
Abstract
Purpose of Review
The balance between inflammation and its resolution plays an important and increasingly appreciated role in heart failure (HF) pathogenesis. In humans, different chronic inflammatory conditions and immune-inflammatory responses to infection can lead to diverse HF manifestations. Reviewing the phenotypic and mechanistic diversity of these HF presentations offers useful clinical and scientific insights.
Recent Findings
HF risk is increased in patients with chronic inflammatory and autoimmune disorders and relates to disease severity. Inflammatory condition–specific HF manifestations exist and underlying pathophysiologic causes may differ across conditions.
Summary
Although inflammatory disease–specific presentations of HF differ, chronic excess in inflammation and auto-inflammation relative to resolution of this inflammation is a common underlying contributor to HF. Further studies are needed to phenotypically refine inflammatory condition–specific HF pathophysiologies and prognoses, as well as potential targets for intervention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
An increasing appreciation of heart failure (HF) phenotypic heterogeneity over the past decade has expanded mechanistic insights related to HF pathophysiology and progression [1, 2]. Augmenting traditional understandings of HF hemodynamics and neurohormonal modulation [3], an increasing body of evidence highlights the role of inflammation in the pathogenesis and progression of various HF phenotypes [4, 5••]. In this context, reviewing diverse HF phenotypes in different chronic inflammatory and infectious conditions may offer insights into how distinct inflammatory pathways impact HF while also providing practical knowledge on inflammatory disease–associated HF for clinicians. We first review the state of the art regarding innate and adaptive immune responses in inflammation; we then discuss HF pathogenesis and presentation in various chronic inflammatory and infectious conditions; finally, we explore scientific gaps and potential therapeutic opportunities.
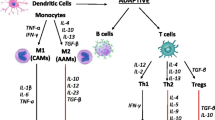
Innate and Adaptive Immune Responses: Focused Overview for the Cardiologist
First, we will briefly provide an overview of innate and adaptive immune responses for the cardiologist; this overview informs the clinically focused discussion later in this article.
Innate Immunity
The human immune system is generally divided into two major components: innate and adaptive immunity [6]. Innate immunity generally acts as the first line of defense and is largely nonspecific, mediated by neutrophils, macrophages, dendritic cells, natural killer cells, monocytes, eosinophils, basophils, and mast cells [7, 8]. Pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) released from damaged cells are recognized by pattern recognition receptors (PPRs), which in turn can also detect endogenous host cellular damage [9]. This triggers downstream signaling cascades that activate nuclear factor-kB, as well as interferon regulatory factor transcription factors, and activator protein 1, leading to transcription of pro-inflammatory cytokines [9]. Innate immune cells enacting these cellular programs demonstrate functional plasticity depending on local conditions and antigenic exposures. Indeed, whereas exposure to certain antigens and adverse metabolic conditions can lead to persistent inflammatory hyperactivation, in non-disease conditions, cells such as macrophages can actively resolve inflammation [10,11,12,13,14].
Trained Immunity: Innate Immune Memory
Recent evidence suggests that innate immune cells have the capacity to build memory [15], an emerging concept termed “trained immunity” or “innate immune memory” [16]. While likely intended to protect against re-infection or other secondary foreign agents, this memory can become maladaptive and harm the host. Indeed, exposure to certain stimuli, such as oxidized cholesterol particles, can lead to epigenetic reprogramming of innate immune cells [16, 17] and drive prolonged inflammatory hyperactivation [16, 18•, 19]. These changes are increasingly implicated in cardiovascular inflammation and, in concert with adaptive immune changes, may serve as important links between antigenic exposures (infectious and noninfectious) and atherosclerosis [18•].
Adaptive Immunity
Adaptive immune responses are relatively specific and mediated largely by B and T cells, which can act independently, in concert with innate immune cells [20], and occasionally with one another [21] to clear pathogens. T cells mature in the thymus, differentiate into memory or effector cells, and are essential to immune memory and homeostasis; T cell receptors (TCRs) recognize a diverse set of environmental and infectious antigens, presented by antigen-presenting cells such as dendritic cells, and can subsequently enact a number of inflammatory and/or inflammation-resolving programs [22,23,24]. This balance between inflammation and its resolution depends on antigen- and milieu-specific triggers, which drive T cell differentiation (e.g., with distinct helper or cytotoxic functions) and related signature cytokine production [22, 25, 26]. B cells, meanwhile, are derived from the bone marrow and, upon encountering antigen epitopes, mature into immunoglobin-secreting plasma cells or memory B cells [27, 28]. Although B cell memory is central to lasting immune protection against pathogens [29], failure of self-tolerance immune checkpoints can result in autoantibody production with related auto-immune conditions such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) [27].
Balance Between Inflammatory Hyperactivation and Resolution: Implications for Myocardial Disease and HF
Relevant to myocardial disease pathogenesis, unresolving inflammation can yield adverse ventricular remodeling, dysfunction, and ultimately HF [5••, 9, 30]. The time course of these varies depending on the underlying pathophysiology. A particularly acute and overt manifestation of immune-mediated HF is myocarditis, in which profound T cell activation plays a central role; in experimental infectious and inflammatory myocarditis, blunting T cell responses [31], particularly CD4+ T cells and their receptors (but perhaps not CD8+ T cells), ameliorates cardiac inflammation, damage, and dysfunction [32, 33]. Apart from acute infectious and inflammatory myocarditis, subtle differences in inflammation persistence vs. resolution impact cardiac function following local injury as occurs in myocardial infarction (MI). For instance, tyrosine kinase receptors expressed on macrophages (such as AXL and MerTK) increase pro-inflammatory responses and result in adverse ventricular remodeling and progression to HF, whereas their blockade ameliorates ischemia–reperfusion MI-induced HF [34]. Post-MI responses from the adaptive immune compartment are likewise essential to dictating the net-reparative vs. harmful impact on myocardial function and HF. T regulatory cells may provide a net-resolving effect related to wound healing after MI (perhaps via modulating inflammatory macrophage responses [35]), although the overall effect of T cells on post-MI inflammation vs. resolution remains under debate [36,37,38]. Meanwhile, B cells appear to be implicated in post-MI adverse remodeling and HF [39], but further study is needed on myocardial-specific effects of B cells as well as the pathophysiologic significance of myocardial autoantibodies [37, 40,41,42,43]. These experimental models, though largely focused on acute/subacute injury, have provided essential mechanistic insights into immune-mediated mechanisms of cardiac injury and dysfunction. Further mechanisms and related models of innate and adaptive immune responses implicated in HF pathogenesis have been discussed in detail elsewhere and are beyond the scope of this review [5••, 44]. With our brief foregoing review of immune response elements as a foundation, the remainder of this review focuses on clinically relevant presentations and underlying pathophysiologies of HF in distinct chronic infectious and inflammatory conditions; an overview of these diverse immunopathogenic mechanisms in the context of other common causes of cardiac injury is provided in Fig. 1.

Diversity of heart failure immunopathogenesis: infection, auto-inflammation, and impaired injury responses
Heart Failure in Chronic Inflammatory Conditions: Clinical Presentations and Mechanistic Correlates
Chronic inflammatory and autoimmune disorders are associated with a range of cardiovascular comorbidities including ASCVD, conduction abnormalities, valvular disease, and HF [45]. The increased risks for CVDs vary depending on the condition and stem from complex interactions between immunity-specific factors, accelerated atherosclerosis, persistent inflammation, microvascular disease, higher prevalence of CVD traditional risk factors, and off-target therapeutic effects [46,47,48]. Given diverse chronic inflammatory disease pathophysiologies, HF manifestations in these conditions are likewise non-uniform. Here, we present the current state of the art—based on admittedly limited existing data—on specific chronic inflammatory disease–associated HF risks and presentations.
