
Abstract
Strong evidence supports the ability of the aldosterone/mineralocorticoid receptor (MR) system to dominate long-term blood pressure control. It is also increasingly recognized as an important mediator of cardiovascular and renal diseases, particularly in the presence of excessive salt intake. In a subgroup of individuals with metabolic syndrome, adipocyte-derived aldosterone-releasing factors cause inappropriate secretion of aldosterone in the adrenal glands during salt loading, resulting in the development of salt-induced hypertension and cardiac and renal damage. On the other hand, emerging data reveal that aldosterone is not a sole regulator of MR activity. We have identified the signaling crosstalk between MR and small GTPase Rac1 as a novel pathway to facilitate MR signaling. Such a local control system for MR can also be relevant to the pathogenesis of salt-sensitive hypertension, and future studies will clarify the detailed mechanism for the intricate regulation of the aldosterone/MR cascade.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although the precise mechanism of hypertension remains unknown, there is ample evidence suggesting the central role of the kidneys in long-term blood pressure control. The cross-transplantation experiments in 1970s using a hereditary model of hypertension afforded strong evidence for the role of kidneys in blood pressure elevation [1]. The primacy of renal salt handling was further strengthened by the concept of pressure natriuresis [2], whereby the kidneys alter sodium and water excretion in response to changes in arterial pressure. It was postulated that almost all forms of hypertension can be explained by impaired renal sodium excretion [3, 4]. In more recent years, its critical role has been established by the molecular identification of rare mendelian disorders that cause high blood pressure [5]. The fact that almost all monogenic forms of hypertension solved to date are caused by altered renal sodium handling—especially dysregulation of the aldosterone/mineralocorticoid receptor (MR) cascade and its downstream targets [5]—highlights the ability of this system to dominate long-term blood pressure control in humans.
Habitual salt intake is closely related to blood pressure levels. An epidemiologic study involving many centers worldwide revealed that the systolic blood pressure slope with age is correlated with dietary salt intake [6]. Experimental data in other terrestrial mammals have largely confirmed the relationship between salt and blood pressure [7], but it is also known that there is heterogeneity in salt sensitivity of blood pressure. Indeed, blood pressure response to high salt intake differs among individuals with essential hypertension [8], and this difference in salt sensitivity independently affects the outcome of cardiovascular diseases [9]. The partnership between aldosterone and MR in the kidneys plays a central role in the physiological response to variations in dietary salt intake. In addition, aldosterone, or activation of MR, is now recognized as an important mediator of hypertension-associated target organ damage. Hence, dysregulation of MR signaling not only causes salt-dependent elevation of arterial pressure but also can explain the increased cardiovascular morbidity and mortality in patients with salt-sensitive hypertension. Moreover, in an effort to identify the mechanism of salt-induced organ damage, we have recently discovered signaling cross-talk between MR and small GTPase Rac1, challenging the prevailing view of the regulation of the aldosterone/MR signaling cascade [10••]. This article reviews recent progress regarding MR function and regulation, in order to address the role of MR signaling in the pathogenesis of salt-sensitive hypertension.
Renal Salt Handling and Aldosterone/MR
One of the major roles of the kidneys is to control fluid homeostasis in the body. About 99% of the filtered sodium and water is reabsorbed in the tubules via a primary active inward transport. Although the sodium handled in the distal nephron is a minor portion of the sodium reabsorbed in the kidneys, the fine tuning of sodium and water regulation occurs in these segments. The aldosterone/MR cascade exerts its effects in the so-called aldosterone-sensitive distal nephron (ASDN), which includes the late distal convoluted tubule, connecting tubule, and collecting duct. Upon binding of aldosterone, MR undergoes conformational changes, dissociation from chaperone proteins, dimerization, and translocation to the nucleus, where it binds to the responsive elements in the promoter regions of target genes to regulate transcription. Among the aldosterone-induced genes, serum/glucocorticoid regulated kinase 1 (SGK1) plays a major role in the control of sodium reabsorption. Studies have clarified the detailed mechanism of epithelial sodium channel (ENaC) regulation by SGK1 (Fig. 1). Nedd4-2 (neural precursor cell expressed, developmentally downregulated 4-2) is a HECT domain–containing E3 ubiquitin ligase that interacts with the C terminus of ENaC subunits and maintains the plasma membrane ENaC at low levels through ubiquitination-dependent mechanisms. Aldosterone-induced SGK1 phosphorylates the Nedd4-2, which disrupts the tonic inhibition of ENaC by Nedd4-2, leading to indirect stimulation of sodium transport. SGK1 phosphorylation of Nedd4-2 results in 14-3-3 binding and suppresses Nedd4-2–ENaC interaction [11]. ENaC mutations in Liddle’s syndrome also affect the Nedd4-2 interaction, leading to constitutive ENaC expression and increased sodium reabsorption [12]. SGK1 may also modulate ENaC activity though a mechanism independent of Nedd4-2 [13].

