
Abstract
Despite ongoing medical advances, cardiovascular disease continues to be a leading health concern. The renin-angiotensin system (RAS) plays an important role in regulating cardiovascular function, and is, therefore, the subject of extensive study. Several drugs currently used to treat hypertension and heart failure are designed to target angiotensin II synthesis and function, but thus far, none have been able to completely block the effects of RAS signaling. This review discusses current and emerging approaches towards inhibiting cardiac RAS function in order to further improve cardiovascular disease outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
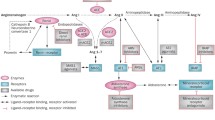
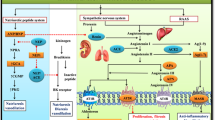
Cardiovascular disease continues to be a leading cause of death and reduced quality of life. The renin-angiotensin system (RAS) and its effector molecule, angiotensin (Ang) II, are known to affect a wide variety of functions in the cardiovascular and renal systems, playing an important role in the pathophysiology of cardiovascular disease. Therefore, there has been considerable interest over the years in understanding exactly how the RAS functions and is regulated. The RAS was once thought to be a fairly simple linear cascade operating systemically, but over the past few decades it has become increasingly clear that the RAS is actually a complex system that includes multiple alternate pathways and feedback mechanisms. The fact that the RAS functions locally as well as systemically, and that this local function may be differently regulated and involve different components than the systemic RAS, is of particular importance in understanding the impact of current or potential therapies designed to inhibit the pathophysiological effects of Ang II. In addition, the discovery of additional Ang peptides, including Ang-(1–5), Ang-(1–7), Ang-(1–9), Ang-(2–8), Ang-(3–8), and Ang-(1–12) [1–3], has made it clear that Ang II can no longer be viewed as acting in isolation.
Nevertheless, Ang II is generally still considered to be the primary effector molecule of the RAS, and its impact on the heart, both directly and as a result of renal and vascular effects, is significant. With roles in vasoconstriction, mitochondrial function [4], thrombosis, fibrosis, hypertrophy [5], apoptosis [6], and autophagy [7••], Ang II is intimately involved in cardiac function and remodeling. Heart failure is associated with RAS upregulation, and acute myocardial ischemia-reperfusion significantly increases expression of several of the major cardiac RAS components, including Ang II [8]. This review will focus on the regulation of Ang II in the heart, in the context of current and emerging modes of intervention.
Formation of Angiotensin II
Angiotensin-Converting Enzyme
In the classic RAS, angiotensinogen produced in the liver enters the bloodstream, where it is cleaved by kidney-derived renin to form the decapeptide, Ang I. Endothelial cell–bound angiotensin-converting enzyme (ACE) then further cleaves Ang I to produce the biologically active octapeptide, Ang II. The ubiquitously expressed ACE exists in both membrane-bound and soluble forms, and has two independent catalytic sites with distinct substrate and inhibitor specificities. ACE is also capable of hydrolyzing a variety of other substrates, including the cardioprotective agents bradykinin, N-acetyl-seryl-aspartyl-lysyl-proline tetrapeptide, and Ang-(1–7) [2, 3, 9]. Furthermore, ACE inhibitors enhance kinin B1 and B2 receptor signaling [10]. Thus, ACE has both Ang II–dependent and Ang II–independent effects on cardiovascular function and is a logical target for RAS regulation.
Indeed, ACE inhibition reduces blood pressure, left ventricular hypertrophy, and cardiac inflammation, possibly through inactivation of NFκB, in spontaneously hypertensive rats [11, 12]. Clinical studies have shown various ACE inhibitors to be effective in treating congestive heart failure, acute myocardial infarction, and coronary artery disease, as well as hypertension [13•]. Several different ACE inhibitors are commercially available, and current guidelines include the use of ACE inhibitors as first-line therapy for patients with heart failure or at risk for heart failure [14]. However, use of ACE inhibitors is associated with a relatively high incidence of adverse effects, presumably owing to their effect on the kallikrein/kinin system.
