
Abstract
Purpose of Review
Neutrophil extracellular trap (NET) formation is a newly discovered, reactive oxygen species-dependent regulated process, whereby neutrophils degranulate and extrude genetic material, after engulfing various infectious or neoplastic antigens, culminating in a measurable serologic footprint. Recent research has highlighted the involvement of NETs in cancer and cancer-related pathologies. We review the role of NET formation in cancer biology, prognosis and potential therapeutic modulators.
Recent Findings
Elevated NET levels are associated with cancer metastasis and may be modified by some anaesthetic-analgesic techniques during tumour resection surgery. It promotes tumour cell migration, angiogenesis and hypercoagulability. Although there are potential anti-NET formation therapeutics available, their role has not been formally assessed in cancer patients.
Summary
Limited available evidence suggests an association between elevated NET expression and cancer metastasis, but its validity as a prognostic indicator for cancer-related outcomes is inconclusive. Further observational and interventional studies are warranted to comprehend the potential prognostic and therapeutic role of NETs in cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neutrophil extracellular trap (NET) formation is a reactive oxygen species (ROS)–dependent regulated process, whereby neutrophils degranulate, releasing intracellular and nuclear contents which form an extracellular snare with a role in bacterial or neoplastic cell trapping and killing. This process bequeaths a serologic footprint which may be detected many years later. The term NETosis has been previously used for describing this phenomenon. Recently, NETosis was defined as being a ROS-dependent regulated cell death, specific to cells of haematopoietic origin and associated with NET release [1]. It is recommended to avoid using the term NETosis in those instances where there is no evidence of cell death associated with NET formation. The term NET extrusion may also be used for NET formation without cell death. These two terms will be used interchangeably for the remaining of this article.
NETs are rich in cell nuclear components (e.g. DNA, histone proteins). They also contain granular and cytoplasmatic proteins or enzymes (myeloperoxidase (MPO), neutrophil elastase (NE)) [2] and antimicrobial and proinflammatory proteins [3]. A defining characteristic of NET generation is the translocation of genetic material from the nucleus to the extracellular environment [4].
Although NET formation was initially described over 20 years ago, it has only emerged as a current topic of interest since 2014 [5, 6]. Since then, it has been identified as a factor in many physiological and pathological processes like immunity, sepsis, inflammation, cancer metastasis, cancer-related thrombosis, and more recently in severe acute respiratory syndrome coronavirus 2 (SARS-CoV 2) infection [7,8,9,10,11].
In this review, we summarise the current understanding of the multifactorial role of NETs in tumour cell biology, metastatic development and venous thromboembolism (VTE) occurrence. We also review pharmacological agents, including anaesthesia technique, with a potential role in NET modulation.
Mechanisms of NET Formations
Classically, neutrophils are recognised as the mainstay of the innate immune system, delivering chemotaxis, endothelial adherence, phagocytosis, oxidative burst and toxic granule dependent microbial killing [4]. Important steps of NET formation include chromatin decondensation, nuclear membrane rupture, nucleoplasm spillage into the cytoplasm, cell membrane destruction and cell death have been described. This has been described both in vitro and in vivo.
Suicidal NET formation is NADPH oxidase (NOX)–dependent and involves chromatin decondensation, nuclear envelope disintegration, plasma membrane perforation, cell lysis and tumour cell or bacteria fixation to the DNA structures [12, 13]. Subsequent work revealed the role of ROS, NE and MPO in NET release [2]. The ROS triggers the release of NE and MPO from the cytoplasmic granules and promotes the MPO-mediated proteolytic activity of NE. The NE and MPO are translocated to the nucleus, which facilitates histone degradation chromatin decondensation [2].
This contrasts with “vital NETosis”, a more rapid process in which neutrophils became anuclear cells still capable of survival and of engulfing antigens [4, 14]. Currently, three types of vital NET formation have been described (Table 1).
Suicidal and vital NET formation terminology is being gradually replaced by NOX-dependent and NOX-independent NET production. NOX-dependent or NOX-independent pathway is activated in response to specific triggers. Lipopolysaccharide (LPS) and bacteria stimulate the NOX-dependent pathway leading to nucleus breakdown [2] and activation of a specific set of kinases (ERK1, ERK2, Akt, P38, Src) [17••].
Bacterial or neoplastic cell products and uric acid crystals activate the NOX-independent pathway, leading to the formation of mitochondrial ROS [18]. This acts on several kinases. The activation of each pathway’s kinases leads to transcriptional firing, which enables chromatin decondensation [6]. In the NOX-independent pathway, peptidyl arginine deaminase type 4 (PAD4)-mediate the production of chromatin networks by the cancer cells themselves [19, 20]. There is also evidence that NET formation can take place in a PAD4-independent fashion [21] particularly in the NOX-dependent pathway [18, 22].
Measuring NET Expression
The production of NETs creates a footprint, by which NET formation can be detected. The various components of NETs (e.g. histones, DNA, enzymes) and the molecules involved in NET expression (e.g. PAD-4) serve as potential targets for measuring this process. NET expression can be quantified directly by microscopy [23], flow-cytometry or real-time imaging or indirectly, using the enzyme-linked immunosorbent assay (ELISA) technique mainly for serological measurements [24]. MPO, NE, and citrullinated histone 3 (citH3), among others, represent measurable components or markers of NETs [25, 26, 27•], but may not be specific. For example, NE and MPO can be released during degranulation without NET formation [28]. It has been proposed that measuring conjugates like MPO/circulating free DNA (cfDNA) and citH3 could be more specific for NETs than cfDNA alone [29].