Mechanism of epithelial sodium channel (ENaC) regulation by the aldosterone/mineralocorticoid receptor (MR) system. Aldosterone-induced SGK1 phosphorylates Nedd4-2, which disrupts the tonic inhibition of ENaC. Glucocorticoid-induced leucine-zipper protein (GILZ) augments ENaC activity through the recruitment of SGK1 to ENaC complex including Nedd4-2, as well as through the inhibition of Raf-1. In addition, SGK1 regulates ENaCα gene transcription through phosphorylation of AF9, leading to the inhibition of Dot1a/AF9 interaction and the histone hypomethylation of ENaCα transcription. Aldosterone/MR signaling also interacts with the WNK kinase family. Nedd4-2—neural precursor cell expressed, developmentally downregulated 4-2; SGK1—serum/glucocorticoid regulated kinase 1
In addition, SGK1 regulates ENaCα gene transcription through epigenetic mechanism. A putative transcription factor, AF9, interacts with Dot1a, a histone H3 Lys-79 methyltransferase, to form a nuclear repressor complex with chromatin at specific regions of the ENaCα promoter, where it regulates histone H3 Lys-79 methylation and thereby represses ENaCα transcription under basal conditions. SGK1 phosphorylates AF9 and impairs the Dot1a-AF9 interaction, leading to histone hypomethylation and de-repression of ENaCα transcription [14].
Glucocorticoid-induced leucine-zipper protein (GILZ) was identified as an aldosterone-induced protein in cortical collecting duct cell lines. Raf-MEK-ERK1/2 forms a regulatory complex with ENaC and inhibits its function. GILZ interacts with the ENaC regulatory complex and augments ENaC activity via the inhibition of Raf-1 [15]. It is also shown to inhibit Nedd4-2 through the recruitment of SGK1 [16].
Recent studies also suggest that aldosterone/MR signaling interacts with the WNK kinase family. Mutations in WNK1 and WNK4 are known to be associated with pseudohypoaldosteronism type II (PHAII), or Gordon’s syndrome [17]. WNK4 has the SGK1 phosphorylation sites, and the WNK4 mutant mimicking SGK1 phosphorylation shows altered effects on the sodium-chloride cotransporter (NCC), ENaC, and the renal outer medullary potassium channel (ROMK), leading to speculation that SGK1 and WNK4 act in concert to regulate the balance between sodium reabsorption and potassium secretion [18, 19]. Conversely, some investigators have reported that SGK1 activity is regulated by WNK family members [20]. In addition, aldosterone can induce the expression of kidney-specific WNK1, a kinase-deficient isoform that lacks the N-terminal 437 residues of WNK1 [21]. These results indicate that the aldosterone/MR system controls electrolyte and fluid homeostasis in part through interaction with WNK kinases. Some studies have shown that a common single nucleotide polymorphism (SNP) in WNK kinases show an association with altered blood pressure [22], although its significance remains to be validated.