Although acute ACE inhibition decreases plasma Ang II, chronic treatment results in an escape phenomenon in which Ang II returns to pretreatment levels. The mechanisms involved in this rebound are not well understood, but there is some evidence that ACE may be involved in cellular signaling, in addition to its enzymatic role [15]. Protein kinase CK2 binds and phosphorylates Ser1270 in the ACE cytoplasmic tail, an interaction that is enhanced in endothelial cells in the presence of bradykinin or an ACE inhibitor. The phosphorylated ACE activates JNK, leading to nuclear accumulation of c-Jun and an increase in ACE expression. Although Kohlstedt et al. [15] suggest that this ACE inhibitor–induced increase in ACE expression may somehow contribute to the beneficial effects of ACE inhibition, this “ACE signaling” may act as a negative feedback mechanism by which ACE inhibition becomes self-limiting. On the other hand, it is also possible that long-term ACE inhibition eventually shifts the balance toward the alternative, chymase-dependent, Ang II synthesis pathway.
Chymase
Although ACE is the main enzyme responsible for systemic Ang II synthesis, chymase is also a major Ang II–forming enzyme in the human heart. Chymase is found mainly in the secretory granules of mast cells, where it remains inactive until its release into surrounding interstitial tissues. It is also strongly inhibited by endogenous serine protease inhibitors when not complexed with heparin proteoglycan. This fact, together with the existence of significant species variability in both the number and types of chymase, has contributed to some controversy about the physiological role of chymase, but there is accumulating evidence that chymase plays an important role in cardiac tissue formation of Ang II, particularly under pathophysiological conditions [16–18]. Recently, Wei and colleagues [19••] demonstrated that, although chymase activity in the interstitial fluid of conscious mice was negligible under basal conditions, chronic ACE inhibition markedly increased transcription and activity of an Ang II–forming chymase via a kinin B2 receptor/mast cell–dependent mechanism, while Ang II levels remained unchanged. Addition of a chymase inhibitor significantly lowered Ang II levels, demonstrating that chymase was, in fact, responsible for the maintained Ang II levels. Dual treatment with both ACE and chymase inhibitors dramatically improved left ventricular function, remodeling, and survival following myocardial infarction in hamsters. Furthermore, recent evidence suggests that Ang-(1–12), a recently discovered biologically active Ang peptide, can act as an alternative precursor for Ang II [20••], and that chymase is responsible for this process in cardiac tissue [21].
In addition to cleaving Ang peptides, chymase also appears to play a role in tissue remodeling and organ damage during heart failure. Chymase activates transforming growth factor-β (TGF-β), which increases fibroblast proliferation and collagen deposition, and matrix metalloprotease-9 (MMP-9), which is involved in extracellular matrix turnover. Thus, as with ACE inhibition, chymase inhibition potentially could provide benefits in patients with cardiovascular disease that are both dependent and independent of Ang II [22].
Renin/Prorenin
Clearly, the existence of alternate pathways for conversion of Ang I to Ang II makes complete inhibition of the RAS problematic at this point. As mentioned above, renin functions further upstream in Ang II formation, converting angiotensinogen to Ang I. Circulating renin originates primarily in the kidneys, where it is synthesized by cleavage of its precursor, prorenin. The source of renin involved in local Ang II synthesis is less clear, as renin expression levels in cardiac tissue are generally very low under basal conditions. However, cardiac renin expression has been detected under certain pathological conditions, such as diabetes [23], and a nonsecreted, truncated active form of renin is induced by myocardial infarction [24]. In addition, circulating prorenin and renin can bind to mannose-6-phosphate and (pro)renin receptors, allowing them to contribute to local Ang II synthesis. Whether cardiac mast cells are a significant source of renin remains controversial and may depend on context, species, or both [25–27].