NETs and Cancer
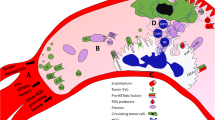
Elevated NET levels are associated with the diagnosis of cancer [4, 10, 17••, 27•]. NETs contribute to primary tumour growth [9], metastasis [30] and cancer-related thrombosis [31] through various mechanisms (Fig. 1). NETs associated enzymes are involved in cancer growth by stimulating phosphatidylinositol-3 kinases (PI3Ks) (e.g. NE) and matrix destruction (e.g. matrix metallopeptidase 9 (MMP-9)) [36, 37]. In breast cancer cells in vitro, NET expression upregulates gene expression for several factors related to pro-metastatic properties of breast cancer [38]. Also, cancer cell biology and NET formation share common elements of ROS signalling pathways [39] and reliance on similar kinases (e.g. p38) [17••, 40]. For example, stimulation of toll-like receptor 4 (TLR4) receptor (e.g. by LPS) induces NET formation [32••] with a role in host immunity. High mobility group box protein 1 (HMGB1) protein is involved in many steps of the protumorigenic pathway [41] and is produced by neutrophils on degranulation/NET formation. This protein seems to be one of the NETs associated chemotactic factors responsible for the NET-induced tumour cell invasion [26]. Other proteins released during NET production like MMP-9 and NE have been shown to be involved in tumour progression, creating an adequate environment for tumour cell seeding and growth [26]. Despite some evidence that in vitro cancer-induced NET formation is a suicidal process [42], it remains unclear if the in vivo process is vital or suicidal [32••].

Schematic representation of the NET formation modulators and the development of the pre-metastatic niche in the perioperative setting [17••, 25, 32••, 33,34,35]. citH3, citrullinated histone 3 protein; CTCs, circulating tumour cells; CXCR1/2, CXC receptor 1/2; EC, endothelial cells; EV, extracellular vesicles; G-CSF, granulocyte colony-stimulating factor; G-CSFR, granulocyte colony-stimulating factor receptor; HMGB1, high mobility group box 1; IL-8, interleukin 8; LAIR1, leucocyte-associated immunoglobulin-like receptor 1; LPS, lipopolysaccharide; MPO, myeloperoxidase; N, neutrophils; NE, neutrophil elastase; P, platelets; PAF, platelet-activating factor; P-SEL, selectin P; PSGL-1, selectin P receptor 1; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; SIGLEC 5/6, sialic acid-binding immunoglobulin-type lectins 5/6; SIRL1, signal inhibitory receptor on leucocytes 1; TF, tissue factor; TLR2, toll-like receptor 2; TLR4, toll-like receptor 4; VTE, venous thromboembolism; £ induces tumour cell proliferation; $ dormant cancer cell activator; % synergistic effect with LPS to exacerbate infection response; * correlates with the metastatic potential of some cancers
In an in vitro model of surgery, it was shown that NETs stimulated Kupffer cells to produce proinflammatory cytokines directly associated with cancer progression (interleukin 6 (IL-6), tumour necrosis factor-alpha (TNF-alpha) and chemokine CXC motif ligand 10 (CXCL-10)) [26, 43]. This was alleviated by DNase and PAD4 inhibition [26]. Although the full list of stimuli of NET production in cancer is unknown, there is evidence for the circulation of pro-NET formation factors. Neutrophils from healthy donors were stimulated to produce NETs when incubated with cancer patient’s plasma [44].
Tumour Cell-Neutrophil Interplay in NET Formation
Tumour cell-neutrophil-platelets-vascular endothelium interactions are essential for cancer cell survival and the development of conditions which facilitate metastasis. These processes have been recently summarised [32••]. Neutrophils are subject to modulation form both circulating tumour cells and factors released by the solid tissue tumours through extracellular vesicles (EVs). Some of the identified modulators produced by the primary tumours are tissue factor (TF), granulocyte colony-stimulating factor (G-CSF), HMGB1, IL-8 and ROS. The interaction between tumour factors, activated platelets and endothelium leads to neutrophil activation, NET formation and adhesion of the neutrophils to the endothelium [45]. In breast cancer, increased G-CSF is associated with increased metastases [46]. In a murine model of breast cancer and insulinoma, it was shown that cancer-associated heart, kidney and vascular dysfunction, which caused increased NETs, was improved by neutrophil depletion, G-CSF inhibition and DNAse administration [47]. In this model, vascular NET development was mediated by increased levels of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion protein 1 (VCAM-1), IL-1Beta, IL-6 and chemokine CXCL1. These observations may explain the distant organ dysfunction seen in cancer.
Extracellular vesicles are membrane-enclosed structures produced by cells (e.g. during cell stress response) and could contain specific cytoplasmic solid and soluble components specific to the parent cell [32••]. Tumour EVs can directly trigger NET formation after they have been phagocytosed by neutrophils. They have been found to be present in patients with breast cancer and to predict therapeutic failure and poor prognosis [48]. Some of the constituents of EVs (e.g. interleukins and G-CSF) are NET formation mediators [49]. In a murine model (BALB/c mice) injected with 4T1 breast cancer cells, the EVs of 4T1 cells triggered NET formation and when administered intravenously contributed to accelerated venous thrombosis [50]. In a mouse model of lung tumours postintravenous B16 melanoma cell injection, the authors showed that the tumour cell EVs were ingested by neutrophils [51] which could have pro-NET generation effects.