Role of MR Signaling in Target Organ Damage
Although abnormal activation of the renin-angiotensin-aldosterone system has long been reported to participate in the occurrence of end-organ damage in hypertensive patients, many studies have focused on the role of angiotensin II, and relatively little attention has been paid to the role of aldosterone. However, two clinical studies, the Randomized Aldactone Evaluation Study (RALES) and the Eplerenone Post-Acute Myocardial Infarction Heart Failure and Survival Study (EPHESUS), have demonstrated that MR antagonists, added to standard therapy including angiotensin-converting enzyme (ACE) inhibitor, substantially improved cardiac mortality [23, 24]; subsequent studies revealed that aldosterone/MR can have an effect on the vasculature, inducing inflammation, oxidative stress, fibrosis, and increased sympathetic tone. Hypertensive kidney damage is an important cause of end-stage renal disease. Accumulating data indicate that aldosterone/MR signaling is also a key mediator of the kidney injury. Although the precise mechanisms remain unclear, aldosterone/MR is shown to possess deleterious effects on non-ASDN cells in the kidneys, such as podocytes. Enhanced MR effector mechanism is closely related to protein leakage from the kidneys [25], a strong risk factor for progression of chronic kidney disease. Clinical studies have also revealed that MR blockade reduces urinary protein excretion even better than ACE inhibition [26]. In addition, experimental studies, including our own, have demonstrated the unequivocal benefits of a selective MR antagonist, eplerenone, in ameliorating proteinuria and glomerular podocyte damage [10••, 27]. It has been postulated that aldosterone/MR induces apoptosis or alters the adhesive capacity of podocytes [28, 29]. Because these cells are highly differentiated and are considered to lack proliferative capacity, the reduced numbers of podocytes cause denudation of the glomerular basement membrane and adhesion to Bowman’s capsule, a common pathway leading to glomerulosclerosis. MR is known to be present in other kidney cells, including mesangial cells and renal fibroblasts, and to modulate their functions via profibrotic action or alteration of cell cycle regulators [30]. In addition, aldosterone activates NF-κB in principal cells of the cortical collecting duct through mechanisms that involve SGK1 [31]. NF-κB regulates inflammation and transepithelial sodium transport, so this observation indicates a convergence of sodium reabsorption and inflammatory stimulation in the collecting duct. In line with these experimental data, subjects with aldosterone overproduction have a high incidence of renal dysfunction [32]. These cellular effects could be relevant to kidney injury in hypertensive patients with increased aldosterone/MR activity.
Aldosterone Excess and Salt-Sensitive Hypertension
Abnormal aldosterone production causes salt-sensitive hypertension. Aldosterone is produced in the zona glomerulosa, the outermost layer of adrenal cortex. Angiotensin II and hyperkalemia promote conversion of cholesterol to pregnenolone via stimulation of steroidogenic acute regulatory protein (StAR), and of corticosterone to aldosterone via aldosterone synthase (CYP11B2). The layer-specific synthesis is ensured by the specific expression of CYP11B2 and by the suppression of 17α hydroxylase (CYP17A1) that is necessary for the production of cortisol and androgen. Glucocorticoid-remediable aldosteronism, a monogenic form of hypertension, results from fusion of the synthetic portion of CYP11B2 and the regulator portion of CYP11B1 that catalyzes the conversion of 11-deoxycortisol to cortisol, leading to aldosterone production in response to ACTH in zona fasciculata [33].
Aldosterone secretion is controlled by circadian rhythm, and disruption of essential clock components is shown to cause abnormal aldosterone synthesis. Doi et al. [34••] reported that mice lacking Cryptochrome-1 (Cry1) and Cryptochrome-2 (Cry2) show salt-sensitive hypertension because of abnormally high synthesis of aldosterone. Further research revealed that Hsd3b6, an enzyme that catalyzes the conversion of Δ5-3β-hydroxysteroids into hormonally active Δ4-3-ketosteroids, is exclusive expressed in mice in the zona glomerulosa of the adrenal gland, and that this enzyme is upregulated in Cry knockout mice, causing enhanced aldosterone production [34••]. They also showed that HSD3B1, a human counterpart of mouse Hsd3b6, is enriched in human zona glomerulosa cells, indicating a role for this enzyme in the layer-specific biosynthesis of aldosterone. Future studies will clarify the role of HSD3B1 in the dysregulation of aldosterone and salt-sensitive hypertension in humans.
Aldosterone overproduction in primary aldosteronism (PA) is a common cause of blood pressure elevation. One study reported that this disorder accounts for more than 10% of cases of hypertension [35]. In addition, patients with PA exhibit cardiac remodeling, endothelial dysfunction, and albuminuria [32, 36], especially when salt intake is not decreased. One of the classic animal models for PA is a rat receiving chronic infusion of aldosterone and a high-salt diet. Using this model, we have shown that albuminuria induced by aldosterone and salt is attributable to podocyte injury [37]. Another disorder that exhibits increased aldosterone production is metabolic syndrome. Metabolic syndrome, in which visceral obesity is clustered with hypertension, impaired glucose tolerance, and dyslipidemia, has become increasingly frequent in Western societies. Obese hypertensive patients exhibit greater depressor response to salt restriction than lean hypertensives, indicating increased salt sensitivity [38]. Several factors may induce abnormal natriuresis in obesity-induced salt-sensitive hypertension, but excessive aldosterone concentrations in plasma have been reported to be common in obese patients [39].
