
Abstract
Purpose of review
SGLT2 inhibitors (SGLT2i) are new drugs for patients with heart failure (HF) irrespective of diabetes. However, the mechanisms of SGLT2i in HF remain elusive. This article discusses the current clinical evidence for using SGLT2i in different types of heart failure and provides an overview about the possible underlying mechanisms.
Recent findings
Clinical and basic data strongly support and extend the use of SGLT2i in HF. Improvement of conventional secondary risk factors is unlikely to explain the prognostic benefits of these drugs in HF. However, different multidirectional mechanisms of SGLT2i could improve HF status including volume regulation, cardiorenal mechanisms, metabolic effects, improved cardiac remodelling, direct effects on cardiac contractility and ion-homeostasis, reduction of inflammation and oxidative stress as well as an impact on autophagy and adipokines.
Summary
Further translational studies are needed to determine the mechanisms of SGLT2i in HF. However, basic and clinical evidence encourage the use of SGLT2i in HFrEF and possibly HFpEF.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction: Clinical Background
SGLT2 Inhibitors and Cardiovascular Outcome Trials
Sodium-glucose-cotransporter 2 inhibitors (SGLT2i) increase the urine glucose excretion by inhibiting SGLT2 in the proximal tubule of the kidney and thereby lower blood glucose levels. Initially, SGLT2i were evaluated for their cardiovascular safety in type 2 diabetes mellitus (T2DM) patients with either established atherosclerotic cardiovascular disease or with multiple cardiovascular risk factors. SGLT2i showed unexpected beneficial effects on the rate of major adverse cardiovascular events (MACE) consisting of cardiovascular events, cardiovascular death and all-cause mortality, with benefit only seen in patients with atherosclerotic cardiovascular disease and not in those without [1,2,3,4,5].
Interestingly, the reduction of cardiovascular events was primarily driven by a prevention of heart failure (HF) hospitalisation (reduction of ~30%) rather than atherothrombotic events [5]. Moreover, the favourable effects on mortality and HF hospitalisation became apparent already after 2–3 months of treatment, which make an improvement of atherosclerotic risk factors e.g. via glucose excretion rather unlikely to account for these effects [6].
Consequently, additional studies displayed that the beneficial effects of SGLT2i are less likely to be explained by refinement of classical secondary cardiovascular risk factors as blood pressure [7], cholesterol [8] or HbA1c levels [9,10,11]. Moreover, SGLT2i have been shown to improve cardiovascular outcomes in diabetic patients independent of cardiovascular risk/established cardiovascular disease [1, 5, 9], HF risk factors (i.e. assessed by the Health ABC HF Risk score) [10], diagnosed HF at baseline [12] or renal function [1, 13]. Therefore, the favourable effects of SGLT2i in these trials seem to be mediated by other mechanisms and raised the question about a particular benefit in HF patients.
SGLT2 Inhibitors in Patients with Heart Failure
The DAPA-HF trial was the first one studying the efficacy of dapagliflozin in patients who suffer from HF with reduced ejection fraction (HFrEF, ejection fraction (EF) ≤40%, NYHA II–IV) with and without T2DM with respect to cardiovascular death or worsening HF [14]. Dapagliflozin therapy was established in addition to guideline-directed HF therapy. In 4744 HF patients, dapagliflozin reduced the primary composite outcome of worsening HF (hospitalisation or urgent intravenous therapy for HF) and death from cardiovascular causes by 26% as well as each component of the primary endpoint alone. Moreover, dapagliflozin caused an improved all-cause mortality and reduced HF symptoms (assessed by the Kansas City Cardiomyopathy Questionnaire, KCCQ) [14]. Likewise, in the EMPEROR-reduced trial enrolling 3730 patients with HFrEF (EF≤40%, NYHA II–IV) with and without T2DM, empagliflozin reduced the primary composite outcome of death from cardiovascular cause and hospitalisation for HF [15]. Most importantly, the beneficial effects on all mentioned end-points of dapagliflozin and empagliflozin in HF patients were achieved in patients with and without T2DM [14,15,16,17]. Of note, in both trials, SGLT2i also significantly reduced adverse renal outcomes [14,15,16,17]. Finally, the SOLOIST-WHF trial investigated the therapy with the combined SGLT2 and SGLT1 inhibitor sotagliflozin in 1222 patients with T2DM hospitalised for HF. Sotagliflozin significantly reduced the primary endpoint of cardiovascular death, HF hospitalisation and urgent visit for HF [18]. Focusing on all of these large randomised controlled trials, only DAPA-HF demonstrated a significantly improved all-cause mortality as a secondary outcome in HF patients. However, a recent meta-analysis combining DAPA-HF and EMPEROR-Reduced clearly indicated a consistent reduction of all-cause mortality in respective HF patients [19].