A number of direct renin inhibitors exist [28•], but the most extensively studied inhibitor is aliskiren. In spontaneously hypertensive rats, aliskiren was equivalent to other RAS inhibitors in improving coronary endothelial function and cardiac hypertrophy, but it provided superior long-term suppression of cardiac angiotensin [29]. Similarly, diabetes-induced intracellular Ang II synthesis in adult rat cardiomyocytes, and the associated oxidative stress and fibrosis, were inhibited more effectively by aliskiren than by other RAS inhibitors [23]. Though approved for antihypertensive use, the relative effectiveness of aliskiren in reducing hypertension is still a matter of debate. Though some studies have shown aliskiren to be superior to other RAS inhibitors, others have shown comparable but not superior effectiveness [28•, 30, 31]. Although direct renin inhibition should more completely inhibit downstream RAS function, and aliskiren has a longer half-life than other RAS inhibitors, the absence of negative feedback from Ang II signaling leads to an increase in plasma renin/prorenin levels. This increase in renin/prorenin concentration may enhance (pro)renin receptor signaling, which has been shown to activate extracellular signal-regulated kinase (ERK) in an Ang II–independent manner even in the presence of aliskiren [32••], although the physiological relevance of this pathway in vivo remains to be determined. Alternatively, Ang-(1–12) may serve as an alternate substrate for Ang II formation in the absence of renin activity [33], thereby limiting Ang II suppression. On the other hand, there is evidence that aliskiren may have cardioprotective effects in patients with left ventricular hypertrophy or heart failure [28•, 30, 31], and ongoing studies should provide additional data on its clinical usefulness against conditions associated with high plasma renin activity.
Angiotensin II Function
Angiotensin II Type 1 Receptor Blockers
Ang II can bind to either of two seven-transmembrane G protein-coupled receptors, the Ang II type 1 receptor (AT1R) or the Ang II type 2 receptor (AT2R). Although it has been suggested that intracellular Ang II may have intracrine functions independent of the AT1R [17], most of the known functions of Ang II involve binding to this receptor, and the AT1R exerts its effects through both G protein signaling and heterodimerization with or transactivation of other receptors [3]. AT1R is found on the cell surface, where it binds extracellular Ang II and is rapidly internalized, as well as on mitochondria [4] and nuclei [34]. AT2R, on the other hand, has very limited expression in adult organisms but is upregulated during certain pathophysiological conditions. Its functions generally oppose those of the AT1R [5, 35].
AT1R blockers (ARBs) are designed to specifically inhibit Ang II-induced AT1R signaling, and, therefore, theoretically should provide more complete RAS blockade than interventions that block only one of the alternative Ang II formation pathways. Moreover, AT2R signaling induced by the resulting accumulated Ang II may further potentiate the protective effects of ARBs. Clinical trials have shown ARBs to be effective in reducing hypertension [36], but reports on the effectiveness of ARBs in treating other cardiovascular disease outcomes have been mixed [37•]. These results may be due in part to the absence of rigorous dosage optimization, as suggested by the differential effects of high and low doses of losartan seen in the HEAAL study [38]. In addition, small differences in the structure of different ARBs have proven to have a significant impact on their functionality, including both AT1R-dependent and AT1R-independent effects [39, 40]. Therefore, the effectiveness of any given ARB may depend on the specific disease condition. Current guidelines recommend the use of ARBs in patients at risk for heart failure and as an alternative treatment for selected heart failure patients who are intolerant of ACE inhibitors [14]. However, a recent meta-analysis found that ARBs may be associated with an increased risk of cancer [41•], suggesting that further investigation of these drugs is warranted.
AT1R-Associated Protein
AT1R-associated protein (ATRAP) is an 18-kDa protein which, upon Ang II stimulation, binds to the carboxyl-terminal cytoplasmic tail of AT1R in cardiovascular cells and promotes receptor internalization, acting as an endogenous AT1R inhibitor [42]. Although the physiological importance of this protein needs further clarification, it appears that the AT1R:ATRAP ratio is important in determining the effect of Ang II on the heart. Ang II–induced cardiac hypertrophy was accompanied by a significant decrease in ATRAP expression in wild-type mice and was completely abolished in transgenic mice with cardiomyocyte-specific overexpression of ATRAP, independent of any effect on blood pressure [43••]. Thus, increasing ATRAP expression could be a novel therapeutic option for inhibiting AT1R signaling.