Rheumatoid Arthritis
Patients with rheumatoid arthritis (RA) are at elevated risk for CVDs in general [49, 50•] and HF in specific, even after adjustment for cardiovascular risk factors and underlying ischemic heart disease [49, 50•, 51•]. Regarding HF phenotypes in RA, RA patients with HF were more likely to have preserved ejection fraction compared to non-RA patients [52, 53]. RA-associated HF risk may be particularly apparent among women; women with RA are 3- to fourfold more likely to have diastolic dysfunction compared with women without RA with similar demographic and clinical profiles [54]. Perhaps not surprisingly, worse disease control and related heightened autoimmune inflammation in RA associate with adverse cardiac remodeling [55] marked by excess fibrosis [56], impaired diastolic function, and HF [51•]. A single-center electronic records–based study suggested that chronic methotrexate treatment is associated with lower risk for HF among RA patients; however, further clarification is needed regarding effects of specific disease-modifying antirheumatic drugs (DMARDs) on HF pathogenesis and progression in RA [51•].
Systemic Lupus Erythematosus
Patients with systemic lupus erythematosus (SLE) are at heightened risk for a variety of CVDs, in particular atherosclerosis and thrombosis; they are likewise at heightened risk for pericarditis, endocarditis, valvular disease, and HF [57]. Prevalence of HF in patients with SLE is estimated to be between 1 and 10% [58] and HF incidence is 3–5 times higher in SLE patients compared to controls [59]. Limited data exist regarding HF phenotype in SLE; in a single-center study evaluating left ventricular ejection fraction (LVEF) trajectory over time in patients with chronic inflammatory diseases, SLE was more commonly associated with heart failure with preserved or midrange ejection fraction (HFpEF, HFmrEF) rather than with reduced ejection fraction (HFrEF) [60•]. A diversity of mechanisms are implicated in SLE-associated HF. These include premature athero-thrombosis and MI (with related myocardial damage), chronic myocardial inflammation (in absence of acute ischemic injury), and microvascular dysfunction/ischemia [58]. Further studies are needed to clarify specific mechanisms underlying SLE-associated HF [58, 59].
Systemic Sclerosis
Systemic sclerosis (SSc) is associated with significantly elevated risk for CVDs including pulmonary hypertension, valvular diseases, arrhythmias, and heart failure [61]. In a single-center study of patients with various chronic inflammatory diseases, patients with SSc had the highest incidence of HF of all chronic inflammatory diseases—and as much as 7- to eightfold higher than controls—even after adjustment for demographic and clinical covariates of relevance [62•]. The phenotypic presentation of HF associated with SSc also appears to be distinct from that of other chronic inflammatory diseases. Isolated right-sided heart failure is quite common in SSc [63], which is perhaps not surprising given the high prevalence of pulmonary arterial hypertension in SSc [64]. Even in SSc patients without overt HF, abnormal cardiac tissue structure and mechanics are common: approximately 1/3rd of SSc patients have late gadolinium enhancement on cardiac magnetic resonance imaging [65] and diastolic dysfunction is highly prevalent [66]. The pathophysiology of SSc-associated myocardial dysfunction and HF involves a deleterious combination of fibrosis and ischemia, with existing fibrosis exacerbating ischemia due to mechanical limitation of blood flow, resulting in further ischemia/injury and fibrosis; epicardial vasospastic phenomena (referred to as “myocardial Raynaud’s phenomenon”) are also implicated [62•, 67,68,69]. Prognosis of SSc-associated HF is poor, and approximately 25% of deaths in patients with SSc are due to cardiac causes, mainly heart failure and arrhythmias [70].
Psoriatic Disease
Psoriatic disease (PsD), which includes psoriasis and psoriatic arthritis, is also associated with increased risk of CVDs, with a considerable literature demonstrating PsD-associated vascular inflammation implicated in atherosclerotic CVDs [71,72,73]. Furthermore, HF incidence appears to be higher among PsD patients than controls [74, 75], although findings have been non-uniform across cohorts [76] and may depend on PsD severity [77], with worse PsD severity conferring higher HF risk.
Despite limited mechanistic data related to PsD-specific HF pathogenesis, there are several potential mechanisms that may be implicated in PsD-associated HF. Persistent uncontrolled inflammation appears to be central: tumor necrosis factor-alpha (TNF-α), profoundly elevated and HF associated with worse prognosis [78], is a key pro-inflammatory mediator in PsD [79]. Moreover, psoriasis is associated with endothelial dysfunction and arterial stiffness, consequently leading to HF by decreasing coronary flow reserve [80, 81]. Patients with PsD have been observed to have coronary microvascular dysfunction (CMVD) due to the systemic inflammation, with one study reporting the prevalence of CMVD to be 61.3% in patients with PsD compared to 38.4% in matched controls [82]. Another study linked PsD to cases of dilated cardiomyopathy (DCM) [83], and hypothesized that an autoimmune process may play a role in development of DCM, as well as cytokine-induced collagen impairment leading to myocardial fibrosis and dysfunction [84].
Inflammatory Bowel Diseases
Findings related to inflammatory bowel disease (IBD) and HF have been inconsistent. One retrospective longitudinal cohort study found that patients with inflammatory bowel disease (IBD) have a two-fold increase in incidence of HF (with ulcerative colitis patients being at higher risk than patients with Crohn’s disease) [85], and another observed an approximately 1.4-fold IBD-associated relative increase in HF hospitalization risk [86]. However, a separate study observed no significant IBD-associated HF risk [77]. Among patients with IBD, disease severity appears to associate with HF risk [86] and women with IBD may have an especially elevated relative risk for HF [85]. Altered collagen metabolism is thought to play a role in pathophysiology of HF in patients with IBD [87]; patients also may develop myocardial fibrosis due to vitamin deficiencies, microvascular endothelial dysfunction, and altered nitric oxide–mediated vasodilation [88, 89].
Infectious Causes of Heart Failure: Role of Inflammation
Innate and adaptive immune responses to foreign pathogens, while essential for pathogen clearance and acute infection resolution, can lead to self-tissue damage in the setting of excess and mistargeted inflammation. In this context, we will discuss distinct infectious triggers of deleterious cardiac inflammation resulting in HF, which highlight both the diversity of infectious causes of HF and the central role of immune responses in each of these conditions.
Acute Infectious-Inflammatory Myocarditis
The clinical manifestation of myocarditis is broad, ranging from mild and virtually asymptomatic to severe, fulminant heart failure resulting in cardiogenic shock. An estimated 1–5% of patients with acute viral infections may develop a form of myocarditis, ranging from asymptomatic myocardial inflammation to overt HF; young men have the highest risk [90, 91]. The most common pathogens implicated in acute myocarditis are viruses, including enteroviruses such as coxsackieviruses, parvovirus B19, Epstein-Barr virus, and human herpesvirus-6 [92], and more recently, SARS-CoV-2 [93,94,95]. Myocarditis can also be caused by non-viral infectious agents including bacteria, Chlamydia, Rickettsia, fungi, and protozoa [96].
Although pathogens may be inciting factors in myocarditis, experimental models suggest that host immune responses to these pathogens—rather than the pathogens themselves—are perhaps the most important determinant of myocarditis pathophysiology and presentation. For instance, in a model of coxsackievirus-induced myocarditis [97, 98•], virus-infected cardiomyocytes die, resulting in damage-associated molecular patterns and related innate immune activation, and after being phagocytosed, have their antigen epitopes presented to T cells and B cells, which perform key effector functions in cardiac injury and development of myocarditis [98•]. This robust immune activation may drive antigenic self-targeting; for instance, studies of cytomegalovirus (CMV) [99] and coxsackievirus [100] models of myocarditis have demonstrated shared epitopes between viral particles and cardiac myosin, resulting in persistent myocardial auto-inflammation and myocarditis. These considerations highlight the overlap between infectious-inflammatory and autoimmune myocarditis, as infections have the potential to generate autoimmune responses. This is perhaps best highlighted by rheumatic heart disease, discussed next.