Further evidence of the favourable impact of SGLT2i in HF is given by the DEFINE-HF trial, which showed improvement in HF symptoms (based on the KCCQ) over 12 weeks in 263 patients with HF (EF≤40%, NYHA II–III) treated with dapagliflozin, irrespective of T2DM [20]. Of note, NT-proBNP was not changed after 12 weeks of treatment [20].
The effects of empagliflozin on 6-min walk tests were further tested in 312 patients with HFrEF in the EMPERIAL-Reduced trial which, however, showed no significant improvement after treatment with empagliflozin [21]. As discussed recently, based on the robust data of the EMPEROR-Reduced trial, different issues could possibly underlie the lack of efficacy in this study including small sample size and the use of the 6-min walk test as primary endpoint [22]. Interestingly, the favourable effects of SGLT2i on HF in the DAPA-HF and EMPEROR-Reduced trials were consistent concerning different subgroups with respect to sex, T2DM, renal function and existing HF therapy including angiotensin receptor-neprilysin inhibitors [14, 23, 24].
Therefore, SGLT2i have been emerged as a new therapy for patients with HFrEF with or without T2DM mellitus. Accordingly, dapagliflozin has recently been approved for the treatment of patients with HFrEF independent of T2DM. Clinical HF guidelines will very likely suggest SGLT2i as basal treatment for patients with HFrEF, as there is a theoretical data basis for a class IA recommendation with two clinical trials. However, the underlying mechanisms of SGLT2i in HF are still a matter of debate. Therefore, in this article, we discuss the evidence and the significance of the mechanism of action of SGLT2i with a special focus on their myocardial effects in healthy and diseased hearts.
Mechanisms of SGLT2 Inhibitors in HF
Translational Aspects for Mechanistic Investigation in HF: Back to Bench
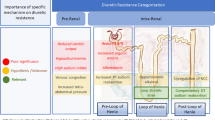
Various mechanisms of SGLT2i have been proposed in different cardiovascular diseases over the last years. Based on clinical evidence with early beneficial effects of SGLT2i, an improvement of conventional secondary risk factors including improved glycaemic control, reduced blood pressure, weight loss or improved cholesterol seems to be unlike to explain the prognostic benefit of SGLT2i in HF [7,8,9,10,11, 16]. Therefore, we specifically review the potential cardiac mechanisms of SGLT2i which potentially play a favourable role in HF pathophysiology contributing thereby to improved clinical outcomes (Fig. 1).