Cell-Penetrating Peptides
Cell-penetrating peptides are typically short cationic peptide sequences, which can be used to deliver proteins into intact cells. A cell-penetrating peptide consisting of the second intracellular loop of the AT1R linked to the HIV-transactivating regulatory protein domain effectively inhibited AT1R functions mediated by the second intracellular loop, but not those previously shown to be unaffected by mutations in this region [44]. Although this approach requires further development and a more complete understanding of the mechanisms involved in AT1R signaling, this study suggests that cell-penetrating peptides potentially may be used to take advantage of the biased agonism observed in AT1R signaling, serving as a powerful tool for selectively regulating Ang II receptor function.
AT2R Stimulation
As mentioned above, Ang II–AT2R signaling generally opposes that of the AT1R, and AT2R signaling triggered by accumulated Ang II may be partially responsible for the antihypertensive effect of ARBs through its induction of bradykinin/nitric oxide–mediated vasodilation [45]. In addition, the AT2R may have ligand-independent protective effects, inhibiting AT1R signaling directly through heterodimerization of the two receptors [3] and constitutively inhibiting cardiomyocyte autophagy [7••]. However, AT2R signaling has also been linked to cardiac hypertrophy and apoptosis [5], and a recent study suggests that AT2R function can change from relaxant to constrictor upon Ang II binding under hypertensive conditions [46••]. Nevertheless, direct AT2R stimulation with a novel nonpeptide AT2R agonist, compound 21, inhibited myocardial infarction–induced apoptosis, inflammation, scar formation, and systolic and diastolic left ventricular dysfunction [47], suggesting that direct AT2R stimulation could be an effective way to prevent cardiac remodeling.
Degradation of Angiotensin II
Like its homolog, ACE, ACE2 is a zinc-metallopeptidase that can exist in both membrane-bound and soluble forms. Whereas ACE synthesizes Ang II from Ang I, however, ACE2 serves the opposite function, degrading Ang II to Ang-(1–7). In addition, ACE2 can cleave Ang I to Ang-(1–9), which can then be further cleaved by ACE to give Ang-(1–7). Although its importance in baseline cardiac function is somewhat controversial [48], several studies in mice and rats have shown ACE2 to be protective against pathologic cardiac remodeling and dysfunction, owing to either the resultant decrease in Ang II levels or the increase in the biologically active Ang-(1–7) [49–52].
Degradation of Ang II is an obviously important part of ACE2 function, but synthesis of biologically active cleavage products also appears to play an important role in mediating its cardioprotective effects. The degradation product of Ang II, Ang-(1–7), binds to the Mas receptor, thereby exerting effects that oppose those of Ang II, including inhibiting cardiac hypertrophy and fibrosis [53, 54••, 55]. Ang-(1–7) has also been shown to bind to ACE, inhibiting bradykinin degradation, as well as to AT1R and AT2R, inhibiting the former and stimulating the latter [2]. Ang-(1–9) is formed by cleavage of Ang I by either ACE2 or carboxypeptidase A. Though Ang-(1–9) was once thought to be simply a stable intermediary in Ang-(1–7) production, it has now been shown to be biologically active in its own right. Administration of Ang-(1–9) following myocardial infarction in rats decreased plasma Ang II, inhibited ACE activity, and prevented cardiomyocyte hypertrophy [56••]. Furthermore, treatment of myocardial infarcted rats with either an ACE inhibitor or ARB led to an increase in circulating Ang-(1–9) but not Ang-(1–7), suggesting that Ang-(1–9) may be an important cardioprotective factor.
ACE2 expression is inhibited by Ang II and is upregulated by treatment with ACE inhibitors and ARBs, suggesting that it may contribute to the effectiveness of these drugs and may itself be a good candidate for therapeutic intervention. Possible approaches for increasing ACE2 activity include using a small-molecule activator, such as XNT ([1{(2-[dimethylamino]ethyl)amino}-4-[hydroxymethyl]-7-[{(4-methylphenyl)sulfonyl}oxy]-9 H-xanthen-9-one]), and gene transfer therapy [53]. Further investigation is warranted, however, as long-term extreme overexpression of ACE2 resulted in severe cardiac fibrosis in stroke-prone spontaneously hypertensive rats [57], and ACE2 has recently been identified as the receptor for the severe acute respiratory syndrome (SARS) virus [58]. As with ACE2, Ang-(1–7) levels are increased by ACE inhibitor and ARB treatment, suggesting that it too plays a role in the beneficial effects of these drugs. However, Ang-(1–7) has a short half-life, making it unsuitable for direct therapeutic use. Alternative Mas agonists, such as AVE0991, as well as approaches to increasing the stability of Ang-(1–7), are currently under investigation [53].
