker K, Tsai P, Fitzgerald S, Shields P, et al. Recurrent loss of heterozygosity correlates with clinical outcome in pancreatic neuroendocrine cancer. NPJ Genom Med. 2018;3:18.
Han X, Chen W, Chen P, Zhou W, Rong Y, Lv Y, et al. Aberration of ARID1A is associated with the tumorigenesis and prognosis of sporadic nonfunctional pancreatic neuroendocrine tumors. Pancreas. 2020;49(4):514–23.
Cao Y, Gao Z, Li L, Jiang X, Shan A, Cai J, et al. Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat Commun. 2013;4:2810.
George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524(7563):47–53.
Flaherty KT, Gray RJ, Chen AP, Li S, McShane LM, Patton D, et al. Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: national cancer institute molecular analysis for therapy choice (NCI-MATCH). J Clin Oncol. 2020;38(33):3883–94.
Venizelos A, Elvebakken H, Perren A, Nikolaienko O, Deng W, Lothe IMB, et al. The molecular characteristics of high-grade gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2021;29(1):1–14.
Puccini A, Poorman K, Salem ME, Soldato D, Seeber A, Goldberg RM, et al. Comprehensive genomic profiling of gastroenteropancreatic neuroendocrine neoplasms (GEP-NENs). Clin Cancer Res. 2020;26(22):5943–51.
Sigal DS, Bhangoo MS, Hermel JA, Pavlick DC, Frampton G, Miller VA, et al. Comprehensive genomic profiling identifies novel NTRK fusions in neuroendocrine tumors. Oncotarget. 2018;9(88):35809–12.
Akhoundova D, Haberecker M, Fritsch R, Höller S, Kiessling MK, Rechsteiner M, et al. Targeting ALK in neuroendocrine tumors of the lung. Front Oncol. 2022;12:911294.
Sigal D, Tartar M, Xavier M, Bao F, Foley P, Luo D, et al. Activity of entrectinib in a patient with the first reported NTRK fusion in neuroendocrine cancer. J Natl Compr Canc Netw. 2017;15(11):1317–22.
de Mestier L, Couvelard A, Blazevic A, Hentic O, de Herder WW, Rebours V, et al. Critical appraisal of MGMT in digestive NET treated with alkylating agents. Endocr Relat Cancer. 2020;27(10):R391-r405.
Della Monica R, Cuomo M, Visconti R, di Mauro A, Buonaiuto M, Costabile D, et al. Evaluation of MGMT gene methylation in neuroendocrine neoplasms. Oncol Res. 2022;28(9):837–45.
Qi Z, Tan H. Association between MGMT status and response to alkylating agents in patients with neuroendocrine neo- plasms: a systematic review and meta-analysis. Biosci Rep. 2020;40(3):BSR20194127
• Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–65. The majority of patients with NEN in this study had TMB below 10 mutations per megabase and this group had low response rate to the immune checpoint inhibitor pembrolizumab.
•• Zakka K, Nagy R, Drusbosky L, Akce M, Wu C, Alese OB, et al. Blood-based next-generation sequencing analysis of neuroendocrine neoplasms. Oncotarget. 2020;11(19):1749–57. Demonstrates the feasibility of panel sequencing of ctDNA in patients with NEN.
Mettler E, Fottner C, Bakhshandeh N, Trenkler A, Kuchen R, 997 Weber MM. Quantitative analysis of plasma cell-free DNA and 998 its DNA integrity and hypomethylation status as biomarkers for 999 tumor burden and disease progression in patients with metastatic 1000 neuroendocrine neoplasias. Cancers (Basel). 2022;14(4):1025
•• Boons G, Vandamme T, Marien L, Lybaert W, Roeyen G, Rondou T, et al. Longitudinal copy-number alteration analysis in plasma cell-free DNA of neuroendocrine neoplasms is a novel specific biomarker for diagnosis, prognosis, and follow-up. Clin Cancer Res. 2022;28(2):338–49. This study suggests a prognostic role of longitudinal circulating tumour DNA in patients with pancreatic NEN. Also suggests that ctDNA copy number profile can differentiate between pancreatic NEN and pancreatic adenocarcinoma.