© Shutterstock/Vasif Maharov; Image human: © Shutterstock/10topvector
Possible mechanisms of SGLT2 inhibitors (SGLT2i) on the heart with respect to rather systemic (left panel), combined (middle panel) or myocardial effects (right panel). Image heart:
Of note, it is important to distinguish between systemic and myocardial effects when investigating the possible mechanisms of SGLT2i in HF, as direct myocardial mechanisms cannot really be elucidated in a systemic setting where secondary factors (i.e. altered blood pressure) influence the cardiac function. Therefore, in vitro isolated hearts and myocardial tissue/cells with well-defined conditions have the advantages to study the possible direct cardiac effects of SGLT2i. Nevertheless, in vivo studies using different disease models are of translational importance to study the effects of SGLT2i in the complex, multidirectional systemic setting. It is noteworthy that the expression of SGLT2 in the heart is negligible [25, 26], while SGLT1 is expressed in the myocardium [25]. Only sotagliflozin has been shown to have a relevant inhibitory effect on SGLT1, while other SGLT2i are high selective for SGLT2 [27,28,29]. Nevertheless, direct cardiac effects on the heart have been demonstrated, which suggest SGLT2 independent actions in the heart.
Volume Regulation, Haemoconcentration and Preload
Blocking SGLT2, which is strongly expressed in the proximal tubule of the kidney, causes glucosuria, natriuresis and osmotic diuresis. Empagliflozin has been demonstrated to cause a reduction in blood and plasma volume after 14 days in a cohort of 20 patients with T2DM and HF [30]. In a randomised controlled trial of 75 diabetic patients, dapagliflozin was associated with reduced blood pressure and plasma volume [31]. Canagliflozin was also shown to transiently diminish plasma volume in the first weeks of treatment [32]. However, after 12 weeks, this effect was largely attenuated [32]. Therefore, a diuretic effect of SGLT2i with consecutive favourable effects on blood volume has been proposed. A study on the diuretic effects of SGLT2i utilised a mathematical model based on data from healthy volunteers, which were treated for 7 days with dapagliflozin or bumetanide. The authors proposed a stronger reduction in electrolyte-free water and thus in interstitial fluid after dapagliflozin compared to bumetanide, which rather reduced intravascular volume [33]. The diuretic effects of SGLT2i have been proposed to regulate plasma volume without an activation of the sympathetic nervous system or the renin-angiotensin-aldosterone axis as it is known for loop diuretics [30]. This differential modulation of reducing interstitial fluid without largely affecting intravascular volume was described as the fluid hypothesis [34]. However, profound evidence is required to fully support this hypothesis.
Potentially driven by a diuretic effect of SGLT2i, systolic blood pressure and body weight were reduced after treatment with SGLT2i [31]. In line with a possible reduced plasma volume, SGLT2i have been shown to increase the haematocrit of individuals with and without T2DM [31, 32], while also having effects on erythropoietin levels (see below) [35, 36]. In reference to the diuretic effects, it has been proposed that SGLT2i reduce the preload which itself leads to improved cardiac loading conditions [34]. In line with that, cardiac magnetic resonance imaging (MRI) data showed reduced end diastolic volume in patients with T2DM after treatment with empagliflozin [37]. The reduction of interstitial fluid would beneficially affect congestion and pulmonary oedema in HF and may improve cardiac function via lowered preload.
However, the impact on plasma volume has only been reported at early time points [30,31,32], and the diuretic effects of SGLT2i have been demonstrated to decrease after weeks [38]. Therefore, relevant diuretic effects of SGLT2i for reducing cardiovascular mortality and HF outcomes are still questionable. Recently, a secondary analysis of the EMPEROR-Reduced trial demonstrated no differences between the effects of empagliflozin on cardiovascular mortality and HF hospitalisation in patients with (clinically assessed 4 weeks before randomisation) volume overload compared with euvolemic patients [39]. Also, markers of plasma volume regulation like haematocrit, body mass index, NT-proBNP and albumin provided no clear support for relevant diuretic properties [39]. Another argument against a prominent diuretic effect is the fact that the dose of the diuretic medication in the DAPA-HF trial could not be lowered as a consequence of dapagliflozin treatment [14, 40]. Therefore, further validation of the proposed effects of SGLT2i on volume regulation and its contribution to the cardiovascular outcomes is warranted.