In addition, NETs act like “fishing nets” capturing circulating tumour cells which generate further endothelial damage facilitating metastasis [17••]. Key ligand/neutrophils receptor interactions (e.g. HMBG1-TLR4/receptor for advanced glycation end products (RAGE)) are essential in this scenario [32••] and could develop into therapeutic targets. Although the IL-8/CXC chemokine receptor 1/2 (CXCR1/2) interaction is beneficial in acute infection [52], it seems to be detrimental in cancer. In a murine model of lung cancer and concomitant sepsis, intravascularly deposited NETs captured lung cancer cells and were associated with gross metastatic burden at 2 weeks [53]. The metastatic burden was attenuated by NET formation inhibitors (e.g. DNAse, neutrophil elastase inhibitor (NEi)). In a murine model of murine colon cancer (CT-26) peritoneal metastasis, decreasing the circulating neutrophils or disrupting NET formation by DNAse markedly decreased the number of metastases [30]. In the same study, mice and humans were shown to have significant NETs in peritoneal metastases from colon cancer, and blocking the integrins abolished NET-induced cancer cell adhesion.
Cancer cell NET formation occurs by two pathways: pro-tumorigenic effects of NETs and cancer cells’ NET-inducing capacity. In a study involving patients with pancreatobiliary malignancy and the pancreatic cancer cell line AsPC-1, cells themselves and their conditioned medium were strong promoters of NET formation. In this study, ROS inhibition did not block NET formation but prostaglandin E1 (PGE1) did, suggesting a role of cyclical adenosine monophosphate (AMP) in NET formation [54]. It was shown that HMGB1 protein is released from NETs and serves as a ligand on the TLR4 receptor on the surface of cancer cells [26, 27•, 55].
The age of the neutrophil is relevant for both NET formation and cancer biology [56]. Interestingly, the more immature neutrophils have a reduced NET formation activity via reduced ROS production and increased mitotic activity [57].
NETs and Cancer-Associated Thrombosis
Endothelial cells and platelets require prior activation before triggering NET formation [58]. Platelets can be activated by the histones in NET structure [59] and can directly stimulate NET formation, interacting with the neutrophil via the selectin P (P-SEL)/selectin P receptor 1 (PSGL-1) [32••]. NETs are rich in citrullinated histone 3 (citH3), NE, and MPO, all triggering further platelet activation and contributing to the thromboembolic phenomenon associated with cancer. In addition to the previously known mechanisms of platelet activation, an in vitro study using AsPC-1 pancreatic cell line suggested platelet activation may be driven by the tumour cells directly via TF expression [60], which could potentially be a constituent of the tumour EVs [32••]. HMBG1 factor and TLR4 receptor are used by the activated human platelets to trigger NET formation [58]. Furthermore, DNA activates the intrinsic coagulation pathway [61]. Jung et al. proposed that the DNA contained in NETs could be the trigger for NET-associated thrombin generation and subsequent cancer-related thrombotic phenomena [27•].
NETs have been identified in patients with thrombotic phenomena (i.e. stroke). In a prospective, observational case-control study, patients were divided into two groups based on the serum troponin levels [62]. High-sensitivity troponin (hsTnT) was associated with a higher risk of malignancy. Autopsies in these patients showed widespread micro-thrombosis containing citH3, a biomarker for NETs. A procoagulant state with increased NET serum levels was seen in patients with elevated hsTnT compared to that in patients with normal levels (p < 0.001). CitH3 was also correlated with thrombin-antithrombin complex and soluble P-SEL providing a further link between NETs and the prothrombotic state in cancer patients.
A similar prothrombotic state was found in a study of patients with pancreatobiliary malignancy, who had a high level of circulating NET markers (histone-DNA complexes and cfDNA) and hypercoagulability markers [27•]. Also, hypercoagulability markers were higher in patients with more advanced cancer (stage IV vs stage I/II). In the same study, using the AsPC-1 pancreatic cancer cell line, the investigators also documented that antithrombin significantly blocks NET formation.
NETs as a Prognostic Marker
Plasma taken from treatment-naive patients with lung or upper GI adenocarcinoma and compared to the plasma of healthy individuals showed an elevation in NET expression (measured as MPO) in the cancer patient cohort compared to that of the control group (p = 0.03). This study also showed significant elevation in NET levels in patients with advanced cancer (stages III & IV oesophagogastric, stages II & III lung) compared to patients with a less advanced diagnosis (stages I & II oesophagogastric and stage I lung) (p = 0.01) [63•]. NET expression was also higher in patients with early-stage head and neck cancer compared to that in healthy control participants of the same age [64•]. As part of the same study, it was shown that neutrophils of these cancer patients released more NETs compared to those of the healthy control cohort. The increased NET expression in cancer patients and the increased release of NETs from neutrophils in vitro support the hypothesis that NETs may be a biomarker for cancer.
Although most evidence points towards a deleterious effect of NETs in cancer, a multi-omics study on ovarian cancer patients found that NET formation correlated with better outcome [25]. Increased CRP is typically associated with poor prognosis in this type of cancer, whilst the S100A8 protein is released by neutrophils upon local NET generation. The S100A8/CRP abundance ratio significantly stratified patients and the increase in the ratio correlated with favourable survival.
The opposite was observed in patients with colorectal cancer, with an in vitro increase in NET expression in response to stimulation of systemic neutrophils. This was associated with more postoperative complications (Clavien-Dindo classification ≥ 3) and prolonged hospital stay (>5 days). [23].