Antibodies
Because patient adherence to a daily medication regimen can be problematic, a vaccine administered only several times a year may be a more effective mode of treatment. Although early attempts to develop vaccines against renin and Ang I were not successful, recent work on an Ang II vaccine, CYT006-AngQb, appears to be more promising. In initial testing, a 300-μg dose significantly reduced blood pressure and appeared to be well tolerated [59]. Further study is required, however, to determine the long-term efficacy and safety of such a vaccine. In a somewhat different approach, injection of antibodies against the C-terminal fragment of the AT1R (kardos) also appears to have an antihypertensive effect and is currently under investigation [60].
Conclusions
Ongoing advances in modern medicine have led to the development of many effective treatment options for hypertension and cardiovascular disease. Nevertheless, morbidity and mortality related to cardiovascular disease remain high. The RAS plays an important role in determining cardiovascular function, and several currently used therapeutic approaches specifically target critical points in the RAS signaling pathway. However, because of the existence of alternative synthesis and signaling pathways and tight regulation via positive and negative feedback loops, the goal of complete inhibition of the effects of Ang II remains elusive. The extent to which Ang II exerts its effects via pathways dependent on blood pressure or independent of it remains a matter of debate, prompting some to argue that the main focus of any treatment regimen should be blood pressure control, rather than specifically targeting the RAS. On the other hand, drugs chosen for effective control of hypertension may not be optimal for preventing organ damage and may require further optimization for treatment of heart failure. Combinations of multiple RAS inhibitors are also being heavily investigated as a means to more completely inhibit RAS function, but these remain controversial. As our understanding of the mechanisms involved in RAS function and its inhibition grows, however, it should become possible to further improve patient outcomes. The ongoing discovery of new functions of current drugs, as well as new potential therapeutic targets, is an indication that there is still much to be learned and great potential for more effective cardiovascular treatment.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ferrario CM: New physiological concepts of the renin-angiotensin system from the investigation of precursors and products of angiotensin I metabolism. Hypertension 2010, 55(2):445–452.
Iusuf D, Henning RH, van Gilst WH, Roks AJ: Angiotensin-(1-7): pharmacological properties and pharmacotherapeutic perspectives. Eur J Pharmacol 2008, 585(2-3):303–312.
Kurdi M, De Mello WC, Booz GW: Working outside the system: an update on the unconventional behavior of the renin-angiotensin system components. Int J Biochem Cell Biol 2005, 37(7):1357–1367.
Re RN, Cook JL: The mitochondrial component of intracrine action. Am J Physiol 2010, 299(3):H577–583.
Schluter KD, Wenzel S: Angiotensin II: a hormone involved in and contributing to pro-hypertrophic cardiac networks and target of anti-hypertrophic cross-talks. Pharmacol Ther 2008, 119(3):311–325.
Burniston JG, Saini A, Tan LB, Goldspink DF: Angiotensin II induces apoptosis in vivo in skeletal, as well as cardiac, muscle of the rat. Exp Physiol 2005, 90(5):755–761.
•• Porrello ER, D’Amore A, Curl CL, et al.: Angiotensin II type 2 receptor antagonizes angiotensin II type 1 receptor-mediated cardiomyocyte autophagy. Hypertension 2009, 53(6):1032–1040. This study describes a previously unknown function of the RAS, providing the first evidence that Ang II and its receptors are involved in regulating autophagy in the heart.
Oyamada S, Bianchi C, Takai S, et al.: Impact of acute myocardial ischemia reperfusion on the tissue and blood-borne renin-angiotensin system. Basic Res Cardiol 2010, 105(4):513–522.