Rheumatic Heart Disease
Rheumatic heart disease (RHD) is a relatively common (0.55 to 11 per thousand individuals worldwide [101]) pan-carditis with prominent valvular involvement, resulting in heart failure in approximately one-third of affected individuals and worse outcomes in women [102, 103]. Molecular mimicry and autoantibodies are hallmarks of rheumatic carditis. Heart-reactive antibodies have been isolated from the serum of patients with RHD for decades, and RHD was recognized as an autoimmune sequalae of group A streptococcal infection [104]. Initially, molecular mimicry was defined as shared identical amino acid sequences between host tissue and the bacterium [105]. However, studies using monoclonal antibodies have helped identify other types of molecular mimicry, such as antibody recognition of alpha-helical coiled-coil molecules, a protein structure shared among different host proteins (cardiac myosin, tropomyosin, vimentin, keratin, and laminin) and streptococcal M protein. A third type of molecular mimicry is cross reactivity between diverse molecules such as DNA, proteins, and carbohydrates [105]. In RHD, molecular mimicry between group A streptococcal protein and cardiac proteins leads to both humoral and cellular autoimmune reactions. Antibodies recognize internal biomarkers which cross-react with the streptococcal antigen. These autoantibodies can then cause inflammation of the endothelium (in particular valvular endothelium), driving vulnerability to autoreactive T cell infiltration. Resulting inflammation causes scarring and damage of the valve, which can worsen with recurrent infection and make penicillin prophylaxis important to prevent worsening [101].
Chronic Chagas Cardiomyopathy
Chagas disease is a sequelae of infection with the intracellular parasite Trypanosoma cruzi [106] that affects 6 to 8 million people worldwide [107] and wide range of presentations. Approximately one-quarter of infected patients progress to chronic Chagas cardiomyopathy (CCC), with one-quarter of these presenting with reduced left ventricular ejection fraction (LVEF) [108]. Parasitic persistence appears to be required for CCC development; a combination of direct parasitic damage and inflammatory T cell responses have been implicated, although the extent to which either is the primary driver of disease remains up for debate [109].
Human Immunodeficiency Virus
The epidemiology, pathogenesis, and presentation of human immunodeficiency virus (HIV)–associated heart failure have evolved considerably over the past two decades. Our group and others have discussed the evolving nature and immunopathogenesis of HIV-associated HF previously [110,111,112,113,114]; an in-depth discussion is beyond the scope of this article. In the modern antiretroviral therapy (ART) era, proportionate CVD mortality [115] and the global CVD burden [116] among people with HIV have increased considerably. People with HIV have significantly higher risks for heart failure, including HFpEF and HFrEF, than HIV-uninfected controls even after adjustment for demographic and clinical confounders [117•, 118•, 119]. Poor viral control and immune progression (marked by circulating viremia and lower CD4 + T cell counts) are associated with particularly elevated HF risk for people with HIV, but individuals on ART without significant immune progression (e.g., CD4 decline) continue to have elevated HF risks [117•, 118•, 119]. In the modern ART era, the putative pathophysiologies of HIV-associated HF are diverse and somewhat less overt/direct than other viral infections; these include impaired myocardial responses to ischemia [120], metabolic dysregulation related to both chronic infection/inflammation and certain antiretrovirals, autonomic dysregulation, and convergence of other factors such as drug toxicity (in particular, methamphetamines and cocaine) and smoking [121]. In reality, multiple co-existing pathophysiologic “hits” are likely involved in HIV-associated HF [122], with underlying immune dysregulation and pro-inflammatory bias [123•] driving maladaptive, tissue-damaging responses to subsequent challenges ranging from hypertension to co-infection. Importantly, HIV-associated HF risks are especially pronounced in women, who have higher rates of HF hospitalizations and CVD mortality [124,125,126].
Hepatitis C Virus
Several studies have described associations between chronic hepatitis C virus (HCV) infection and HF [127, 128], yet limited pathophysiologic knowledge exists regarding this association. Perhaps the most likely mechanism by which HCV contributes to CVD and HF relates to chronic inflammation and immune dysregulation, similar to chronic HIV infection [129]. Patients with HCV have been observed to have higher levels of pro-inflammatory cytokines, including TNF-α and IL-6 [130,131,132,133]. HCV infection is also associated with intestinal bacterial overgrowth, which can lead to bacterial translocation into the bloodstream, increasing LPS levels, triggering toll-like receptor (TLR) activation, and resulting in systemic inflammation [134, 135]. Importantly, effective treatment of HCV appears to decrease CVD risks [129, 136,137,138].
Leveraging Insights from Chronic Inflammatory and Infectious Conditions: Immune Targeting in Heart Failure
Fortunately, for most chronic inflammatory and infectious causes of myocardial dysfunction and HF, treatment of the underlying inflammatory/infectious condition decreases associated HF risks to some extent. Yet, HF risks remain elevated in many of these conditions despite effective therapy; accordingly, exploring existing and potential therapies offers the potential to yield disease-specific and broader insights into immune-targeted HF therapy.
In chronic inflammatory conditions, effects of immunomodulatory medications on HF risk appear to depend on the medication, its target(s), and the underlying population in which medications are used. Although corticosteroids reduce systemic inflammation, they can induce dyslipidemia, insulin resistance, and obesity, known CVD risk factors, and increase risk of CVDs including HF [139]. Immunosuppressants, such as mycophenolate mofetil, cyclophosphamide, and azathioprine, have shown inconsistent associations with ASCVD risk, without as clearly defined effects on HF [129, 140,141,142,143]. Perhaps the clearest demonstration of the barriers between inflammatory disease–specific efficacy and broader application is with TNF-α antagonists: whereas observational studies of RA patients suggested a benefit of TNF-α antagonists in reducing rates of HF hospitalization [144], this HF-reducing benefit was not replicated when TNF-α antagonists were tested in the general HF population without RA [145, 146]. Perhaps more promising are the results of a sub-analysis of the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS), in which treatment of post-MI patients with canakinumab, an IL-1β antagonist, led to a dose-dependent reduction in HF hospitalization and a composite of HF hospitalization and mortality [147••]. A separate, smaller trial of IL-1 receptor antagonist anakinra in post-MI patients likewise yielded a reduction in incident HF [148]. Effects of colchicine on HF are less clear, as randomized trials have focused largely on post-MI recurrence of athero-thrombotic CVD rather than HF and variably suggested benefit [149] or neutrality/possible harm [150]. The reason behind the heterogeneous response to immunomodulatory agents in different HF populations is likely related to the unique mechanism underlying HF development in various diseases. Once we better define the pathophysiology of HF in these diseases, we will be able to find better targets for intervention specific to each population.
Several novel targets for immunomodulation in HF treatment are under exploration. One such proof-of-concept example is the use of transient antifibrotic chimeric antigen receptor (CAR) T cells to reduce myocardial fibrosis in mouse models of HF [151••]. Other novel approaches include engineered biomaterials used to induce macrophage-mediated inflammation resolution in cardiac injury models [152, 153], as well as engineered cardioprotective T regulatory cell administration or induction [154, 155]. While these immune-targeted therapies remain largely in the proof-of-concept stage, considerable interest in further development of immunomodulatory therapies for HF exists with the goal of fundamentally altering problematic myocardial tissue substrates in HF.