Vascular Function and Afterload
Another hypothesis by which SGLT2i could improve cardiac function is a reduction in afterload [34]. In HF, endothelial and vascular dysfunctions are present [41]. Optimising endothelial function and thus vascular stiffness might improve hemodynamic and cardiac function via lowering afterload. Interestingly, SGLT2i were suggested to improve vascular function. In a post-hoc analysis of patients with T2DM and hypertension, empagliflozin reduced blood pressure and improved markers of arterial stiffness and vascular resistance [42]. Accordingly, a small study investigating 16 T2DM patients suggested that dapagliflozin acutely improved endothelial function and vascular stiffness independent of blood pressure alterations [43]. A mechanistic study in aortic preparations from animals additionally showed that dapagliflozin exerts vasodilatory effects via protein kinase G (PKG) and voltage-gated K channels [44].
SGLT2i caused modest antihypertensive effects in different trials [7]. Effects of SGLT2i on volume regulation, arterial stiffness, natriuresis and weight loss could underlie the lowered blood pressure. However, as blood pressure would expect to improve cardiovascular risk in the long term, it appears unlikely that the modest reduction in blood pressure by SGLT2i significantly contributes to the observed early clinical outcomes [45]. Moreover, one would expect a reduction in atherothrombotic events via reduced blood pressure, but the risk for stroke was not influenced by SGLT2i [46]. Conclusively, a meta-analysis in patients with T2DM receiving SGLT2i showed no relationship between blood pressure reduction and cardiovascular events [7]. Therefore, effects of SGLT2i on blood pressure are unlikely to explain the favourable outcomes of the drugs in HF patients. Potential differential effects of SGLT2i on vascular function i.e. with respect to pulmonary circulation require further investigation.
Cardiorenal Effects
While this review focuses on the impact of SGLT2i in HF, some potential HF-relevant mechanisms on the cardiorenal axis should be discussed as they clearly determine mortality in HF. The tremendously important prognostic significance of chronic kidney disease for cardiovascular endpoints and HF has been addressed for years and is well demonstrated [47]. A causal therapy for cardiorenal syndrome that can help improving the prognosis of patients with chronic kidney disease and cardiovascular disease beyond standard therapy with ACE inhibitors and angiotensin receptor blockers has long been lacking. SGLT2i could fill an important gap here. In this context, the DAPA CKD trial can be seen as a real milestone [48]. Patients with stage 2–4 chronic kidney diseases were included. Sixty-seven patients of the patients were diabetic, and nearly all of them were on ACE inhibitors or angiotensin receptor blockers [49]. In these patients, it was shown that taking SGLT2i not only significantly improved the prognosis of important renal parameters with respect to renal endpoints (eGFR decline above 50%, end-stage chronic kidney disease or death from renal causes), but also reduced the combined risk of death from cardiovascular causes or hospitalisation for HF by 29% in these patients. Finally, there was even a 31% reduction in all-cause mortality. These data not only point the way to an apparently highly effective therapy for chronic kidney disease, but also refer once again to the close connection between possible common pathophysiological mechanisms of cardiac and renal failure. Possible cardiorenal effects of SGLT2i will therefore be presented here.
As mentioned before, effects of glucosuria via glycaemic control and weight loss are unlikely to contribute to clinical outcomes in HF patients without T2DM. However, besides glucosuria, SGLT2i cause a natriuretic effect in the kidney. Via increased natriuresis, SGLT2 have shown to increase the amount of Na+ detected by the juxtaglomerular apparatus at the distal renal tubules, which causes a vasoconstriction of the afferent arteriolar vessel. As under hyperglycaemic conditions, Na+ transportation to the juxtaglomerular apparatus is reduced via SGLT2, the tone of the afferent arteriolar vessel is inadequately regulated [50]. Therefore, SGLT2i restore the impaired tubuloglomerular feedback mechanism [50]. Consecutively, the reduced blood flow stimulates the release of erythropoietin [51].