NET formation has been predictive for decreased disease-free survival. In a cohort of 50 colorectal patients undergoing curative liver metastases resection, it was observed that increased postoperative NET formation was associated with over fourfold reduction in disease-free survival [26]. Similar findings occurred in a murine model (C57BL/6J WT mice or PAD4−/− mice) of surgical stress employing liver ischemia-reperfusion, where NET expression was associated with an accelerated development of metastatic disease (MC38 or MC38/Luc cancer cells) [26]. These effects were mitigated by inhibiting NET formation with DNAse (68% reduction in hepatic tumour burden) or PAD4 inhibition.
In a study of 60 patients with various adenocarcinomas, squamous cell carcinomas, melanomas, and glioblastomas, there was a threefold increase in the median plasma citH3 (NET biomarker) levels in patients with advanced cancer compared with age-matched healthy individuals or severely ill patients without cancer [65]. Furthermore, plasma levels of IL-8, IL-6, TNF-alpha, IL-1beta and G-CSF (all involved in NET production) were all elevated in cancer patients. Levels of citH3 above the 75th percentile were associated with a twofold short increased short-term mortality. Besides, these patients had a significantly higher number of peripheral neutrophils, neutrophils positive for intracellular citH3, plasma MPO, and MPO-DNA complexes, suggestive of neutrophil activation and NET extrusion. In a systematic review of 30 studies where the authors examined the relationship between perioperative inflammation, NET expression and metastasis in colorectal cancer, the presence of a preoperative systemic inflammatory state but not sepsis was associated with increased cancer recurrence [66].
Cancer cells can hijack neutrophil function, inducing NET formation independently of direct tumour cell-neutrophil interaction (via G-CSF priming) and could be a marker of tumour aggression. In an in vitro study, NETs were found in 16/20 of the primary breast tumours and in 13/19 metastatic lung lesions. Lung metastases of triple-negative breast cancer had the highest level of NET expression when compared to luminal breast cancer [42]. Interestingly, these in vitro experiments revealed a lytic (i.e. suicidal) NET formation pathway and concluded that NETs do not promote tumour cell extravasation, but rather mediate the expansion of already disseminated cells
NETs Promote Angiogenesis
NETs are promoters of inflammation via NE and MPO, which could damage the endothelium, triggering further IL-8, ROS and platelet activation, in turn further stimulating neutrophil recruitment and NET release [32••]. In addition, in vitro, histones are stimulators of angiogenesis when incubated with endothelial cells [27•]. As they are a major component of NETs, it is reasonable to conclude that NETs can promote angiogenesis too. The entire effect was blocked by histone chelating agents (e.g., heparin, polysialic acid (PSA)).
Does Anaesthesia Technique and the Perioperative Surgical Stress Impact on NET Formation?
The effects of surgical stress and anaesthetic technique on NET expression have been assessed in vivo and clinically, with intriguing results.
In an observational cohort study evaluating the metastatic potential of surgical stress-induced NET formation, liver ischemia-reperfusion (I/R) injury increased NET expression. Serum was obtained day 1 postoperatively for patients who underwent hepatectomy for metastatic colorectal cancer. NET expression (measured by serum MPO-DNA complexes) was significantly increased in patients who underwent major liver resection (3 or more segments removed) with unavoidable liver I/R injury when compared to patients who underwent smaller resection or to healthy controls. Patients with elevated NET levels post-operatively, experienced a fourfold reduction in disease-free survival when compared to the cohorts with lower NET expression (95% CI: 1.39–12.81, p = 0.01). In the same study, using a murine model of liver I/R injury and MC38 cancer cells, the ischaemic group showed increased serum citH3, when compared to controls. After 2 weeks, the liver I/R mice showed greater hepatic metastases compared with mice who did not receive liver I/R injury [26].
The impact of the anaesthetic technique on perioperative NET expression was tested in the first randomised controlled clinical trial in this field on women undergoing primary breast tumour resection [67••]. Patients were randomly assigned to receive one of the following 4 anaesthetic techniques: inhalational sevoflurane (S), sevoflurane plus IV lidocaine (SL), intravenous propofol (P) or propofol plus IV lidocaine (PL). Although there was no difference between the S and P groups, the addition of lidocaine significantly decreased citH3 expression (109 ± 23 vs 125 ± 22 ng ml−1, p = 0.01 for SL and S and 98 ± 14 vs 130 ± 32 ng ml−1, p = 0.007, for PL and P respectively). MPO, another NET formation biomarker, was also decreased by lidocaine. Lidocaine further decreased expression of MMP-3 but not MMP-9, regardless of which anaesthetic technique was used. Therefore, an inhalational versus intravenous anaesthetic technique has little effect on NET expression, but the addition of lidocaine to either technique significantly reduced these markers, consistent with the hypothesis that intravenous lidocaine during cancer surgery of curative intent might reduce recurrence. However, in a parallel-group randomised control trial (RCT) [68••], which evaluated the effects of different anaesthetic techniques among women undergoing breast cancer surgery, NET expression was not influenced by the anaesthetic technique. Forty women partaking in a larger clinical trial (NCT-00418457), undergoing breast cancer surgery, were randomly assigned to receive volatile general anaesthesia (GA) or propofol GA plus paravertebral regional anaesthesia (PPA). NET levels were measured using the MPO and citH3 biomarkers. There was no difference between the two groups in either MPO concentration (10.5 ± 6.6 vs 11.5 ± 4.7, ng ml−1, p = 0.60) or citH3 concentration (3.6 ± 2.3 vs 4.0 ± 5.9, ng ml−1, p = 0.80) respectively.