Adipose tissue is now recognized as an endocrine organ that secretes a variety of adipokines. Components of the renin-angiotensin system are present in adipose tissues and are increased in obese patients. In addition, studies have demonstrated that adipocytes secrete as-yet-undefined factors that stimulate aldosterone synthesis in adrenal gland [40]. We have shown that serum aldosterone levels were increased in obese, spontaneously hypertensive rats (SHR), a rat model of metabolic syndrome, as compared with those in nonobese SHR [27]. Moreover, the aldosterone secretagogue activity of fat-cell–conditioned medium derived from obese SHR was greater than that from nonobese SHR. Metabolic syndrome is associated with the increased risk of albuminuria [41]. In obese SHR, there was an age-dependent increase in urinary protein excretion accompanied by podocyte injury, whereas no such findings were noted in nonobese SHR. Moreover, administration of an MR blocker markedly decreased the podocyte damage and proteinuria. Thus, increased aldosterone excretion can also contribute to the renal injury in metabolic syndrome.
Factors That Affect Aldosterone/MR Signaling and Its Relevance to Salt-Sensitive Hypertension
Without doubt, aldosterone is the central regulator of MR-mediated gene transcription, but it is not the sole determinant of MR signaling activity. Indeed, recent studies have led to a paradigm shift in our understanding of the regulation of the aldosterone/MR cascade. MR belongs to a superfamily of nuclear receptors, and the mechanisms that regulate other nuclear receptors can largely be applied to MR. Nuclear receptor signaling is modulated by several factors, including the receptor expression levels (through either altered transcription or stability), ligands, post-translational modifications, interaction with other signaling pathways, and recruitment of co-regulator molecules. Studies have indicated that these mechanisms are also involved in the regulation of MR activity [42, 43•].
The human MR gene (NR3C2) is located at chromosome 4q31 and consists of 10 exons. NR3C2 has two different promoters, termed P1 and P2, which generate alternative transcription products. In addition, a variant that lacks the hinge region and ligand binding domain of MR (hMRΔ5,6) is shown to be present in humans [44]. This variant possesses transcriptional activity in vitro, which is independent of the ligand. The presence of these human MR transcript variants may be involved in the context-dependent modulation of MR expression. In support of this idea, Krug et al. [43•] recently reported that MR expression is increased in aorta from aged rats, which causes enhanced MR signaling and age-associated cardiovascular damage. Aging is one of the important factors that modulate sodium excretion capacity, and the same mechanism may be applicable to age-related salt-sensitive hypertension.
Glucocorticoid can also act as a ligand for MR. Endogenous glucocorticoids—cortisol in humans and corticosterone in rodents—manifest the same affinity for MR as aldosterone in vitro, and the cortisol concentration is approximately 1,000 times higher than the concentration of aldosterone. In the kidneys, the ligand selectivity for MR is at least in part explained by the presence of 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2), which converts cortisol to inactive cortisone. Mutation or inactivation of this enzyme leads to glucocorticoid-induced MR activation and hypertension [5]. In contrast, MR is considered to be occupied by glucocorticoids in cardiomyocytes. Nonetheless, their agonistic activity is considered to be least potent in vivo, and the artificial overexpression of 11βHSD2 in the heart causes cardiac hypertrophy due to aldosterone-induced MR activation. Therefore, cortisol can prevent aldosterone binding and MR activation in physiologic conditions. However, it is postulated that the ability of cortisol to activate MR is influenced by the intracellular redox state, with increased oxidative stress resulting in cortisol-induced MR activation [45].
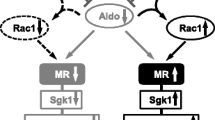
Small GTPase Rac1, a member of the Rho family GTPases, regulates diverse biologic processes, including cell migration, cell cycle, activation of NADPH oxidase, and cell adhesion [46]. In addition, recent evidence suggests that Rac1 serves as a key regulator of nuclear transcription factors [47]. Recently, the interaction between MR and Rac1 was shown to be an alternative pathway that potentiates MR activity, both in vitro and in vivo [10••]. In an in vitro study assessing nuclear trafficking using green fluorescent protein (GFP)–tagged MR, transfection of active Rac1 could induce nuclear translocation of MR-GFP even in the absence of aldosterone, and it further increased nuclear accumulation in the presence of aldosterone. In a luciferase reporter assay, overexpression of active Rac1 significantly potentiated aldosterone-induced MR transcriptional activity. This effect was not observed in other Rho GTPases, including RhoA and Cdc42. Rac1 induced MR activation even without aldosterone, but when aldosterone was added, the level of transcriptional activity was 20 times higher, suggesting that aldosterone increases Rac1-stimulated MR activation. In vitro experiments using cultured podocytes have had identical findings.