SGLT2i further possibly increase erythropoietin levels via hypoxia-inducible factors and utilisation of ketone bodies [51]. The SGLT2i-mediated increase in erythropoietin leads to enhanced red blood cell mass and haematocrit [31, 52], potentially associated with a reduction of renal stress and also sympathetic hyperactivity [53]. In an analysis of potential explanations of the reduced cardiovascular death in the EMPA-REG OUTCOME trial, the authors proposed that changes in haematocrit and haemoglobin, possibly related to change in volume status, mediate the diminished risk for cardiovascular death [54]. Based on the increased haematocrit, oxygen supply of the heart may be improved [51], which could theoretically increase the function of the failing heart. Of note, different renal effects of SGLT2i have been comprehensively reviewed elsewhere [55].
Uric Acid
Uric acid is a product of the degradation of purine nucleotides and has been shown to be associated with cardiovascular diseases [56, 57]. Interestingly, uric acid is associated with elevated levels of inflammation and oxidative stress as well as reduced NO bioavailability and thus endothelial dysfunction [58]. Moreover, the renin-angiotensin-aldosterone system has been shown to be activated by uric acid [58]. SGLT2i proved a relatively robust evidence to reduce uric acid in patients with T2DM as indicated by a meta-analysis of 62 randomised control trials of SGLT2i [59]. While the effect persisted during long-term treatment, patients with chronic kidney disease (eGFR <60 mL/min) however showed no decline in serum uric acid [59]. SGLT2i reduce serum uric acid via increasing urine glucose levels which are reported to suppress urate reabsorption via SLC2A9 [56]. Although further causal mechanistic and clinical data are needed, lowering uric acid via SGLT2i might have potential cardiac benefits.
Metabolic Effects and the Fuel Hypothesis
Since glycaemic control has no significant influence on the prognostic effects of SGLT2i, at least in the DAPA-HF and the EMPEROR-Reduced trials, it is unlikely that improvement of T2DM and hyperglycaemia-related metabolic abnormalities significantly contribute to the positive HF outcomes. However, different metabolic effects of SGLT2i have been proposed, which itself might influence cardiac function in a T2DM-independent manner.
As glucose availability is reduced upon SGLT2i, lipolysis and ketogenesis have shown to be increased in animals and patients with T2DM [60, 61]. After treatment with dapagliflozin, a reduction of visceral adipose tissue and subcutaneous adipose tissue has been shown in patients with T2DM and left ventricular (LV) hypertrophy [62]. Moreover, a shift from carbohydrate usage to lipid usage could be demonstrated in patients with T2DM [61] accompanied by an increase in circulating ketone levels such as β-hydroxybutyrate [63, 64].
As ketone bodies produce more ATP per consumed oxygen atom compared to glucose or free fatty acids, energy efficacy of the heart is higher when ketone bodies are utilised [65, 66]. Therefore, it has been postulated that the increased ketone body utilisation upon SGLT2i treatment could fuel the metabolic state of the heart and thus improve energetics [66]. Interestingly, in end-stage human HF patients, ketone utilisation is also enhanced, independent of T2DM [67]. In an interesting small, randomised trial, application of the ketone body 3-hydroxybutyrate caused an increase in EF in patients with HF without lowering myocardial external energy efficiency [68]. Moreover, metabolic regulation of branched chain amino acids (BCAAs), which is altered in HF [69], has also been reported to be affected by empagliflozin in an untargeted metabolomics approach in 25 patients with T2DM and cardiovascular disease [70]. In a porcine non-diabetic ischemic HF model (LAD occlusion), empagliflozin increased the utilisation of BCAA, free fatty acids and ketone bodies, which was associated with a mitigation of HF remodelling and improved LV function [71]. Therefore, ‘fuelling’ the heart via enhanced energy substrates upon SGLT2i might improve cardiac performance. However, the full effects of SGLT2i on ketone body utilisation, its interference with other potentially adverse pathways and finally its relevance for cardiac function and cardiovascular benefits are still controversial [72,73,74]. In addition to that, it has also recently been hypothesised that SGLT2i rather induce an energy-saving ‘dormancy’ programme than ‘super-fuelling’ the heart [75].