Peng H, Carretero OA, Liao TD, et al.: Role of N-acetyl-seryl-aspartyl-lysyl-proline in the antifibrotic and anti-inflammatory effects of the angiotensin-converting enzyme inhibitor captopril in hypertension. Hypertension 2007, 49(3):695–703.
Erdos EG, Tan F, Skidgel RA: Angiotensin I-converting enzyme inhibitors are allosteric enhancers of kinin B1 and B2 receptor function. Hypertension 2010, 55(2):214–220.
Miguel-Carrasco JL, Zambrano S, Blanca AJ, et al.: Captopril reduces cardiac inflammatory markers in spontaneously hypertensive rats by inactivation of NF-kB. J Inflamm 2010, 7:21.
Simko F, Pechanova O, Pelouch V, et al.: Effect of melatonin, captopril, spironolactone and simvastatin on blood pressure and left ventricular remodelling in spontaneously hypertensive rats. J Hypertens Suppl 2009, 27(6):S5–10.
• Katragadda S, Arora RR: Role of angiotensin-converting enzyme inhibitors in vascular modulation: beyond the hypertensive effects. Am J Ther 2010, 17(1):e11–e23. This article is a good review of the data obtained from randomized controlled trials evaluating ACE inhibitors. However, it does not include the recent, much discussed ONTARGET study, which suggested that dual blockade with ACE inhibitors and ARBs may have little clinical benefit and may actually be detrimental in some patients.
Jessup M, Abraham WT, Casey DE, et al.: 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009, 119(14):1977–2016.
Kohlstedt K, Brandes RP, Muller-Esterl W, et al.: Angiotensin-converting enzyme is involved in outside-in signaling in endothelial cells. Circ Res 2004, 94(1):60–67.
Dell’Italia LJ, Husain A: Dissecting the role of chymase in angiotensin II formation and heart and blood vessel diseases. Curr Opin Cardiol 2002, 17(4):374–379.
Kumar R, Singh VP, Baker KM: The intracellular renin-angiotensin system in the heart. Curr Hypertens Rep 2009, 11(2):104–110.
Miyazaki M, Takai S, Jin D, Muramatsu M: Pathological roles of angiotensin II produced by mast cell chymase and the effects of chymase inhibition in animal models. Pharmacol Ther 2006, 112(3):668–676.
•• Wei CC, Hase N, Inoue Y, et al.: Mast cell chymase limits the cardiac efficacy of Ang I-converting enzyme inhibitor therapy in rodents. J Clin Invest 2010, 120(4):1229–1239. This study suggests that, rather than inhibiting local generation of Ang II, chronic ACE inhibition actually plays a direct role in maintaining Ang II levels in the cardiac interstitium by inducing chymase-dependent Ang II synthesis.
•• Trask AJ, Jessup JA, Chappell MC, Ferrario CM: Angiotensin-(1-12) is an alternate substrate for angiotensin peptide production in the heart. Am J Physiol 2008, 294(5):H2242–2247. This study provides the first evidence that Ang-(1–12) can serve as an alternative precursor for Ang II synthesis.
Prosser HC, Forster ME, Richards AM, Pemberton CJ: Cardiac chymase converts rat proAngiotensin-12 (PA12) to angiotensin II: effects of PA12 upon cardiac haemodynamics. Cardiovasc Res 2009, 82(1):40–50.
Takai S, Jin D, Miyazaki M: New approaches to blockade of the renin-angiotensin-aldosterone system: chymase as an important target to prevent organ damage. J Pharmacol Sci 2010, 113(4):301–309.
Singh VP, Le B, Khode R, et al.: Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008, 57(12):3297–3306.
Wanka H, Kessler N, Ellmer J, et al.: Cytosolic renin is targeted to mitochondria and induces apoptosis in H9c2 rat cardiomyoblasts. J Cell Mol Med 2009, 13(9A):2926–2937.