Rho GDP dissociation inhibitor (RhoGDI) is a negative regulator of Rho GTPases. In mice lacking the alpha subtype of RhoGDI (RhoGDIαKO), no morphologic abnormality of the kidneys was noted at E18.5, indicating that the nephrogenesis is normal. However, the knockout mice exhibited severe proteinuria and glomerulosclerosis that progressed with age [10••]. Electron microscopy revealed extensive foot process effacement, suggesting that severe proteinuria in this model could be attributable to podocyte damage. Rac1 activity was found to be upregulated exclusively in the kidneys, whereas RhoA activity was not. Consistent with this finding, renal injury in RhoGDIαKO mice was inhibited by the administration of a Rac-specific inhibitor, but Rho-kinase inhibitor had no protective effect. In this model, nuclear MR protein in the kidneys, as well as its downstream mediator Sgk1, was upregulated despite comparable serum aldosterone levels in RhoGDIαKO mice and their wild-type littermates. Moreover, inhibition of Rac1 activity antagonized the increased MR signaling, indicating an interaction between Rac1 and MR in vivo. In support of this idea, an MR blocker markedly reduced the nephrotic-range proteinuria and histologic change in the glomeruli of the RhoGDIαKO mice. In sum, we conclude that the crosstalk between Rac1 and MR activity is a novel mechanism that potentiates MR activity.
Aldosterone/MR signaling is closely related to dietary salt intake. Excessive salt intake can cause MR activation through an unidentified mechanism [48], leading to the unfavorable synergistic action of aldosterone and high salt intake. In relation to this unresolved mystery, we have obtained evidence suggesting that Rac1 plays a key role in salt-induced MR activation and hypertension. The Dahl rat is a well-established model of salt-sensitive hypertension and glomerular injury. In this model, the MR downstream mediator SGK1 is paradoxically elevated in response to high sodium loading [49] despite appropriate suppression of plasma aldosterone, an observation implicated in the pathogenesis of salt-dependent elevation of blood pressure. The role of salt-induced MR activation in this model is further supported by the protective effects of eplerenone [27]. In our experiments, we have found that renal Rac1 was also activated by a high-salt diet in Dahl salt-sensitive rats [10••], and that Rac1 inhibitor considerably attenuated hypertension and glomerular damage, along with the SGK1 repression. Thus, changes in Rac1 activity, as well as in plasma aldosterone concentrations, can modulate MR activation, and the Rac1 activity can be modulated by sodium status, causing inappropriate MR activity in a subpopulation of subjects with salt-sensitive hypertension. We consider that these two mechanisms can be interdependent. Our preliminary experiments revealed that an inhibitor of the Rho/Rho-kinase pathway had no depressor effects in our model, in contrast to the effect of Rac1 blockade. Of note, however, a recent study revealed that angiotensin II enhances the Rho-kinase pathway through the regulation of the Rho guanine exchange factor Arhgef1, which is responsible for angiotensin II-dependent, salt-independent hypertension through vasoconstriction [50]. Taking these observations together, it is interesting to speculate that vascular Rho/Rho-kinase plays a role in angiotensin II-dependent hypertension via vasoconstriction, whereas renal Rac1 is involved in salt-sensitive hypertension through MR-mediated sodium retention.
Conclusions
Accumulating studies reveal that aldosterone/MR plays a significant role in the development of salt-sensitive hypertension and associated end-organ damage. In addition, recent data indicate that besides systemic aldosterone, MR signaling activity is regulated locally and can be involved in the pathogenesis of salt-sensitive hypertension. For terrestrial tetrapods, the aldosterone/MR system is the principal regulatory mechanism maintaining water and electrolyte homeostasis necessary for the independent existence on land. The multiple regulatory system of aldosterone/MR axis may have served primarily to ensure efficient sodium and water homeostasis in a low-salt environment. Excessive salt intake in modern industrialized societies results in inappropriate suppression of the aldosterone/MR axis, causing hypertension, cardiovascular damage, and kidney dysfunction. Future studies will clarify the detailed mechanism for the interdependent regulation of salt homeostasis and the aldosterone/MR system, and its relevance in salt-sensitive hypertension.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Dahl LK, Heine M: Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res 1975, 36:692–696.