Modlin IM, Kidd M, Bodei L, Drozdov I, Aslanian H. The clinical utility of a novel blood-based multi-transcriptome assay for the diagnosis of neuroendocrine tumors of the gastrointestinal tract. Am J Gastroenterol. 2015;110(8):1223–32.
Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–63.
Rinke A, Wittenberg M, Schade-Brittinger C, Aminossadati B, Ronicke E, Gress TM, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors (PROMID): results of long-term survival. Neuroendocrinology. 2017;104(1):26–32.
Caplin ME, Pavel M, Cwikla JB, Phan AT, Raderer M, Sedlackova E, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224–33.
Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 Trial of (177)Lu-dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125–35.
•• Strosberg JR, Caplin ME, Kunz PL, Ruszniewski PB, Bodei L, Hendifar A, et al. (177)Lu-dotatate plus long-acting octreotide versus highdose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021;22(12):1752–63. The final reports of the first randomised phase III trial to demonstrate the efficacy of PRRT in midgut NENs in terms of PFS. OS was not impacted by cross over.
Starr JS, Sonbol MB, Hobday TJ, Sharma A, Kendi AT, Halfdanarson TR. Peptide receptor radionuclide therapy for the treatment of pancreatic neuroendocrine tumors: recent insights. Onco Targets Ther. 2020;13:3545–55.
•• Satapathy S, Mittal BR. 177Lu-DOTATATE peptide receptor radionuclide therapy versus Everolimus in advanced pancreatic neuroendocrine tumors: a systematic review and meta-analysis. Nucl Med Commun. 2019;40(12):1195–203. The first meta-analysis of trials demonstrating the superioroty of PRRT over everolimus in patients with pancreatic NENs.
•• Baudin E, Walter T, Beron A, Smith D, Hadoux J, Lachach C, et al. First multicentric randomized phase II trial investigating the antitumor efficacy of peptide receptor radionucleide therapy with 177lutetium–octreotate (OCLU) in unresectable progressive neuroendocrine pancreatic tumor: results of the OCLURANDOM trial. ESMO Congress 2022, Abstract 887O. Annals of Oncology 2022;33 (suppl_7): S410-S416. The intial report of the first trial evaluating PRRT versus sunitinib in patients in pancreatic NENs- demonstrating the superiority of PRRT.
•• Strosberg J, Leeuwenkamp O, Siddiqui MK. Peptide receptor radiotherapy re-treatment in patients with progressive neuroendocrine tumors: A systematic review and meta-analysis. Cancer Treat Rev. 2021;93: 102141. The first meta-analysis reporting the efficacy of re-treatment PRRT post initial induction therapy.
Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514–23.
Yao JC, Fazio N, Singh S, Buzzoni R, Carnaghi C, Wolin E, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968–77.
Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–13.
• Das S, Al-Toubah T, Strosberg J. Chemotherapy in neuroendocrine tumors. Cancers (Basel). 2021;13(19):4872. Very good review of systemic therapy in patients with NENs.
•• Kunz PL, Graham NT, Catalano PJ, Nimeiri HS, Fisher GA, Longacre TA, et al. A randomized study of temozolomide or temozolomide and capecitabine in patients with advanced pancreatic neuroendocrine tumors (ECOG-ACRIN E2211). J Clin Oncol. 2022. https://doi.org/10.1200/JCO.22.01013 (published online). The first randomisd phase II trial demonstrating the high activity of capecitabine and temozolomide in patients with grade 1 and 2 pancreatic NENs.
•• Salazar R, Tafuto S, Krogh M, Teule A, Garcia-Carbonero R, Klumpen H, et al. Randomized open label phase III study comparing the efficacy and safety of everolimus followed by chemotherapy (CT) with streptozotocin (STZ)-5FU upon progression or the reverse sequence, in advanced progressive panNETs: the SEQTOR study (GETNE 1206). Annals of Oncology. 2022;33, Supplement 7, September 2022, Page S1412.The first randomised trial evaluating the optimal sequential systemic therapy in patients with pancreatic NETs- both sequence arms were of equivalent efficacy.