Ventricular Remodelling and Fibrosis
Different reports suggest that SGLT2i may be involved in LV remodelling. The placebo-controlled, randomized EMPA-HEART trial studying 97 patients with T2DM, coronary artery disease, and preserved EF reported a reduction in LV mass index as obtained with cardiac MRI after 6 months of treatment [76]. Interestingly, in additional exploratory analyses, the changes in LV mass were not associated with blood pressure alterations after 6 months [76]. Likewise, empagliflozin has been demonstrated to reduce LV mass and to improve diastolic function in echocardiographic data concerning a small uncontrolled trial in T2DM patients [77]. Moreover, dapagliflozin significantly reduced LV mass in patients with T2DM [62]. In a retrospective study of diabetic patients with and without HF, SGLT2i caused an improved echocardiographic LV end-diastolic-diameter [78]. On top of that, in HFrEF patients, improved LV EF and diastolic function were suggested [78]. However, in a small randomised, double blind, placebo-controlled study of 56 diabetic patients with a previously reported reduced EF, dapagliflozin had no effects on LV end-systolic volume, LV end-diastolic volume or LV mass index obtained by cardiac MRI after 1 year [79]. Yet, the authors discussed that the study could have been limited by a small sample size and less severe HF in the patients [79]. A possible explanation is given by experimental data, in which dapagliflozin mitigated cardiac hypertrophy, apoptosis as well as fibrosis and improved LV EF in mice with pressure-induced HF [80].
Taken together, compelling evidence indicates that SGLT2i favourably affect LV remodelling in T2DM patients and in HF patients, which could be a central mechanism of the beneficial cardiac effects of SGLT2i. However, further data on potential differential effects in HFrEF and HFpEF and on the underlying mechanisms are needed.
Fibrosis plays a central role in the context of structural HF-remodelling. Interestingly, dapagliflozin exerted antifibrotic properties in a rat model of myocardial infarction by reducing reactive nitrogen and oxygen species, thereby regulating myofibroblast and M2 macrophages infiltration [81]. SGLT2i also reduced cardiac fibrosis in experimental animal models of hypertension [82] and T2DM [83]. Conversely, in 35 patients with T2DM, 6 months of treatment with empagliflozin did not change fibrosis indices as obtained by cardiac MRI. However, LV function was normal in these patients and, hence, could not be influenced positively by empagliflozin [84]. LV remodelling represents a final common pathway of different cardiac diseases. While a SGLT2i-induced mitigation of pathologic remodelling in HF shed light onto important effects in cardiovascular disease, the underlying mechanisms still need to be elucidated.
Myocardial Contraction and Relaxation
Improving myocardial relaxation has often been proposed to be a potential prognostic mechanism of SGLT2i. Indeed, in vivo studies described effects of SGLT2i on diastolic function. In a clinical study in patients with T2DM and established cardiovascular disease (discussed above), empagliflozin improved diastolic function assessed via echocardiography [77]. Likewise, canagliflozin improved echocardiographic diastolic function in 38 patients with T2DM in a prospective observational study after 3 months of treatment [85]. Moreover, in a prospective trial in patients with T2DM and stable HF irrespective of EF, dapagliflozin was proposed to cause an improvement in echocardiographic diastolic parameters after 6 months of treatment [86] as well as in patients with T2DM [87]. MRI studies showed decreased LV end-diastolic volumes upon empagliflozin treatment in HFrEF patients with and without T2DM, while the effects on systolic parameters are controversial [37, 88, 89].