Guyton AC: Blood pressure control—special role of the kidneys and body fluids. Science 1991, 252:1813–1816.
Hall JE, Mizelle HL, Hildebrandt DA, Brands MW: Abnormal pressure natriuresis. A cause or a consequence of hypertension? Hypertension 1990, 15:547–559.
Kimura G, Saito F, Kojima S, et al.: Renal function curve in patients with secondary forms of hypertension. Hypertension 1987, 10:11–15.
Lifton RP, Gharavi AG, Geller DS: Molecular mechanisms of human hypertension. Cell 2001, 104:545–556.
Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. Intersalt Cooperative Research Group. BMJ 1988, 297:319–328.
Denton D, Weisinger R, Mundy NI, et al.: The effect of increased salt intake on blood pressure of chimpanzees. Nat Med 1995, 1:1009–1016.
Fujita T, Henry WL, Bartter FC, et al.: Factors influencing blood pressure in salt-sensitive patients with hypertension. Am J Med 1980, 69:334–344.
Morimoto A, Uzu T, Fujii T, et al.: Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet 1997, 350:1734–1737.
•• Shibata S, Nagase M, Yoshida S, et al.: Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med 2008, 14:1370–1376. This study identified the signaling interaction between Rac1 and MR as a mechanism to potentiate MR signaling both in vitro and in vivo, indicating that the aldosterone/MR axis can be regulated by other mechanisms in addition to the ligand.
Bhalla V, Soundararajan R, Pao AC, et al.: Disinhibitory pathways for control of sodium transport: regulation of ENaC by SGK1 and GILZ. Am J Physiol Renal Physiol 2006, 291:F714–721.
Shimkets RA, Warnock DG, Bositis CM, et al.: Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 1994, 79:407–414.
Diakov A, Korbmacher C: A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel’s alpha-subunit. J Biol Chem 2004, 279:38134–38142.
Zhang W, Xia X, Reisenauer MR, et al.: Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. J Clin Invest 2007, 117:773–783.
Ayroldi E, Zollo O, Macchiarulo A, et al.: Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol 2002, 22:7929–7941.
Soundararajan R, Melters D, Shih IC, et al.: Epithelial sodium channel regulated by differential composition of a signaling complex. Proc Natl Acad Sci U S A 2009, 106:7804–7809.
Wilson FH, Disse-Nicodeme S, Choate KA, et al.: Human hypertension caused by mutations in WNK kinases. Science 2001, 293:1107–1112.
Ring AM, Leng Q, Rinehart J, et al.: An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci U S A 2007, 104:4025–4029.
Rozansky DJ, Cornwall T, Subramanya AR, et al.: Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest 2009, 119:2601–2612.
Heise CJ, Xu BE, Deaton SL, et al.: Serum and glucocorticoid-induced kinase (SGK) 1 and the epithelial sodium channel are regulated by multiple with no lysine (WNK) family members. J Biol Chem 2010, 285:25161–25167.
Naray-Fejes-Toth A, Snyder PM, Fejes-Toth G: The kidney-specific WNK1 isoform is induced by aldosterone and stimulates epithelial sodium channel-mediated Na+ transport. Proc Natl Acad Sci U S A 2004, 101:17434–17439.
Newhouse SJ, Wallace C, Dobson R, et al.: Haplotypes of the WNK1 gene associate with blood pressure variation in a severely hypertensive population from the British Genetics of Hypertension study. Hum Mol Genet, 2005, 14:1805–1814.
Pitt B, Zannad F, Remme WJ, et al.: The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999, 341:709–717.
Pitt B, Remme W, Zannad F, et al.: Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003, 348:1309–1321.
Quinkler M, Zehnder D, Eardley KS, et al.: Increased expression of mineralocorticoid effector mechanisms in kidney biopsies of patients with heavy proteinuria. Circulation 2005, 112:1435–1443.
Williams GH, Burgess E, Kolloch RE, et al.: Efficacy of eplerenone versus enalapril as monotherapy in systemic hypertension. Am J Cardiol 2004, 93:990–996.