•• Sorbye H, Baudin E, Borbath I, Caplin M, Chen J, Cwikla JB, et al. Unmet needs in high-grade gastroenteropancreatic neuroendocrine neoplasms (WHO G3). Neuroendocrinology. 2019;108(1):54–62. Excellent review of the biology and management approaches of G3 NENs.
Garcia-Carbonero R, Sorbye H, Baudin E, Raymond E, Wiedenmann B, Niederle B, et al. ENETS consensus guidelines for high-grade gastroenteropancreatic neuroendocrine tumors and neuroendocrine carcinomas. Neuroendocrinology. 2016;103(2):186–94.
Sorbye H, Welin S, Langer SW, Vestermark LW, Holt N, Osterlund P, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann Oncol. 2013;24(1):152–60.
• Thomas KEH, Voros BA, Boudreaux JP, Thiagarajan R, Woltering EA, Ramirez RA. Current treatment options in gastroenteropancreatic neuroendocrine carcinoma. Oncologist. 2019;24(8):1076–88. Very good review of the systemic treatment approaches for GEP NENs.
Moertel CG, Kvols LK, O’Connell MJ, Rubin J. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer. 1991;68(2):227–32.
Patta A, Fakih M. First-line cisplatin plus etoposide in high-grade metastatic neuroendocrine tumors of colon and rectum (MCRC NET): review of 8 cases. Anticancer Res. 2011;31(3):975–8.
Dasari A, Mehta K, Byers LA, Sorbye H, Yao JC. Comparative study of lung and extrapulmonary poorly differentiated neuroendocrine carcinomas: a SEER database analysis of 162,983 cases. Cancer. 2018;124(4):807–15.
Zhang P, Li J, Li J, Zhang X, Zhou J, Wang X, et al. Etoposide and cisplatin versus irinotecan and cisplatin as the first-line therapy for patients with advanced, poorly differentiated gastroenteropancreatic neuroendocrine carcinoma: a randomized phase 2 study. Cancer. 2020;126(Suppl 9):2086–92.
Morizane C, Machida N, Honma Y, Okusaka T, Boku N, Kato K, et al. Effectiveness of etoposide and cisplatin vs irinotecan and cisplatin therapy for patients with advanced neuroendocrine carcinoma of the digestive system: the TOPIC-NEC phase 3 randomized clinical trial. JAMA Oncol. 2022;8(10):1447–55. The first randomised phase III trial comparing IE versus EP in patients with G3NEC- demonstrating equivalence in terms of activity between the arms but with greater tolerance for the IE arm.
Mitry E, Baudin E, Ducreux M, Sabourin JC, Rufie P, Aparicio T, et al. Treatment of poorly differentiated neuroendocrine tumours with etoposide and cisplatin. Br J Cancer. 1999;81(8):1351–5.
Raj N, Valentino E, Capanu M, Tang LH, Basturk O, Untch BR, et al. Treatment response and outcomes of grade 3 pancreatic neuroendocrine neoplasms based on morphology: well differentiated versus poorly differentiated. Pancreas. 2017;46(3):296–301.
Velayoudom-Cephise FL, Duvillard P, Foucan L, Hadoux J, Chougnet CN, Leboulleux S, et al. Are G3 ENETS neuroendocrine neoplasms heterogeneous? Endocr Relat Cancer. 2013;20(5):649–57.
Rogowski W, Wachula E, Gorzelak A, Lebiedzinska A, Sulzyc-Bielicka V, Izycka-Swieszewska E, et al. Capecitabine and temozolomide combination for treatment of high-grade, well-differentiated neuroendocrine tumour and poorly-differentiated neuroendocrine carcinoma - retrospective analysis. Endokrynol Pol. 2019;70(4):313–7.
Liu AJ, Ueberroth BE, McGarrah PW, Buckner Petty SA, Kendi AT, Starr J, et al. Treatment outcomes of well-differentiated high-grade neuroendocrine tumors. Oncologist. 2021;26(5):383–8.
Owen DH, Alexander AJ, Konda B, Wei L, Hemminger JA, Schmidt CR, et al. Combination therapy with capecitabine and temozolomide in patients with low and high grade neuroendocrine tumors, with an exploratory analysis of O(6)-methylguanine DNA methyltransferase as a biomarker for response. Oncotarget. 2017;8(61):104046–56.