In addition to that, preclinical data support the findings of an improved diastolic function in vivo. In obese diabetic rats, which are characterised by a prolonged isovolumetric relaxation time (IVRT), acute treatment with empagliflozin mitigated diastolic dysfunction without showing effects on the LV EF [90]. In a HFpEF mouse model, empagliflozin has been demonstrated to improve diastolic function without altering systolic force, and it was associated with improved hemodynamics [91]. Moreover, in chronically treated T2DM mice models, empagliflozin beneficially affected diastolic parameters as measured by echocardiography [92, 93] or pressure catheter [94]. Conversely, another animal study reported a preserved systolic function in mice with pressure-overload-induced HF after treatment with empagliflozin, without showing effects on diastolic function [95].
However, the question about the potential mechanisms underlying the improved diastolic function upon SGLT2i arises. As discussed above, some studies reported that the improvement in diastolic function was associated with hemodynamic, metabolic or structural changes. However, these findings were not consistent in the different studies. A recent study in diabetic patients indeed showed that empagliflozin improved echocardiographic parameters of diastolic function without affecting hemodynamic parameters [96]. In vitro data (which exclude systemic confounder like plasma volume regulation or blood pressure) of direct SGLT2i-related effects on contractility are of translational importance to further clarify the underlying (cardiac) mechanisms.
Our groups provided first evidence of SGLT2i-mediated effects on myocardial contractility in human ventricular trabecula from patients with HFrEF, irrespective of T2DM [90]. Empagliflozin caused a significant acute reduction of the pathologically enhanced diastolic tension in twitching human HFrEF trabecula without altering the systolic contractile force (Fig. 2) [90]. We have elucidated the underlying mechanisms of an improved diastolic function by demonstrating that empagliflozin directly reduces passive myofilament stiffness in human HFpEF myocardium (Fig. 3). These effects were mediated by an enhanced phosphorylation, typically dephosphorylated in HFpEF, of titin and other myofilament regulatory proteins via improvement of the nitric oxide (NO)-soluble guanylyl cyclase (sGC)-cGMP-dependent protein kinase (PKG) signalling pathway by empagliflozin [90, 97]. These changes were associated with altered oxidative and inflammatory pathways in human and rodent HFpEF myocardium as discussed below [97]. Also, in studies of diabetic mice, empagliflozin beneficially impacted cardiac function via NO-sGC-PKG pathway after 8 weeks of treatment [98]. As altered NO-sGC-PKG signalling is a key mechanism of diastolic dysfunction and HFpEF [99, 100], further investigations of this pathway upon SGLT2i in HFpEF appear promising [101]. Moreover, the reduced fibrotic content upon SGLT2i or changes in cardiomyocyte ion-homeostasis (see below) could also favourably affect HFpEF patients.

Contractility of isolatedhuman HFrEF trabecula upon empagliflozin treatment. A Original twitches of stimulated human trabecula before and after wash-in of increasing concentrations of empagliflozin. B Normalised developed (systolic) force (Tdev) and C normalised diastolic tension (Tdia). Raw data before and after wash-in of empagliflozin are provided in the inlay scatter. With permission from Pabel et al. [90]

Effects of empagliflozin on the passive stiffness of (skinned) cardiomyocytes from HFpEF patients and from healthy donors. A The original recordings of force response during stepwise cell stretches. B Normalised passive stiffness of HFpEF and non-failing (NF) cardiomyocytes upon empagliflozin measured at different sarcomere lengths. With permission from Pabel et al. [90]
What to Expect from SGLT2 Inhibitors in HFpEF
As dapagliflozin and empagliflozin have shown to be effective for the prognostic and symptomatic treatment of HFrEF-patients independent of T2DM [102], the question about potential benefits in HFpEF arises. Current therapeutic options for HFpEF patients are limited, and no drug has yet been proven to exert prognostic relevant effects in HFpEF patients. Interestingly, a meta-analysis based on limited data of the DECLARE-TMI 58 trial and the VERTIS CV study suggests effects of SGLT2 on HF hospitalisation in patients with HFpEF [19]. In general, it can be assumed that a not insignificant number of HFpEF patients were included in the studies due to the inclusion criteria and the patient risk profile. Moreover, preclinical data on the effects of SGLT2i on diastolic function and pathways involved in HFpEF pathophysiology might also provide a rationale for the efficacy of these drugs in this complex disease [101]. The DELIVER trial (NCT03619213) investigates the effects of dapagliflozin in patients with HFpEF (EF >40%, structural heart disease, elevated NT-pro BNP levels, NYHA II–IV) on cardiovascular death or HF events. Accordingly, the EMPEROR-Preserved trial (NCT03057951) studies the effect of empagliflozin in patients with HFpEF (EF >40%, NYHA II–IV, elevated NT-proBNP, structural heart disease or HF hospitalisation) on cardiovascular death or hospitalisation due to HF. Both clinical trials will be of utmost importance, since HFpEF is still an unmet need in cardiology.