Conclusion
Several chronic inflammatory and infectious conditions confer increased risks for HF. The pathophysiologies and clinical presentations of these condition-specific HF etiologies differ; however, an imbalance between persistent inflammation and resolution thereof is centrally implicated in myocardial tissue damage, dysfunction, and resultant HF. Given limited clinical and mechanistic data, further studies are needed to define condition-specific HF pathophysiologies. These studies offer the dual potential of impacting the immediate, real-world care of patients with HF-associated inflammatory and infectious conditions, as well as informing novel developmental immune-targeted therapies in HF.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Shah SJ, Katz DH, Selvaraj S, et al. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation. 2015;131(3):269–79. https://doi.org/10.1161/circulationaha.114.010637.
Ahmad T, Pencina MJ, Schulte PJ, et al. Clinical implications of chronic heart failure phenotypes defined by cluster analysis. J Am Coll Cardiol. 2014;64(17):1765–74. https://doi.org/10.1016/j.jacc.2014.07.979.
Dick SA, Epelman S. Chronic heart failure and inflammation: what do we really know? Circ Res. 2016;119(1):159–76. https://doi.org/10.1161/CIRCRESAHA.116.308030.
Van Linthout S, Tschöpe C. Inflammation - cause or consequence of heart failure or both? Curr Heart Fail Rep. 2017;14(4):251–65. https://doi.org/10.1007/s11897-017-0337-9.
•• Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. 2020;17(5):269–285. https://doi.org/10.1038/s41569-019-0315-xIn-depth review on the role of the immune system and inflammation in heart failure, with a focus on immune cell subsets. Emerging therapies to target inflammation in heart failure are also discussed.
Medzhitov R, Janeway C Jr. Innate immunity. N Engl J Med. 2000;343(5):338–44. https://doi.org/10.1056/nejm200008033430506.
Passos LSA, Lupieri A, Becker-Greene D, Aikawa E. Innate and adaptive immunity in cardiovascular calcification. Atherosclerosis. 2020;306:59–67. https://doi.org/10.1016/j.atherosclerosis.2020.02.016.
Chávez-Sánchez L, Espinosa-Luna JE, Chávez-Rueda K, Legorreta-Haquet MV, Montoya-Díaz E, Blanco-Favela F. Innate immune system cells in atherosclerosis. Arch Med Res. 2014;45(1):1–14. https://doi.org/10.1016/j.arcmed.2013.11.007.
Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015;116(7):1254–68. https://doi.org/10.1161/CIRCRESAHA.116.302317.
Baratin M, Simon L, Jorquera A, et al. T cell zone resident macrophages silently dispose of apoptotic cells in the lymph node. Immunity. 2017;47(2):349–362 e5. https://doi.org/10.1016/j.immuni.2017.07.019
Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. 2020;20(4):254–67. https://doi.org/10.1038/s41577-019-0240-6.
Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14(3):166–80. https://doi.org/10.1038/nri3607.
Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe-/- mice. Arterioscler Thromb Vasc Biol. 2008;28(8):1421–8. https://doi.org/10.1161/ATVBAHA.108.167197.
Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. 2009;86(5):1089–95. https://doi.org/10.1189/jlb.0209115.
Divangahi M, Aaby P, Khader SA, et al. Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat Immunol. 2021;22(1):2–6. https://doi.org/10.1038/s41590-020-00845-6.
Netea MG, Joosten LA, Latz E, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352(6284):aaf1098. https://doi.org/10.1126/science.aaf1098
Zhong C, Yang X, Feng Y, Yu J. Trained immunity: an underlying driver of inflammatory atherosclerosis. Front Immunol. 2020;11:284. https://doi.org/10.3389/fimmu.2020.00284.
• Leentjens J, Bekkering S, Joosten LAB, Netea MG, Burgner DP, Riksen NP. Trained innate immunity as a novel mechanism linking infection and the development of atherosclerosis. Circ Res. 2018;122(5):664–669. https://doi.org/10.1161/CIRCRESAHA.117.312465Important experimental study which proposes that trained immunity, an emerging new concept, mediates the link between infections and atherosclerotic cardiovascular disease.
Flores-Gomez D, Bekkering S, Netea MG, Riksen NP. Trained immunity in atherosclerotic cardiovascular disease. Arterioscler Thromb Vasc Biol. 2021;41(1):62–9. https://doi.org/10.1161/ATVBAHA.120.314216.
Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7(1):9–18. https://doi.org/10.1038/nri1994.
Petersone L, Edner NM, Ovcinnikovs V, et al. T cell/B cell collaboration and autoimmunity: an intimate relationship. Front Immunol. 2018;9:1941. https://doi.org/10.3389/fimmu.2018.01941.
Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. 2018;48(2):202–13. https://doi.org/10.1016/j.immuni.2018.01.007.
Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. 2008;123(3):326–38. https://doi.org/10.1111/j.1365-2567.2007.02719.x.
Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445–89. https://doi.org/10.1146/annurev-immunol-030409-101212.
Perez-Hernandez J, Chiurchiu V, Perruche S, You S. Regulation of T-cell immune responses by pro-resolving lipid mediators. Front Immunol. 2021;12: 768133. https://doi.org/10.3389/fimmu.2021.768133.
Gascoigne NR. Do T cells need endogenous peptides for activation? Nat Rev Immunol. 2008;8(11):895–900. https://doi.org/10.1038/nri2431.
Cyster JG, Allen CDC. B cell responses: cell interaction dynamics and decisions. Cell. 2019;177(3):524–40. https://doi.org/10.1016/j.cell.2019.03.016.
LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570–80. https://doi.org/10.1182/blood-2008-02-078071.
Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol. 2020;20(4):229–38. https://doi.org/10.1038/s41577-019-0244-2.
Mann DL, Topkara VK, Evans S, Barger PM. Innate immunity in the adult mammalian heart: for whom the cell tolls. Trans Am Clin Climatol Assoc. 2010;121:34–50; discussion 50–1.
Liu P, Aitken K, Kong YY, et al. The tyrosine kinase p56lck is essential in coxsackievirus B3-mediated heart disease. Nat Med. 2000;6(4):429–34. https://doi.org/10.1038/74689.
Opavsky MA, Penninger J, Aitken K, et al. Susceptibility to myocarditis is dependent on the response of alphabeta T lymphocytes to coxsackieviral infection. Circ Res. 1999;85(6):551–8. https://doi.org/10.1161/01.res.85.6.551.
Klingel K, Schnorr JJ, Sauter M, Szalay G, Kandolf R. Beta2-microglobulin-associated regulation of interferon-gamma and virus-specific immunoglobulin G confer resistance against the development of chronic coxsackievirus myocarditis. Am J Pathol. 2003;162(5):1709–20. https://doi.org/10.1016/s0002-9440(10)64305-2.
DeBerge M, Glinton K, Subramanian M, et al. Macrophage AXL receptor tyrosine kinase inflames the heart after reperfused myocardial infarction. J Clin Invest. 2021;131(6) https://doi.org/10.1172/JCI139576
Saxena A, Dobaczewski M, Rai V, et al. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol. 2014;307(8):H1233–42. https://doi.org/10.1152/ajpheart.00328.2014.
Hofmann U, Beyersdorf N, Weirather J, et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125(13):1652–63. https://doi.org/10.1161/CIRCULATIONAHA.111.044164.
Hofmann U, Frantz S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res. 2015;116(2):354–67. https://doi.org/10.1161/CIRCRESAHA.116.304072.
Ramos GC, van den Berg A, Nunes-Silva V, et al. Myocardial aging as a T-cell-mediated phenomenon. Proc Natl Acad Sci U S A. 2017;114(12):E2420–9. https://doi.org/10.1073/pnas.1621047114.