Bispo-da-Silva LB, Sivieri-Jr DO, Prado CM, et al.: Cardiac mast cell proteases do not contribute to the regulation of the rat coronary vascular responsiveness to arterial delivered angiotensin I and II. Vascul Pharmacol 2010, 53(1-2):22–27.
Krop M, van Veghel R, Garrelds IM, et al.: Cardiac Renin levels are not influenced by the amount of resident mast cells. Hypertension 2009, 54(2):315–321.
Mackins CJ, Kano S, Seyedi N, et al.: Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest 2006, 116(4):1063–1070.
• Israili ZH, Velasco M, Bermudez V: Direct renin inhibitors as antihypertensive agents. Am J Ther 2010, 17(3):237–254. This review article gives a good overview of the function and effectiveness of currently available direct renin inhibitors.
van Esch JH, Moltzer E, van Veghel R, et al.: Beneficial cardiac effects of the renin inhibitor aliskiren in spontaneously hypertensive rats. J Hypertens 2010, 28(10):2145–2155.
McMurray JJ, Pitt B, Latini R, et al.: Effects of the oral direct renin inhibitor aliskiren in patients with symptomatic heart failure. Circ Heart Fail 2008, 1(1):17–24.
Solomon SD, Appelbaum E, Manning WJ, et al.: Effect of the direct Renin inhibitor aliskiren, the Angiotensin receptor blocker losartan, or both on left ventricular mass in patients with hypertension and left ventricular hypertrophy. Circulation 2009, 119(4):530–537.
•• Feldt S, Batenburg WW, Mazak I, et al.: Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension 2008, 51(3):682–688. This study provides evidence that binding of renin or prorenin to the (pro)renin receptor can trigger Ang II–independent cellular signaling that is not blocked by renin inhibition.
Ferrario CM, Varagic J, Habibi J, et al.: Differential regulation of angiotensin-(1-12) in plasma and cardiac tissue in response to bilateral nephrectomy. Am J Physiol 2009, 296(4):H1184–1192.
Tadevosyan A, Maguy A, Villeneuve LR, et al.: Nuclear-delimited angiotensin receptor-mediated signaling regulates cardiomyocyte gene expression. J Biol Chem 2010, 285(29):22338–22349.
Steckelings UM, Kaschina E, Unger T: The AT2 receptor—a matter of love and hate. Peptides 2005, 26(8):1401–1409.
Weber M: Achieving blood pressure goals: should angiotensin II receptor blockers become first-line treatment in hypertension? J Hypertens Suppl 2009, 27(5):S9–14.
• Siragy HM: Comparing angiotensin II receptor blockers on benefits beyond blood pressure. Adv Ther 2010, 27(5):257–284. This article is a good review of clinical data on the effectiveness of ARBs in treating cardiovascular diseases.
Konstam MA, Neaton JD, Dickstein K, et al.: Effects of high-dose versus low-dose losartan on clinical outcomes in patients with heart failure (HEAAL study): a randomised, double-blind trial. Lancet 2009, 374(9704):1840–1848.
Miura SI, Karnik SS, Saku K: Angiotensin II type 1 receptor blockers: class effects versus molecular effects. J Renin Angiotensin Aldosterone Syst 2010.
Sadoshima J: Novel AT(1) receptor-independent functions of losartan. Circ Res 2002, 90(7):754–756.
• Sipahi I, Debanne SM, Rowland DY, et al.: Angiotensin-receptor blockade and risk of cancer: meta-analysis of randomised controlled trials. Lancet Oncol 2010, 11(7):627–636. In this meta-analysis, Sipahi et al. suggest that ARB use may be associated with an increased risk of cancer.
Tamura K, Tanaka Y, Tsurumi Y, et al.: The role of angiotensin AT1 receptor-associated protein in renin-angiotensin system regulation and function. Curr Hypertens Rep 2007, 9(2):121–127.
•• Wakui H, Tamura K, Tanaka Y, et al.: Cardiac-specific activation of angiotensin II type 1 receptor-associated protein completely suppresses cardiac hypertrophy in chronic angiotensin II-infused mice. Hypertension 2010, 55(5):1157–1164. This transgenic mouse study provides evidence that the endogenous AT1R inhibitor, ATRAP, may be an effective way to suppress Ang II–induced cardiac hypertrophy.