NET Formation Modulators
The perioperative period represents a window of opportunity for metastasis. The therapeutic options during this phase should not interfere with immunity, wound healing and decrease host-defence [26]. A key aspect of anti-NET therapies is represented by appropriate patient selection. For example, expression of some pro-metastatic factors (e.g. G-CSF) by some tumours and the presence of NETs in those tumours have been proposed as a screening tool to identify patients who may benefit from anti-NET therapies [42]. Although drugs which affect NET formation are clinically available (e.g. dornase alpha, lidocaine, PGE-1, etc.), the concept of NET inhibition in cancer is not currently widely supported.
Some potential drugs are experimental (e.g. gasdermin D inhibitor LDC7559 ), whilst others will have to be relicensed from other pathologies (e.g. HMG-CoA reductase inhibitors, dornase alpha, lidocaine). In comparison, analgesics are readily available and may play a double role in the perioperative setting (analgesia/NET inhibition). Of these, cyclooxygenase inhibitors (COX) are the most promising. As the platelets-neutrophil interaction is paramount for NET formation, aspirin (COX inhibitor/antiplatelet agent) has shown promising results, reducing NET production [69]. Although COX blockade leads to prostaglandin generation inhibition, not all COX inhibitors have the same effect on NET expression. In opposition to aspirin, indomethacin (another non-selective COX inhibitor) and diclofenac (COX-2 preferential inhibitor) stimulate NET production by prostaglandin E2 (PGE2) inhibition [70]. In this murine model of transplanted bone marrow neutrophils, indomethacin led to neutrophils’ bactericidal function recovery post-transplant (in vivo) and diclofenac (in vitro), suggesting PGE2 is an inhibitor of NET production [70]. Although various reviews have suggested either an equivocal or slightly positive signal for the use of non-steroidal anti-inflammatory drugs (NSAIDs) in cancer surgery to improve oncological outcomes [33, 71,72,73], it remains unclear what is the role of NSAIDs in cancer-induced NET expression. The potential NET formation inhibitors are summarised in Table 2.
Conclusion
Since its discovery, NETs have been shown to be strongly associated with increased cancer progression with potential value as a prognostic indicator and a marker for increased risk of cancer-related pathologies such as metastasis and thrombus formation. The limited observational nature of most of the studies and the paucity of clinical trials preclude a causal link being made. Two recent RCTs investigating the impact of anaesthetic technique showed that whilst the addition of lidocaine infusion decreased NET formation, regional anaesthesia had no effect. Whether modulation of NETs perioperatively translates into improved longer-term oncologic outcomes warrants further evaluation in observational and interventional studies.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. https://doi.org/10.1038/s41418-017-0012-4.
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–91. https://doi.org/10.1083/jcb.201006052.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5. https://doi.org/10.1126/science.1092385.
Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784–94. https://doi.org/10.1182/blood-2013-04-457671.
Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol. 1996;59(2):229–40. https://doi.org/10.1002/jlb.59.2.229.
Khan MA, Palaniyar N. Transcriptional firing helps to drive NETosis. Sci Rep. 2017;7:41749. https://doi.org/10.1038/srep41749.
Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. 2020;217(6). https://doi.org/10.1084/jem.20200652.
Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21(7):815–9. https://doi.org/10.1038/nm.3887.
Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology. 2016;5(5):e1134073. https://doi.org/10.1080/2162402X.2015.1134073.
Cedervall J, Zhang Y, Olsson AK. Tumor-induced NETosis as a risk factor for metastasis and organ failure. Cancer Res. 2016;76(15):4311–5. https://doi.org/10.1158/0008-5472.CAN-15-3051.
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–53. https://doi.org/10.1038/nm.4027.
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–41. https://doi.org/10.1083/jcb.200606027.
Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE. 2007;2007(379):pe11. https://doi.org/10.1126/stke.3792007pe11.
Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18(9):1386–93. https://doi.org/10.1038/nm.2847.
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16(11):1438–44. https://doi.org/10.1038/cdd.2009.96.
Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185(12):7413–25. https://doi.org/10.4049/jimmunol.1000675.
Ravindran M, Khan MA, Palaniyar N. Neutrophil extracellular trap formation: physiology, pathology, and pharmacology. Biomolecules. 2019;9(8). https://doi.org/10.3390/biom9080365This paper offers an up-to-date review of the current understanding on NET formation physiology and its role in various pathologies.
Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A. 2015;112(9):2817–22. https://doi.org/10.1073/pnas.1414055112.
Yuzhalin AE, Gordon-Weeks AN, Tognoli ML, Jones K, Markelc B, Konietzny R, et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat Commun. 2018;9(1):4783. https://doi.org/10.1038/s41467-018-07306-7.
Shi L, Yao H, Liu Z, Xu M, Tsung A, Wang Y. Endogenous PAD4 in breast cancer cells mediates cancer extracellular chromatin network formation and promotes lung metastasis. Mol Cancer Res. 2020;18(5):735–47. https://doi.org/10.1158/1541-7786.MCR-19-0018.
Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23(3):279–87. https://doi.org/10.1038/nm.4294.
Tatsiy O, McDonald PP. Physiological stimuli induce PAD4-dependent, ROS-independent NETosis, with early and late events controlled by discrete signaling pathways. Front Immunol. 2018;9:2036. https://doi.org/10.3389/fimmu.2018.02036.
Richardson JJR, Hendrickse C, Gao-Smith F, Thickett DR. Neutrophil extracellular trap production in patients with colorectal cancer in vitro. Int J Inf Secur. 2017;2017:4915062–11. https://doi.org/10.1155/2017/4915062.