Zouggari Y, Ait-Oufella H, Bonnin P, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19(10):1273–80. https://doi.org/10.1038/nm.3284.
Kaya Z, Leib C, Katus HA. Autoantibodies in heart failure and cardiac dysfunction. Circ Res. 2012;110(1):145–58. https://doi.org/10.1161/CIRCRESAHA.111.243360.
Lazzerini PE, Capecchi PL, Laghi-Pasini F, Boutjdir M. Autoimmune channelopathies as a novel mechanism in cardiac arrhythmias. Nat Rev Cardiol. 2017;14(9):521–35. https://doi.org/10.1038/nrcardio.2017.61.
Neumann DA, Burek CL, Baughman KL, Rose NR, Herskowitz A. Circulating heart-reactive antibodies in patients with myocarditis or cardiomyopathy. J Am Coll Cardiol. 1990;16(6):839–46. https://doi.org/10.1016/s0735-1097(10)80331-6.
Konstadoulakis MM, Kroumbouzou H, Tsiamis E, Trikas A, Toutouzas P. Clinical significance of antibodies against tropomyosin, actin and myosin in patients with dilated cardiomyopathy. J Clin Lab Immunol. 1993;40(2):61–7.
DeBerge M, Shah SJ, Wilsbacher L, Thorp EB. Macrophages in heart failure with reduced versus preserved ejection fraction. Trends Mol Med. 2019;25(4):328–40. https://doi.org/10.1016/j.molmed.2019.01.002.
Prasad M, Hermann J, Gabriel SE, et al. Cardiorheumatology: cardiac involvement in systemic rheumatic disease. Nat Rev Cardiol. 2015;12(3):168–76. https://doi.org/10.1038/nrcardio.2014.206.
Agca R, Smulders Y, Nurmohamed M. Cardiovascular disease risk in immune-mediated inflammatory diseases: recommendations for clinical practice. Heart. 2022;108(1):73–9. https://doi.org/10.1136/heartjnl-2019-316378.
Mason JC, Libby P. Cardiovascular disease in patients with chronic inflammation: mechanisms underlying premature cardiovascular events in rheumatologic conditions. Eur Heart J. 2015;36(8):482–9. https://doi.org/10.1093/eurheartj/ehu403.
Weber BN, Stevens E, Perez-Chada LM, et al. Impaired coronary vasodilator reserve and adverse prognosis in patients with systemic inflammatory disorders. JACC Cardiovasc Imaging. 2021;14(11):2212–20. https://doi.org/10.1016/j.jcmg.2020.12.031.
Nicola PJ, Maradit-Kremers H, Roger VL, et al. The risk of congestive heart failure in rheumatoid arthritis: a population-based study over 46 years. Arthritis Rheum. 2005;52(2):412–20. https://doi.org/10.1002/art.20855.
• Khalid U, Egeberg A, Ahlehoff O, et al. Incident heart failure in patients with rheumatoid arthritis: a nationwide cohort study. J Am Heart Assoc. 2018;7(2) https://doi.org/10.1161/jaha.117.007227This nationwide study found an association between rheumatoid arthritis and incident heart failure.
• Ahlers MJ, Lowery BD, Farber-Eger E, et al. Heart failure risk associated with rheumatoid arthritis-related chronic inflammation. J Am Heart Assoc. 2020;9(10):e014661. https://doi.org/10.1161/JAHA.119.014661This retrospective study explored the relationship between rheumatoid arthritis and risk of heart failure. It also identified a subgroup of rheumatoid arthritis patients that were at higher and lower risk of developing heart failure.
Park E, Griffin J, Bathon JM. Myocardial dysfunction and heart failure in rheumatoid arthritis. Arthritis Rheumatol. 2022;74(2):184–99. https://doi.org/10.1002/art.41979.
Davis JM 3rd, Roger VL, Crowson CS, Kremers HM, Therneau TM, Gabriel SE. The presentation and outcome of heart failure in patients with rheumatoid arthritis differs from that in the general population. Arthritis Rheum. 2008;58(9):2603–11. https://doi.org/10.1002/art.23798.
Kim GH, Park YJ. Accelerated diastolic dysfunction in premenopausal women with rheumatoid arthritis. Arthritis Res Ther. 2021;23(1):247. https://doi.org/10.1186/s13075-021-02629-1.
Mavrogeni S, Karabela G, Stavropoulos E, et al. Imaging patterns of heart failure in rheumatoid arthritis evaluated by cardiovascular magnetic resonance. Int J Cardiol. 2013;168(4):4333–5. https://doi.org/10.1016/j.ijcard.2013.05.085.
Błyszczuk P, Szekanecz Z. Pathogenesis of ischaemic and non-ischaemic heart diseases in rheumatoid arthritis. RMD Open. 2020;6(1) https://doi.org/10.1136/rmdopen-2019-001032
Fairweather D, Petri MA, Coronado MJ, Cooper LT. Autoimmune heart disease: role of sex hormones and autoantibodies in disease pathogenesis. Expert Rev Clin Immunol. 2012;8(3):269–84. https://doi.org/10.1586/eci.12.10.
Dhakal BP, Kim CH, Al-Kindi SG, Oliveira GH. Heart failure in systemic lupus erythematosus. Trends Cardiovasc Med. 2018;28(3):187–97. https://doi.org/10.1016/j.tcm.2017.08.015.
Kim CH, Al-Kindi SG, Jandali B, Askari AD, Zacharias M, Oliveira GH. Incidence and risk of heart failure in systemic lupus erythematosus. Heart. 2017;103(3):227–33. https://doi.org/10.1136/heartjnl-2016-309561.
• Rivera AS, Sinha A, Ahmad FS, et al. Long-term trajectories of left ventricular ejection fraction in patients with chronic inflammatory diseases and heart failure: an analysis of electronic health records. Circ Heart Failure. 2021;14(8):e008478. https://doi.org/10.1161/circheartfailure.121.008478This study investigated trajectories in cardiac mechanical function over time in patients with different chronic inflammatory diseases, highlighting the heterogeneity of heart failure subtypes in this patient population.
Butt SA, Jeppesen JL, Torp-Pedersen C, et al. Cardiovascular manifestations of systemic sclerosis: a Danish nationwide cohort study. J Am Heart Assoc. 2019;8(17): e013405. https://doi.org/10.1161/jaha.119.013405.
• Prasada S, Rivera A, Nishtala A, et al. Differential associations of chronic inflammatory diseases with incident heart failure. JACC Heart Fail. 2020;8(6):489–498. https://doi.org/10.1016/j.jchf.2019.11.013This study explored the risk of incident heart failure across several chronic inflammatory and infectious diseases, providing estimates of the relative extent of elevated HF risk depending on specific conditions of interest (ranging from systemic sclerosis to rheumatoid arthritis to HIV).
Underberg DL, Rivera AS, Sinha A, Feinstein MJ. Phenotypic presentations of heart failure among patients with chronic inflammatory diseases. Front Cardiovasc Med. 2022;9: 784601. https://doi.org/10.3389/fcvm.2022.784601.
Caetano J, Batista F, Amaral MC, Oliveira S, Alves JD. Acute hospitalization in a cohort of patients with systemic sclerosis: a 10-year retrospective cohort study. Rheumatol Int. 2021:1–10. https://doi.org/10.1007/s00296-021-04983-4
Lee DC, Hinchcliff ME, Sarnari R, et al. Diffuse cardiac fibrosis quantification in early systemic sclerosis by magnetic resonance imaging and correlation with skin fibrosis. J Scleroderma Relat Disord. 2018;3(2):159–69. https://doi.org/10.1177/2397198318762888.
de Groote P, Gressin V, Hachulla E, et al. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis. 2008;67(1):31–6. https://doi.org/10.1136/ard.2006.057760.