Nagase M, Shibata S, Yoshida S, et al.: Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension 2006, 47:1084–1093.
Lee SH, Yoo TH, Nam BY, et al.: Activation of local aldosterone system within podocytes is involved in apoptosis under diabetic conditions. Am J Physiol Renal Physiol 2009, 297:F1381–1390.
Lin S, Li D, Jia J, et al.: Spironolactone ameliorates podocytic adhesive capacity via restoring integrin alpha 3 expression in streptozotocin-induced diabetic rats. J Renin Angiotensin Aldosterone Syst 2010, 11:149–157.
Nagai Y, Miyata K, Sun GP, et al. Aldosterone stimulates collagen gene expression and synthesis via activation of ERK 1/2 in rat renal fibroblasts. Hypertension 2005, 46:1039–1045.
Leroy V, De Seigneux S, Agassiz V, et al.: Aldosterone activates NF-kappaB in the collecting duct. J Am Soc Nephrol 2009, 20:131–144.
Rossi GP, Bernini G, Desideri G, et al.: Renal damage in primary aldosteronism: results of the PAPY Study. Hypertension 2006, 48:232–238.
Lifton RP, Dluhy RG, Powers M, et al.: A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992, 355:262–265.
•• Doi M, Takahashi Y, Komatsu R, et al.: Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med 2010, 16:67–74. Using Cry knockout mice, the investigators provide evidence that aldosterone secretion is controlled by essential clock components, which can be relevant to the circadian control of blood pressure and the pathogenesis of salt-sensitive hypertension. The authors also indicate a role for HSD3B1 in the layer-specific biosynthesis of aldosterone.
Rossi GP, Bernini G, Caliumi C, et al.: A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 2006, 48:2293–2300.
Farquharson CA, Struthers AD: Aldosterone induces acute endothelial dysfunction in vivo in humans: evidence for an aldosterone-induced vasculopathy. Clin Sci (Lond) 2002, 103:425–431.
Shibata S, Nagase M, Yoshida S, et al.: Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension 2007, 49:355–364.
Chen J, Gu D, Huang J, et al.: Metabolic syndrome and salt sensitivity of blood pressure in non-diabetic people in China: a dietary intervention study. Lancet 2009, 373:829–835.
Bentley-Lewis R, Adler GK, Perlstein T, et al.: Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J Clin Endocrinol Metab 2007, 92:4472–4475.
Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A, et al.: Human adipocytes secrete mineralocorticoid-releasing factors. Proc Natl Acad Sci U S A 2003, 100:14211–14216.
Chen J, Muntner P, Hamm LL, et al.: The metabolic syndrome and chronic kidney disease in U.S. adults. Ann Intern Med 2004, 140:167–174.
Yokota K, Shibata H, Kurihara I, et al.: Coactivation of the N-terminal transactivation of mineralocorticoid receptor by Ubc9. J Biol Chem 2007, 282:1998–2010.
• Krug AW, Allenhofer L, Monticone R, et al.: Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension 2010, 55:1476–1483. The investigators showed that enhanced MR sensitivity caused by the increased expression in aorta is involved in age-associated cardiovascular damage.
Zennaro MC, Souque A, Viengchareun S, et al.: A new human MR splice variant is a ligand-independent transactivator modulating corticosteroid action. Mol Endocrinol 2001, 15:1586–1598.
Mihailidou AS, Loan Le TY, Mardini M, Funder JW: Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension 2009, 54:1306–1312.
Takai Y, Sasaki T, Matozaki T: Small GTP-binding proteins. Physiol Rev 2001, 81:153–208.
Wu X, Tu X, Joeng KS, et al.: Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell 2008, 133:340–353.
Nagase M, Matsui H, Shibata S, et al.: Salt-induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: role of oxidative stress. Hypertension 2007, 50:877–883.
Farjah M, Roxas BP, Geenen DL, Danziger RS: Dietary salt regulates renal SGK1 abundance: relevance to salt sensitivity in the Dahl rat. Hypertension 2003, 41:874–878.
Guilluy C, Bregeon J, Toumaniantz G, et al.: The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med 2010, 16:183–190.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Shibata, S., Fujita, T. The Kidneys and Aldosterone/Mineralocorticoid Receptor System in Salt-Sensitive Hypertension. Curr Hypertens Rep 13, 109–115 (2011). https://doi.org/10.1007/s11906-010-0175-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-010-0175-6