Yu J, Taylor L, Mierke D, et al.: Limiting angiotensin II signaling with a cell-penetrating peptide mimicking the second intracellular loop of the angiotensin II type-I receptor. Chem Biol Drug Des 2010, 76(1):70–76.
Yayama K, Okamoto H: Angiotensin II-induced vasodilation via type 2 receptor: role of bradykinin and nitric oxide. Int Immunopharmacol 2008, 8(2):312–318.
•• Moltzer E, Verkuil AV, van Veghel R, et al.: Effects of angiotensin metabolites in the coronary vascular bed of the spontaneously hypertensive rat: loss of angiotensin II type 2 receptor-mediated vasodilation. Hypertension 2010, 55(2):516–522. This study provides evidence that AT2R function may switch from relaxant to constrictor under hypertensive conditions, suggesting that further investigation of its signaling mechanisms is required to better understand what effects AT2R agonists may have in a clinical setting.
Kaschina E, Grzesiak A, Li J, et al.: Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 2008, 118(24):2523–2532.
Gurley SB, Coffman TM: Angiotensin-converting enzyme 2 gene targeting studies in mice: mixed messages. Exp Physiol 2008, 93(5):538–542.
Der Sarkissian S, Grobe JL, Yuan L, et al.: Cardiac overexpression of angiotensin converting enzyme 2 protects the heart from ischemia-induced pathophysiology. Hypertension 2008, 51(3):712–718.
Kassiri Z, Zhong J, Guo D, et al.: Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail 2009, 2(5):446–455.
Trask AJ, Groban L, Westwood BM, et al.: Inhibition of angiotensin-converting enzyme 2 exacerbates cardiac hypertrophy and fibrosis in Ren-2 hypertensive rats. Am J Hypertens 2010, 23(6):687–693.
Zhong J, Basu R, Guo D, et al.: Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 2010, 122(7):717–728, 718 p following 728.
Ferreira AJ, Santos RA, Bradford CN, et al.: Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension 2010, 55(2):207–213.
•• Mercure C, Yogi A, Callera GE, et al.: Angiotensin(1-7) blunts hypertensive cardiac remodeling by a direct effect on the heart. Circ Res 2008, 103(11):1319–1326. This study suggests that the beneficial effects of ACE2 activity in the heart may be due in part to the resultant increase in Ang-(1–7) levels, rather than simply the degradation of Ang II.
Wang Y, Qian C, Roks AJ, et al.: Circulating rather than cardiac angiotensin-(1-7) stimulates cardioprotection after myocardial infarction. Circ Heart Fail 2010, 3(2):286–293.
•• Ocaranza MP, Lavandero S, Jalil JE, et al.: Angiotensin-(1-9) regulates cardiac hypertrophy in vivo and in vitro. J Hypertens 2010, 28(5):1054-1064. This study is the first to demonstrate a role for Ang-(1–9) in the regulation of cardiac hypertrophy.
Masson R, Nicklin SA, Craig MA, et al.: Onset of experimental severe cardiac fibrosis is mediated by overexpression of Angiotensin-converting enzyme 2. Hypertension 2009, 53(4):694–700.
Lambert DW, Clarke NE, Turner AJ: Not just angiotensinases: new roles for the angiotensin-converting enzymes. Cell Mol Life Sci 2010, 67(1):89–98.
Maurer P, Bachmann MF: Immunization against angiotensins for the treatment of hypertension. Clin Immunol 2010, 134(1):89–95.
Petrov VI, Nedogoda SV, Epshtein OI, et al.: The use of ultralow doses of antibodies to C-terminal fragment of angiotensin II AT1 receptor (kardos) in the therapy of arterial hypertension. Bull Exp Biol Med 2009, 148(2):332–334.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zablocki, D., Sadoshima, J. Knocking Out Angiotensin II in the Heart. Curr Hypertens Rep 13, 129–135 (2011). https://doi.org/10.1007/s11906-011-0180-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-011-0180-4