Stronati G, Manfredi L, Ferrarini A, et al. Subclinical progression of systemic sclerosis-related cardiomyopathy. Eur J Prev Cardiol. 2020;27(17):1876–86. https://doi.org/10.1177/2047487320916591.
Kurmann RD, Sandhu AS, Crowson CS, et al. Cardiovascular risk factors and atherosclerotic cardiovascular events among incident cases of systemic sclerosis: results from a population-based cohort (1980–2016). Mayo Clin Proc. 2020;95(7):1369–78. https://doi.org/10.1016/j.mayocp.2019.12.015.
Lambova S. Cardiac manifestations in systemic sclerosis. World J Cardiol. 2014;6(9):993–1005. https://doi.org/10.4330/wjc.v6.i9.993.
Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809–15. https://doi.org/10.1136/ard.2009.114264.
Mehta NN, Yu Y, Saboury B, et al. Systemic and vascular inflammation in patients with moderate to severe psoriasis as measured by [18F]-fluorodeoxyglucose positron emission tomography-computed tomography (FDG-PET/CT): a pilot study. Arch Dermatol. 2011;147(9):1031–9. https://doi.org/10.1001/archdermatol.2011.119.
Naik HB, Natarajan B, Stansky E, et al. Severity of psoriasis associates with aortic vascular inflammation detected by FDG PET/CT and neutrophil activation in a prospective observational study. Arterioscler Thromb Vasc Biol. 2015;35(12):2667–76. https://doi.org/10.1161/ATVBAHA.115.306460.
Schwartz DM, Burma AM, Kitakule MM, Luo Y, Mehta NN. T cells in autoimmunity-associated cardiovascular diseases. Front Immunol. 2020;11: 588776. https://doi.org/10.3389/fimmu.2020.588776.
Khalid U, Ahlehoff O, Gislason GH, et al. Psoriasis and risk of heart failure: a nationwide cohort study. Eur J Heart Fail. 2014;16(7):743–8. https://doi.org/10.1002/ejhf.113.
Koppikar S, Colaco K, Harvey P, et al. Incidence of and risk factors for heart failure in patients with psoriatic disease - a cohort study. Arthritis Care Res (Hoboken). 2021. https://doi.org/10.1002/acr.24578.
Polachek A, Touma Z, Anderson M, Eder L. Risk of cardiovascular morbidity in patients with psoriatic arthritis: a meta-analysis of observational studies. Arthritis Care Res (Hoboken). 2017;69(1):67–74. https://doi.org/10.1002/acr.22926.
Prasada S, Rivera A, Nishtala A, et al. Differential associations of chronic inflammatory diseases with incident heart failure. JACC Heart failure. 2020;8(6):489–98. https://doi.org/10.1016/j.jchf.2019.11.013.
Blum A, Miller H. Pathophysiological role of cytokines in congestive heart failure. Annu Rev Med. 2001;52:15–27. https://doi.org/10.1146/annurev.med.52.1.15.
Dowlatshahi EA, van der Voort EA, Arends LR, Nijsten T. Markers of systemic inflammation in psoriasis: a systematic review and meta-analysis. Br J Dermatol. 2013;169(2):266–82. https://doi.org/10.1111/bjd.12355.
Brezinski EA, Follansbee MR, Armstrong EJ, Armstrong AW. Endothelial dysfunction and the effects of TNF inhibitors on the endothelium in psoriasis and psoriatic arthritis: a systematic review. Curr Pharm Des. 2014;20(4):513–28. https://doi.org/10.2174/138161282004140213123852.
Gisondi P, Fantin F, Del Giglio M, et al. Chronic plaque psoriasis is associated with increased arterial stiffness. Dermatology. 2009;218(2):110–3. https://doi.org/10.1159/000182256.
Weber B, Perez-Chada LM, Divakaran S, et al. Coronary microvascular dysfunction in patients with psoriasis. J Nucl Cardiol. 2022;29(1):37–42. https://doi.org/10.1007/s12350-020-02166-5.
Alfraji N, Douedi S, Alshami A, Kuzyshyn H, Tang X. Nonischemic dilated cardiomyopathy in untreated long-term psoriatic arthritis: a newly recognized association: a case report with mini review. Am J Case Rep. 2021;22: e930041. https://doi.org/10.12659/ajcr.930041.
Hashim T, Ahmad A, Chaudry A, Khouzam R. Psoriasis and cardiomyopathy: a review of the literature. South Med J. 2017;110(2):97–100. https://doi.org/10.14423/smj.0000000000000603.
Aniwan S, Pardi DS, Tremaine WJ, Loftus EV Jr. Increased risk of acute myocardial infarction and heart failure in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2018;16(10):1607-1615.e1. https://doi.org/10.1016/j.cgh.2018.04.031.
Kristensen SL, Ahlehoff O, Lindhardsen J, et al. Inflammatory bowel disease is associated with an increased risk of hospitalization for heart failure: a Danish Nationwide Cohort study. Circ Heart Fail. 2014;7(5):717–22. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001152.
De Simone M, Cioffi U, Contessini-Avesani E, et al. Elevated serum procollagen type III peptide in splanchnic and peripheral circulation of patients with inflammatory bowel disease submitted to surgery. BMC Gastroenterol. 2004;4:29. https://doi.org/10.1186/1471-230X-4-29.
Hatoum OA, Binion DG, Otterson MF, Gutterman DD. Acquired microvascular dysfunction in inflammatory bowel disease: loss of nitric oxide-mediated vasodilation. Gastroenterology. 2003;125(1):58–69. https://doi.org/10.1016/s0016-5085(03)00699-1.
Wasko-Czopnik D, Paradowski L. The influence of deficiencies of essential trace elements and vitamins on the course of Crohn’s disease. Adv Clin Exp Med. 2012;21(1):5–11.
Fung G, Luo H, Qiu Y, Yang D, McManus B. Myocarditis. Circ Res. 2016;118(3):496–514. https://doi.org/10.1161/CIRCRESAHA.115.306573.
Kyto V, Sipila J, Rautava P. Gender differences in myocarditis: a nationwide study in Finland. Eur Heart J. 2013;34(suppl_1) https://doi.org/10.1093/eurheartj/eht309.3505
Martens CR, Accornero F. Viruses in the heart: direct and indirect routes to myocarditis and heart failure. Viruses. 2021;13(10) https://doi.org/10.3390/v13101924
Oleszak F, Maryniak A, Botti E, et al. Myocarditis Associated With COVID-19. Am J Med Case Rep. 2020;8(12):498–502. https://doi.org/10.12691/ajmcr-8-12-19.
Haussner W, DeRosa AP, Haussner D, et al. COVID-19 associated myocarditis: a systematic review. Am J Emerg Med. 2022;51:150–5. https://doi.org/10.1016/j.ajem.2021.10.001.
Abou Hassan OK, Sheng CC, Wang TKM, Cremer PC. SARS-CoV-2 myocarditis: insights into incidence, prognosis, and therapeutic implications. Curr Cardiol Rep. 2021;23(9):129. https://doi.org/10.1007/s11886-021-01551-x.
Kindermann I, Barth C, Mahfoud F, et al. Update on myocarditis. J Am Coll Cardiol. 2012;59(9):779–92. https://doi.org/10.1016/j.jacc.2011.09.074.
Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol. 2008;99:95–114. https://doi.org/10.1016/S0065-2776(08)00604-4.
• Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18(12):733-744. https://doi.org/10.1038/s41577-018-0065-8This review focuses on the role of the immune system in several cardiac disorders including myocarditis.