Chan DL, Bergsland EK, Chan JA, Gadgil R, Halfdanarson TR, Hornbacker K, et al. Temozolomide in grade 3 gastroenteropancreatic neuroendocrine neoplasms: a multicenter retrospective review. Oncologist. 2021;26(11):950–5.
Jeong H, Shin J, Jeong JH, Kim KP, Hong SM, Kim YI, et al. Capecitabine plus temozolomide in patients with grade 3 unresectable or metastatic gastroenteropancreatic neuroendocrine neoplasms with Ki-67 index <55%: single-arm phase II study. ESMO Open. 2021;6(3):100119.
Wang W, Zhang Y, Peng Y, Jin KZ, Li YL, Liang Y, et al. A Ki-67 Index to predict treatment response to the capecitabine/temozolomide regimen in neuroendocrine neoplasms: a retrospective multicenter study. Neuroendocrinology. 2021;111(8):752–63.
Caplin ME, Pavel M, Ruszniewski P. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(16):1556–7.
Pellat A, Coriat R. Well differentiated grade 3 neuroendocrine tumors of the digestive tract: a narrative review. J Clin Med. 2020;9(6):1677
Panzuto F, Rinzivillo M, Spada F, Antonuzzo L, Ibrahim T, Campana D, et al. Everolimus in pancreatic neuroendocrine carcinomas G3. Pancreas. 2017;46(3):302–5.
Pellat A, Dreyer C, Couffignal C, Walter T, Lombard-Bohas C, Niccoli P, et al. Clinical and biomarker evaluations of sunitinib in patients with grade 3 digestive neuroendocrine neoplasms. Neuroendocrinology. 2018;107(1):24–31.
Hentic O, Hammel P, Couvelard A, Rebours V, Zappa M, Palazzo M, et al. FOLFIRI regimen: an effective second-line chemotherapy after failure of etoposide-platinum combination in patients with neuroendocrine carcinomas grade 3. Endocr Relat Cancer. 2012;19(6):751–7.
Bardasi C, Spallanzani A, Benatti S, Spada F, Laffi A, Antonuzzo L, et al. Irinotecan-based chemotherapy in extrapulmonary neuroendocrine carcinomas: survival and safety data from a multicentric Italian experience. Endocrine. 2021;74(3):707–13.
Sugiyama K, Shiraishi K, Sato M, Nishibori R, Nozawa K, Kitagawa C. Salvage chemotherapy by FOLFIRI regimen for poorly differentiated gastrointestinal neuroendocrine carcinoma. J Gastrointest Cancer. 2021;52(3):947–51.
Ferrarotto R, Testa L, Riechelmann RP, Sahade M, Siqueira LT, Costa FP, et al. Combination of capecitabine and oxaliplatin is an effective treatment option for advanced neuroendocrine tumors. Rare Tumors. 2013;5(3):e35.
Bajetta E, Catena L, Procopio G, De Dosso S, Bichisao E, Ferrari L, et al. Are capecitabine and oxaliplatin (XELOX) suitable treatments for progressing low-grade and high-grade neuroendocrine tumours? Cancer Chemother Pharmacol. 2007;59(5):637–42.
Hadoux J, Malka D, Planchard D, Scoazec JY, Caramella C, Guigay J, et al. Post-first-line FOLFOX chemotherapy for grade 3 neuroendocrine carcinoma. Endocr Relat Cancer. 2015;22(3):289–98.
McNamara MG, Frizziero M, Jacobs T, Lamarca A, Hubner RA, Valle JW, et al. Second-line treatment in patients with advanced extra-pulmonary poorly differentiated neuroendocrine carcinoma: a systematic review and meta-analysis. Ther Adv Med Oncol. 2020;12:1758835920915299.
Bongiovanni A, Liverani C, Pusceddu S, Leo S, Di Meglio G, Tamberi S, et al. Randomised phase II trial of CAPTEM or FOLFIRI as SEcond-line therapy in NEuroendocrine CArcinomas and exploratory analysis of predictive role of PET/CT imaging and biological markers (SENECA trial): a study protocol. BMJ Open. 2020;10(7):e034393.