Kasprzycka W, Homa-Mlak I, Mlak R, Małecka-Massalska T. Direct and indirect methods of evaluating the NETosis process. J Pre-Clin Clin Res. 2019;13(1):50–6. https://doi.org/10.26444/jpccr/105563.
Muqaku B, Pils D, Mader JC, Aust S, Mangold A, Muqaku L, et al. Neutrophil extracellular trap formation correlates with favorable overall survival in high grade ovarian cancer. Cancers (Basel). 2020;12(2). https://doi.org/10.3390/cancers12020505.
Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016;76(6):1367–80. https://doi.org/10.1158/0008-5472.CAN-15-1591.
Jung HS, Gu J, Kim JE, Nam Y, Song JW, Kim HK. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS One. 2019;14(4):e0216055. https://doi.org/10.1371/journal.pone.0216055This paper investigated the determinants of pancreatobilliary cancer–associated venous thrombosis. Pancreatic cancer is known to be associated with a high incidence of venous thrombosis. It found high levels of NETs in acancer patients, associated with high hypercoagilability markers.
Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev. 2010;62(4):726–59. https://doi.org/10.1124/pr.110.002733.
Zhu L, Liu L, Zhang Y, Pu L, Liu J, Li X, et al. High level of neutrophil extracellular traps correlates with poor prognosis of severe influenza A infection. J Infect Dis. 2018;217(3):428–37. https://doi.org/10.1093/infdis/jix475.
Al-Haidari AA, Algethami N, Lepsenyi M, Rahman M, Syk I, Thorlacius H. Neutrophil extracellular traps promote peritoneal metastasis of colon cancer cells. Oncotarget. 2019;10(12):1238–49. https://doi.org/10.18632/oncotarget.26664.
Hisada Y, Grover SP, Maqsood A, Houston R, Ay C, Noubouossie DF, et al. Neutrophils and neutrophil extracellular traps enhance venous thrombosis in mice bearing human pancreatic tumors. Haematologica. 2020;105(1):218–25. https://doi.org/10.3324/haematol.2019.217083.
Snoderly HT, Boone BA, Bennewitz MF. Neutrophil extracellular traps in breast cancer and beyond: current perspectives on NET stimuli, thrombosis and metastasis, and clinical utility for diagnosis and treatment. Breast Cancer Res. 2019;21(1):145. https://doi.org/10.1186/s13058-019-1237-6A review paper summarizing the role of NETs in breast cancer, proposing a model for the NETs driven cancer progression.
Wall T, Sherwin A, Ma D, Buggy DJ. Influence of perioperative anaesthetic and analgesic interventions on oncological outcomes: a narrative review. Br J Anaesth. 2019;123(2):135–50. https://doi.org/10.1016/j.bja.2019.04.062.
Bornhofft KF, Galuska SP. Glycans as Modulators for the formation and functional properties of neutrophil extracellular traps: used by the forces of good and evil. Front Immunol. 2019;10:959. https://doi.org/10.3389/fimmu.2019.00959.
Van Avondt K, Hartl D. Mechanisms and disease relevance of neutrophil extracellular trap formation. Eur J Clin Investig. 2018;48(Suppl 2):e12919. https://doi.org/10.1111/eci.12919.
Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. 2010;16(2):219–23. https://doi.org/10.1038/nm.2084.
Acuff HB, Carter KJ, Fingleton B, Gorden DL, Matrisian LM. Matrix metalloproteinase-9 from bone marrow-derived cells contributes to survival but not growth of tumor cells in the lung microenvironment. Cancer Res. 2006;66(1):259–66. https://doi.org/10.1158/0008-5472.CAN-05-2502.
Martins-Cardoso K, Almeida VH, Bagri KM, Rossi MID, Mermelstein CS, Konig S, et al. Neutrophil Extracellular Traps (NETs) Promote pro-metastatic phenotype in human breast cancer cells through epithelial-mesenchymal transition. Cancers (Basel). 2020;12(6). https://doi.org/10.3390/cancers12061542.
Kirchner T, Moller S, Klinger M, Solbach W, Laskay T, Behnen M. The impact of various reactive oxygen species on the formation of neutrophil extracellular traps. Mediat Inflamm. 2012;2012:849136–10. https://doi.org/10.1155/2012/849136.
Zhao M, Howard EW, Parris AB, Guo Z, Zhao Q, Yang X. Alcohol promotes migration and invasion of triple-negative breast cancer cells through activation of p38 MAPK and JNK. Mol Carcinog. 2017;56(3):849–62. https://doi.org/10.1002/mc.22538.
Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J Hepatol. 2015;63(1):114–21. https://doi.org/10.1016/j.jhep.2015.02.009.
Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8(361):361ra138. https://doi.org/10.1126/scitranslmed.aag1711.
Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62(2):600–14. https://doi.org/10.1002/hep.27841.
Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res. 2016;22(15):3924–36. https://doi.org/10.1158/1078-0432.CCR-15-2463.
Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A. 2012;109(32):13076–81. https://doi.org/10.1073/pnas.1200419109.
Mouchemore KA, Anderson RL, Hamilton JA. Neutrophils, G-CSF and their contribution to breast cancer metastasis. FEBS J. 2018;285(4):665–79. https://doi.org/10.1111/febs.14206.
Cedervall J, Zhang Y, Huang H, Zhang L, Femel J, Dimberg A, et al. Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res. 2015;75(13):2653–62. https://doi.org/10.1158/0008-5472.CAN-14-3299.
Konig L, Kasimir-Bauer S, Bittner AK, Hoffmann O, Wagner B, Santos Manvailer LF, et al. Elevated levels of extracellular vesicles are associated with therapy failure and disease progression in breast cancer patients undergoing neoadjuvant chemotherapy. Oncoimmunology. 2017;7(1):e1376153. https://doi.org/10.1080/2162402X.2017.1376153.
Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. 2016;30(6):836–48. https://doi.org/10.1016/j.ccell.2016.10.009.
Leal AC, Mizurini DM, Gomes T, Rochael NC, Saraiva EM, Dias MS, et al. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: implications for the establishment of cancer-associated thrombosis. Sci Rep. 2017;7(1):6438. https://doi.org/10.1038/s41598-017-06893-7.
Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 2016;531(7595):513–7. https://doi.org/10.1038/nature16985.
Xu R, Bao C, Huang H, Lin F, Yuan Y, Wang S, et al. Low expression of CXCR1/2 on neutrophils predicts poor survival in patients with hepatitis B virus-related acute-on-chronic liver failure. Sci Rep. 2016;6:38714. https://doi.org/10.1038/srep38714.
Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123:3446–58. https://doi.org/10.1172/JCI67484.
Wu K, Zhang H, Fu Y, Zhu Y, Kong L, Chen L, et al. TLR4/MyD88 signaling determines the metastatic potential of breast cancer cells. Mol Med Rep. 2018;18(3):3411–20. https://doi.org/10.3892/mmr.2018.9326.
Li Z, Block MS, Vierkant RA, Fogarty ZC, Winham SJ, Visscher DW, et al. The inflammatory microenvironment in epithelial ovarian cancer: a role for TLR4 and MyD88 and related proteins. Tumour Biol. 2016;37(10):13279–86. https://doi.org/10.1007/s13277-016-5163-2.
Mackey JBG, Coffelt SB, Carlin LM. Neutrophil maturity in cancer. Front Immunol. 2019;10:1912. https://doi.org/10.3389/fimmu.2019.01912.
Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361(6409). https://doi.org/10.1126/science.aao4227.
Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12(12):2074–88. https://doi.org/10.1111/jth.12710.
Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118(7):1952–61. https://doi.org/10.1182/blood-2011-03-343061.
Abdol Razak N, Elaskalani O, Metharom P. Pancreatic cancer-induced neutrophil extracellular traps: a potential contributor to cancer-associated thrombosis. Int J Mol Sci. 2017;18(3). https://doi.org/10.3390/ijms18030487.
Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SH, Weitz JI, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol. 2014;34(9):1977–84. https://doi.org/10.1161/ATVBAHA.114.304114.
Thalin C, Demers M, Blomgren B, Wong SL, von Arbin M, von Heijne A, et al. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb Res. 2016;139:56–64. https://doi.org/10.1016/j.thromres.2016.01.009.
Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S, et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight. 2019;5. https://doi.org/10.1172/jci.insight.128008This paper expands on the role of NETs as prognostic tool in cancer. Patients with more advance cancer showed significant elevation in NET levels compared to patients with a less advanced diagnosis.
Decker AS, Pylaeva E, Brenzel A, Spyra I, Droege F, Hussain T, et al. Prognostic role of blood NETosis in the progression of head and neck cancer. Cells. 2019;8(9). https://doi.org/10.3390/cells8090946This article revealed that neutrophils from patients with head and neck cancer were more potent in triggering NET formation when compared to that from healthy controls.
Thalin C, Lundstrom S, Seignez C, Daleskog M, Lundstrom A, Henriksson P, et al. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS One. 2018;13(1):e0191231. https://doi.org/10.1371/journal.pone.0191231.
Carroll GM, Burns GL, Petit JA, Walker MM, Mathe A, Smith SR, et al. Does postoperative inflammation or sepsis generate neutrophil extracellular traps that influence colorectal cancer progression? A systematic review. Surg Open Sci. 2020;2(2):57–69. https://doi.org/10.1016/j.sopen.2019.12.005.
Galos EV, Tat TF, Popa R, Efrimescu CI, Finnerty D, Buggy DJ, et al. Neutrophil extracellular trapping and angiogenesis biomarkers after intravenous or inhalation anaesthesia with or without intravenous lidocaine for breast cancer surgery: a prospective, randomised trial. Br J Anaesth. 2020;125(5):712–21. https://doi.org/10.1016/j.bja.2020.05.003First RCT to test the impact of various anaesthetic techniques on NET expression. Also, the first study to use lidocaine for NET expression modulation. Although there was no difference between the propofol and vapor anaesthesia groups, the addition of lidocaine significantly decreased citH3 and MPO expression.
Aghamelu O, Buggy P, Smith G, Inzitari R, Wall T, Buggy DJ. Serum NETosis expression and recurrence risk after regional or volatile anaesthesia during breast cancer surgery: a pilot, prospective, randomised single-blind clinical trial. Acta Anaesthesiol Scand. 2020. https://doi.org/10.1111/aas.13745This paper which evaluated the impact of volatile general anaesthesia and regional anaesthesia plus propofol found no difference in the expression of NETs markers: citH3 and MPO.
Tilgner J, von Trotha KT, Gombert A, Jacobs MJ, Drechsler M, Doring Y, et al. Aspirin, but not tirofiban displays protective effects in endotoxin induced lung injury. PLoS One. 2016;11(9):e0161218. https://doi.org/10.1371/journal.pone.0161218.
Domingo-Gonzalez R, Martinez-Colon GJ, Smith AJ, Smith CK, Ballinger MN, Xia M, et al. Inhibition of neutrophil extracellular trap formation after stem cell transplant by prostaglandin E2. Am J Respir Crit Care Med. 2016;193(2):186–97. https://doi.org/10.1164/rccm.201501-0161OC.