Lawson CM, O’Donoghue HL, Reed WD. Mouse cytomegalovirus infection induces antibodies which cross-react with virus and cardiac myosin: a model for the study of molecular mimicry in the pathogenesis of viral myocarditis. Immunology. 1992;75(3):513–9.
Maisch B. Cardio-immunology of myocarditis: focus on immune mechanisms and treatment options. Front Cardiovasc Med. 2019;6:48. https://doi.org/10.3389/fcvm.2019.00048.
Guilherme L, Kalil J, Cunningham M. Molecular mimicry in the autoimmune pathogenesis of rheumatic heart disease. Autoimmunity. 2006;39(1):31–9. https://doi.org/10.1080/08916930500484674.
Zühlke L, Engel ME, Karthikeyan G, et al. Characteristics, complications, and gaps in evidence-based interventions in rheumatic heart disease: the Global Rheumatic Heart Disease Registry (the REMEDY study). Eur Heart J. 2015;36(18):1115–22. https://doi.org/10.1093/eurheartj/ehu449.
Mujib M, Desai RV, Ahmed MI, et al. Rheumatic heart disease and risk of incident heart failure among community-dwelling older adults: a prospective cohort study. Ann Med. 2012;44(3):253–61. https://doi.org/10.3109/07853890.2010.530685.
Guilherme L, Kalil J. Rheumatic heart disease: molecules involved in valve tissue inflammation leading to the autoimmune process and anti-S. pyogenes vaccine. Front Immunol. 2013;4:352. https://doi.org/10.3389/fimmu.2013.00352
Cunningham MW. Molecular mimicry, autoimmunity, and infection: the cross-reactive antigens of group A streptococci and their sequelae. Microbiol Spectr. 2019;7(4) https://doi.org/10.1128/microbiolspec.GPP3-0045-2018
Pérez-Molina JA, Molina I. Chagas disease. Lancet. 2018;391(10115):82–94. https://doi.org/10.1016/s0140-6736(17)31612-4.
Lee BY, Bacon KM, Bottazzi ME, Hotez PJ. Global economic burden of Chagas disease: a computational simulation model. Lancet Infect Dis. 2013;13(4):342–8. https://doi.org/10.1016/S1473-3099(13)70002-1.
Sabino EC, Ribeiro AL, Salemi VM, et al. Ten-year incidence of Chagas cardiomyopathy among asymptomatic Trypanosoma cruzi-seropositive former blood donors. Circulation. 2013;127(10):1105–15. https://doi.org/10.1161/CIRCULATIONAHA.112.123612.
Rassi A Jr, Rassi A, Marin-Neto JA. Chagas disease. Lancet. 2010;375(9723):1388–402. https://doi.org/10.1016/S0140-6736(10)60061-X.
Sinha A, Feinstein M. Epidemiology, pathophysiology, and prevention of heart failure in people with HIV. Prog Cardiovasc Dis. 2020;63(2):134–41. https://doi.org/10.1016/j.pcad.2020.01.002.
Sinha A, Feinstein MJ. Immune dysregulation in myocardial fibrosis, steatosis, and heart failure: current insights from HIV and the general population. Curr HIV/AIDS Rep. 2021;18(1):63–72. https://doi.org/10.1007/s11904-020-00536-9.
Erqou S, Lodebo BT, Masri A, et al. Cardiac dysfunction among people living with HIV: a systematic review and meta-analysis. JACC Heart failure. 2019;7(2):98–108. https://doi.org/10.1016/j.jchf.2018.10.006.
Toribio M, Neilan TG, Zanni MV. Heart failure among people with HIV: evolving risks, mechanisms, and preventive considerations. Curr HIV/AIDS Rep. 2019;16(5):371–80. https://doi.org/10.1007/s11904-019-00458-1.
Feinstein MJ, Bogorodskaya M, Bloomfield GS, et al. Cardiovascular complications of HIV in endemic countries. Curr Cardiol Rep. 2016;18(11):113. https://doi.org/10.1007/s11886-016-0794-x.
Feinstein MJ, Bahiru E, Achenbach C, et al. Patterns of cardiovascular mortality for HIV-infected adults in the United States: 1999 to 2013. Am J Cardiol. 2016;117(2):214–20. https://doi.org/10.1016/j.amjcard.2015.10.030.
Shah ASV, Stelzle D, Lee KK, et al. Global burden of atherosclerotic cardiovascular disease in people living with the human immunodeficiency virus: a systematic review and meta-analysis. Circulation. 2018. https://doi.org/10.1161/CIRCULATIONAHA.117.033369.
• Feinstein MJ, Steverson AB, Ning H, et al. Adjudicated heart failure in HIV-infected and uninfected men and women. J Am Heart Assoc. 2018;7(21):e009985. https://doi.org/10.1161/jaha.118.009985Interesting analysis comparing incident heart failure in people living with HIV and uninfected controls.
• Freiberg MS, Chang CH, Skanderson M, et al. Association between HIV infection and the risk of heart failure with reduced ejection fraction and preserved ejection fraction in the antiretroviral therapy era: results from the Veterans aging cohort study. JAMA Cardiol. 2017;2(5):536–546. https://doi.org/10.1001/jamacardio.2017.0264This study investigated whether HIV infection increases the risk of future heart failure subtypes (HFrEF and HFpEF).
Chen Y, Gao Y, Zhou Y, et al. Human immunodeficiency virus infection and incident heart failure: a meta-analysis of prospective studies. J Acquir Immune Defic Syndr. 2021;87(1):741–9. https://doi.org/10.1097/qai.0000000000002629.
Feinstein MJ, Mitter SS, Yadlapati A, et al. HIV-related myocardial vulnerability to infarction and coronary artery disease. J Am Coll Cardiol. 2016;68(18):2026–7. https://doi.org/10.1016/j.jacc.2016.07.771.
Feinstein MJ, Hsue PY, Benjamin LA, et al. Characteristics, prevention, and management of cardiovascular disease in people living with HIV: a scientific statement from the American Heart Association. Circulation. 2019;140(2):e98–124. https://doi.org/10.1161/cir.0000000000000695.
Feinstein MJ. Multihit interactions of antigens, immune responses, and comorbidities in cardiovascular disease pathogenesis: methods and potential mechanisms. Arterioscler Thromb Vasc Biol. 2021;41(1):523–5. https://doi.org/10.1161/atvbaha.120.315569.
• Tracy RP, Doyle MF, Olson NC, et al. T-helper type 1 bias in healthy people is associated with cytomegalovirus serology and atherosclerosis: the Multi-Ethnic Study of Atherosclerosis. J Am Heart Assoc. 2013;2(3):e000117. https://doi.org/10.1161/JAHA.113.000117Important study discussing immunologic pro-inflammatory bias.
Yen YF, Ko MC, Yen MY, et al. Human immunodeficiency virus increases the risk of incident heart failure. J Acquir Immune Defic Syndr. 2019;80(3):255–63. https://doi.org/10.1097/QAI.0000000000001917.
Womack JA, Chang CC, So-Armah KA, et al. HIV infection and cardiovascular disease in women. J Am Heart Assoc. 2014;3(5): e001035. https://doi.org/10.1161/JAHA.114.001035.
Janjua SA, Triant VA, Addison D, et al. HIV infection and heart failure outcomes in women. J Am Coll Cardiol. 2017;69(1):107–8. https://doi.org/10.1016/j.jacc.2016.11.013.
Younossi ZM, Stepanova M, Nader F, Younossi Z, Elsheikh E. Associations of chronic hepatitis C with metabolic and cardiac outcomes. Aliment Pharmacol Ther. 2013;37(6):647–52. https://doi.org/10.1111/apt.12234.