Raj N, Coffman K, Le T, Do RKG, Rafailov J, Choi Y, et al. Treatment response and clinical outcomes of well differentiated high grade neuroendocrine tumors to lutetium-177 DOTATATE. Neuroendocrinology. 2022;112(12):1177–86
Zhang J, Kulkarni HR, Singh A, Niepsch K, Muller D, Baum RP. Peptide receptor radionuclide therapy in grade 3 neuroendocrine neoplasms: safety and survival analysis in 69 patients. J Nucl Med : Off Publ, Soc Nucl Med. 2019;60(3):377–85.
Waseem N, Aparici CM, Kunz PL. Evaluating the role of theranostics in grade 3 neuroendocrine neoplasms. J Nucl Med : Off Publ, Soc Nucl Med. 2019;60(7):882–91.
Sorbye H, Kong G, Grozinsky-Glasberg S. PRRT in high-grade gastroenteropancreatic neuroendocrine neoplasms (WHO G3). Endocr Relat Cancer. 2020;27(3):R67–77. Excellent review of studies evaluating PRRT in G3 NENs—confirming greater efficacy in G3NETs versus G3NEC.
Taboada R, Claro L, Felismino T, de Jesus VH, Barros M, Riechelmann RP. Clinicopathological and molecular profile of grade 3 gastroenteropancreatic neuroendocrine neoplasms. J Neuroendocrinol. 2022;34(4):e13099.
Park EJ, Park HJ, Kim KW, Suh CH, Yoo C, Chae YK, et al. Efficacy of immune checkpoint inhibitors against advanced or metastatic neuroendocrine neoplasms: a systematic review and meta-analysis. Cancers (Basel). 2022;14(3):794
Chan DL, Rodriguez-Freixinos V, Doherty M, Wasson K, Iscoe N, Raskin W, et al. Avelumab in unresectable/metastatic, progressive, grade 2–3 neuroendocrine neoplasms (NENs): combined results from NET-001 and NET-002 trials. Eur J Cancer. 2022;169:74–81.
Vijayvergia N, Dasari A, Deng M, Litwin S, Al-Toubah T, Alpaugh RK, et al. Pembrolizumab monotherapy in patients with previously treated metastatic high-grade neuroendocrine neoplasms: joint analysis of two prospective, non-randomised trials. Br J Cancer. 2020;122(9):1309–14.
Klein O, Kee D, Markman B, Michael M, Underhill C, Carlino MS, et al. Immunotherapy of ipilimumab and nivolumab in patients with advanced neuroendocrine tumors: a subgroup analysis of the CA209-538 clinical trial for rare cancers. Clin Cancer Res. 2020;26(17):4454–9. Phase II trial demonstrating combined activity of CTL4 and PD1 blockade in a cohort of patients with high grade NENs.
Patel SP, Mayerson E, Chae YK, Strosberg J, Wang J, Konda B, et al. A phase II basket trial of dual anti-CTLA-4 and anti-PD-1 blockade in rare tumors (DART) SWOG S1609: high-grade neuroendocrine neoplasm cohort. Cancer. 2021;127(17):3194–201. Phase II trial demonstrating combined activity of CTL4 and PD1 blockade in a cohort of patients with high grade NENs.
Leyden S, Kolarova T, Bouvier C, Caplin M, Conroy S, Davies P, et al. Unmet needs in the international neuroendocrine tumor (NET) community: assessment of major gaps from the perspective of patients, patient advocates and NET health care professionals. Int J Cancer. 2020;146(5):1316–23.
Acknowledgements
Dr. Catherine Mitchell reported the histopathology displayed in Fig. 1.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors declare no conflict of interest. GK has received research funding from Pfizer and Cyclotek, had consulting or advisory role for ITM, and received a Clinical Fellowship Award from the Peter MacCallum Foundation.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kong, G., Boehm, E., Prall, O. et al. Integrating Functional Imaging and Molecular Profiling for Optimal Treatment Selection in Neuroendocrine Neoplasms (NEN). Curr Oncol Rep 25, 465–478 (2023). https://doi.org/10.1007/s11912-023-01381-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-023-01381-w