Cata JP, Guerra CE, Chang GJ, Gottumukkala V, Joshi GP. Non-steroidal anti-inflammatory drugs in the oncological surgical population: beneficial or harmful? A systematic review of the literature. Br J Anaesth. 2017;119(4):750–64. https://doi.org/10.1093/bja/aex225.
Evans MT, Wigmore T, Kelliher LJS. The impact of anaesthetic technique upon outcome in oncological surgery. BJA Educ. 2019;19(1):14–20. https://doi.org/10.1016/j.bjae.2018.09.008.
Cairat M, Al Rahmoun, M., Gunter, M.J. et al,. Use of nonsteroidal anti-inflammatory drugs and breast cancer risk in a prospective cohort of postmenopausal women. Breast Cancer Res 22, 118 2020. https://doi.org/10.1186/s13058-020-01343-1.
Khan MA, D’Ovidio A, Tran H, Palaniyar N. Anthracyclines suppress both NADPH oxidase-dependent and -independent NETosis in human neutrophils. Cancers (Basel). 2019;11(9). https://doi.org/10.3390/cancers11091328.
Hollmen M, Karaman S, Schwager S, Lisibach A, Christiansen AJ, Maksimow M, et al. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. Oncoimmunology. 2016;5(3):e1115177. https://doi.org/10.1080/2162402X.2015.1115177.
Ishikawa M, Yamashita H, Oka N, Ueda T, Kohama K, Nakao A, et al. Antithrombin III improved neutrophil extracellular traps in lung after the onset of endotoxemia. J Surg Res. 2017;208:140–50. https://doi.org/10.1016/j.jss.2016.09.041.
Segoviano-Ramirez JC, Lopez-Altamirano DF, Garcia-Juarez J, Aguirre-Garza JES, Cardenas-Estrada E, Ancer-Rodriguez J. The diethylcarbamazine delays and decreases the NETosis of polymorphonuclear cells of humans with DM type 2. J Diabetes Res. 2020;2020:4827641–8. https://doi.org/10.1155/2020/4827641.
Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673. https://doi.org/10.1038/ncomms7673.
Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26). https://doi.org/10.1126/sciimmunol.aar6689.
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107(36):15880–5. https://doi.org/10.1073/pnas.1005743107.
Sapey E, Patel JM, Greenwood H, Walton GM, Grudzinska F, Parekh D, et al. Simvastatin improves neutrophil function and clinical outcomes in pneumonia. A pilot randomized controlled clinical trial. Am J Respir Crit Care Med. 2019;200(10):1282–93. https://doi.org/10.1164/rccm.201812-2328OC.
Chow OA, von Kockritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe. 2010;8(5):445–54. https://doi.org/10.1016/j.chom.2010.10.005.
Yang Y, Kim HJ, Woo KJ, Cho D, Bang SI. Lipo-PGE1 suppresses collagen production in human dermal fibroblasts via the ERK/Ets-1 signaling pathway. PLoS One. 2017;12(6):e0179614. https://doi.org/10.1371/journal.pone.0179614.
Naffah de Souza C, LCD B, Khan MA, de Almeida SR, NOS C, Sweezey N, et al. Alkaline pH promotes NADPH oxidase-independent neutrophil extracellular trap formation: a matter of mitochondrial reactive oxygen species generation and citrullination and cleavage of histone. Front Immunol. 2017;8:1849. https://doi.org/10.3389/fimmu.2017.01849.
Persi E, Duran-Frigola M, Damaghi M, Roush WR, Aloy P, Cleveland JL, et al. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat Commun. 2018;9(1):2997. https://doi.org/10.1038/s41467-018-05261-x.
Sauter DR, Sorensen CE, Rapedius M, Bruggemann A, Novak I. pH-sensitive K(+) channel TREK-1 is a novel target in pancreatic cancer. Biochim Biophys Acta. 2016;1862(10):1994–2003. https://doi.org/10.1016/j.bbadis.2016.07.009.
Khan MA, Philip LM, Cheung G, Vadakepeedika S, Grasemann H, Sweezey N, et al. Regulating NETosis: increasing pH promotes NADPH oxidase-dependent NETosis. Front Med (Lausanne). 2018;5:19. https://doi.org/10.3389/fmed.2018.00019.
Arpinati L, Shaul ME, Kaisar-Iluz N, Mali S, Mahroum S, Fridlender ZG. NETosis in cancer: a critical analysis of the impact of cancer on neutrophil extracellular trap (NET) release in lung cancer patients vs. mice. Cancer Immunol Immunother. 2020;69(2):199–213. https://doi.org/10.1007/s00262-019-02474-x.
Acknowledgements
We would like to express our very great appreciation to Dr Gillian Crowe and http://smart.servier.com/ for providing the graphic art for Fig. 1.
Author information
Authors and Affiliations
Contributions
CIE and PMB equally contributed to writing the manuscript; DJB conceived the idea, supervised the writing process and edited the final draft of the manuscript. All authors approved the final version to be published.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Anesthesiology and Critical Care
Supplementary material
Table 1S
Summary of relevant articles (DOCX 60 kb)
Rights and permissions
About this article

Cite this article
Efrimescu, C.I., Buggy, P.M. & Buggy, D.J. Neutrophil Extracellular Trapping Role in Cancer, Metastases, and Cancer-Related Thrombosis: a Narrative Review of the Current Evidence Base. Curr Oncol Rep 23, 118 (2021). https://doi.org/10.1007/s11912-021-01103-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s11912-021-01103-0