Tsui JI, Whooley MA, Monto A, Seal K, Tien PC, Shlipak M. Association of hepatitis C virus seropositivity with inflammatory markers and heart failure in persons with coronary heart disease: data from the Heart and Soul study. J Card Fail. 2009;15(5):451–6. https://doi.org/10.1016/j.cardfail.2008.12.003.
Moran CA, Collins LF, Beydoun N, et al. Cardiovascular implications of immune disorders in women. Circ Res. 2022;130(4):593–610. https://doi.org/10.1161/circresaha.121.319877.
Sevastianos VA, Voulgaris TA, Dourakis SP. Hepatitis C, systemic inflammation and oxidative stress: correlations with metabolic diseases. Expert Rev Gastroenterol Hepatol. 2020;14(1):27–37. https://doi.org/10.1080/17474124.2020.1708191.
Durante-Mangoni E, Zampino R, Marrone A, et al. Hepatic steatosis and insulin resistance are associated with serum imbalance of adiponectin/tumour necrosis factor-alpha in chronic hepatitis C patients. Aliment Pharmacol Ther. 2006;24(9):1349–57. https://doi.org/10.1111/j.1365-2036.2006.03114.x.
Tawadrous GA, Aziz AA, Amin DG, Eldemery A, Mostafa MA. RANTES, TNF-α, oxidative stress, and hematological abnormalities in hepatitis C virus infection. J Investig Med. 2012;60(6):878–82. https://doi.org/10.2310/JIM.0b013e318254519e.
Oliveira CP, Kappel CR, Siqueira ER, et al. Effects of hepatitis C virus on cardiovascular risk in infected patients: a comparative study. Int J Cardiol. 2013;164(2):221–6. https://doi.org/10.1016/j.ijcard.2011.07.016.
Zampino R, Marrone A, Restivo L, et al. Chronic HCV infection and inflammation: clinical impact on hepatic and extra-hepatic manifestations. World J Hepatol. 2013;5(10):528–40. https://doi.org/10.4254/wjh.v5.i10.528.
Fan C, Zhang X, Zhang P, et al. LPS stimulation during HCV infection induces MMP/TIMP1 imbalance in macrophages. J Med Microbiol. 2020;69(5):759–66. https://doi.org/10.1099/jmm.0.001185.
Innes HA, McDonald SA, Dillon JF, et al. Toward a more complete understanding of the association between a hepatitis C sustained viral response and cause-specific outcomes. Hepatology. 2015;62(2):355–64. https://doi.org/10.1002/hep.27766.
Hsu YC, Ho HJ, Huang YT, et al. Association between antiviral treatment and extrahepatic outcomes in patients with hepatitis C virus infection. Gut. 2015;64(3):495–503. https://doi.org/10.1136/gutjnl-2014-308163.
Butt AA, Xiaoqiang W, Budoff M, Leaf D, Kuller LH, Justice AC. Hepatitis C virus infection and the risk of coronary disease. Clin Infect Dis. 2009;49(2):225–32. https://doi.org/10.1086/599371.
Pujades-Rodriguez M, Morgan AW, Cubbon RM, Wu J. Dose-dependent oral glucocorticoid cardiovascular risks in people with immune-mediated inflammatory diseases: a population-based cohort study. PLoS Med. 2020;17(12): e1003432. https://doi.org/10.1371/journal.pmed.1003432.
Sazliyana S, Mohd Shahrir MS, Kong CT, Tan HJ, Hamidon BB, Azmi MT. Implications of immunosuppressive agents in cardiovascular risks and carotid intima media thickness among lupus nephritis patients. Lupus. 2011;20(12):1260–6. https://doi.org/10.1177/0961203311411347.
Kiani AN, Magder LS, Petri M. Mycophenolate mofetil (MMF) does not slow the progression of subclinical atherosclerosis in SLE over 2 years. Rheumatol Int. 2012;32(9):2701–5. https://doi.org/10.1007/s00296-011-2048-y.
Burgos PI, Vilá LM, Reveille JD, Alarcón GS. Peripheral vascular damage in systemic lupus erythematosus: data from LUMINA, a large multi-ethnic U.S. cohort (LXIX). Lupus. 2009;18(14):1303–8. https://doi.org/10.1177/0961203309105877
Haque S, Gordon C, Isenberg D, et al. Risk factors for clinical coronary heart disease in systemic lupus erythematosus: the Lupus and Atherosclerosis Evaluation of Risk (LASER) study. J Rheumatol. 2010;37(2):322–9. https://doi.org/10.3899/jrheum.090306.
Bernatsky S, Hudson M, Suissa S. Anti-rheumatic drug use and risk of hospitalization for congestive heart failure in rheumatoid arthritis. Rheumatology. 2005;44(5):677–80. https://doi.org/10.1093/rheumatology/keh610.
Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107(25):3133–40. https://doi.org/10.1161/01.Cir.0000077913.60364.D2.
Mann DL, McMurray JJ, Packer M, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. 2004;109(13):1594–602. https://doi.org/10.1161/01.Cir.0000124490.27666.B2.
•• Everett BM, Cornel JH, Lainscak M, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139(10):1289-1299. https://doi.org/10.1161/circulationaha.118.038010This study is a sub-analysis of the landmark CANTOS trial. It shows a reduction in heart failure hospitalization in post-MI patients treated with canakinumab.
Del Buono MG, Damonte JI, Chiabrando JG, et al. Effect of IL-1 blockade with anakinra on heart failure outcomes in patients with anterior versus non-anterior STEMI. J Cardiovasc Pharmacol. 2022. https://doi.org/10.1097/FJC.0000000000001240.
Tardif JC, Kouz S, Waters DD, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381(26):2497–505. https://doi.org/10.1056/NEJMoa1912388.
Tong DC, Quinn S, Nasis A, et al. Colchicine in patients with acute coronary syndrome: the Australian COPS Randomized Clinical Trial. Circulation. 2020;142(20):1890–900. https://doi.org/10.1161/CIRCULATIONAHA.120.050771.
•• Rurik JG, Tombácz I, Yadegari A, et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375(6576):91-96. https://doi.org/10.1126/science.abm0594. In this study, the authors observed a reduction in myocardial fibrosis in mouse models of heart failure using a programmed therapeutic T cell population capable of ablating activated fibroblasts.
Julier Z, Park AJ, Briquez PS, Martino MM. Promoting tissue regeneration by modulating the immune system. Acta Biomater. 2017;53:13–28. https://doi.org/10.1016/j.actbio.2017.01.056.
Vasconcelos DP, Costa M, Amaral IF, Barbosa MA, Aguas AP, Barbosa JN. Modulation of the inflammatory response to chitosan through M2 macrophage polarization using pro-resolution mediators. Biomaterials. 2015;37:116–23. https://doi.org/10.1016/j.biomaterials.2014.10.035.
Ramjee V, Li D, Manderfield LJ, et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J Clin Invest. 2017;127(3):899–911. https://doi.org/10.1172/JCI88759.
Weirather J, Hofmann UD, Beyersdorf N, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115(1):55–67. https://doi.org/10.1161/CIRCRESAHA.115.303895.
Funding
N.B. is supported by the Stimulating Access to Research in Residency of the National Institutes of Health under Award Number R38AI140299. M.J.F. is supported by the National Institutes of Health Award Numbers R01HL156792 and R01HL154862. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Comorbidities
Rights and permissions
About this article

Cite this article
Beydoun, N., Feinstein, M.J. Heart Failure in Chronic Infectious and Inflammatory Conditions: Mechanistic Insights from Clinical Heterogeneity. Curr Heart Fail Rep 19, 267–278 (2022). https://doi.org/10.1007/s11897-022-00560-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-022-00560-3