Cardiac Ion-Homeostasis
Cardiomyocyte Ca2+ and Na+ handling fundamentally regulates excitation-contraction (EC) coupling and thus cardiac contractility [103]. Dysregulation of Ca2+ and Na2+ homeostasis is typically present in HF and contributes to contractile dysfunction and arrhythmias [104, 105]. Interestingly, direct cardiac effects of SGLT2i on cardiomyocyte ion-homeostasis have been reported. In healthy rabbit and rat cardiomyocytes, empagliflozin has been reported to acutely reduce cytosolic Ca2+ and Na+ by inhibiting the Na+/H+ exchanger 1 (NHE1) in an elevated glucose environment [106]. Interestingly, NHE1 expression is increased in human HF myocardium [107]. The inhibitory effect of SGLT2i on NHE1 was also demonstrated in human atrial tissue [108] and murine myocardium [108, 109]. Disturbed cellular Ca2+ and Na+ homeostasis in HF consisting of reduced systolic Ca2+ transients and elevated diastolic Ca2+ and Na+ levels in the cytosol adversely affects systolic and diastolic contractile function [110,111,112], impairs mitochondrial function [113], triggers proarrhythmic activity and induces electrophysiological signalling changes fuelling detrimental remodelling [105]. Therefore, by reducing cytosolic Na+ (and consecutively cytosolic Ca2+), NHE1 inhibition could favourably affect cardiomyocyte function.
Influences of SGLT2i on cardiomyocyte Na+ homeostasis have also recently been proposed in a study showing inhibitory effects of SGLT2i on the late Na+ current in murine HF cardiomyocytes [114]. The late Na+ current constitutes a persistent Na+ influx throughout the action potential and contributes to adverse electric remodelling in HF [110, 115]. Molecular docking simulations indicated that empagliflozin binds at the major cardiac Na+ channel isoform NaV1.5 and thereby reduces late Na+ current with little effects on peak Na+ current [114]. A reduction in late Na+ current may diminish reverse mode NCX-mediated Ca2+ overload and may thus reduce proarrhythmic triggers and contractile dysfunction [110]. Therefore, the inhibition of this mechanism could be a promising target in HF.
Another way how empagliflozin might counteract disturbed Ca2+ handling in HF has been proposed in failing ventricular murine and human cardiomyocytes. Empagliflozin treatment for 24 h reduced the activity of Ca2+/calmodulin-dependent protein kinase IIδ (CaMKII) [116], which is centrally involved in adverse myocardial remodelling in cardiac disease and contributes to arrhythmias [117,118,119]. In this study, reduction of CaMKII mitigated diastolic Ca2+ leak from the sarcoplasmic reticulum [116], which contributes to contractile dysfunction and arrhythmias [118]. While the reduction of CaMKII activity could be a secondary effect of empagliflozin in the (cultured) cells, this mechanism may nevertheless inhibit the vicious circle of SR Ca2+ leak-dependent CaMKII activation and may thereby soothe adverse electric remodelling in HF.
Yet, other studies on the impact of SGLT2i on cardiomyocyte Ca2+ homeostasis reported contrasting evidence which may be explained by different experimental protocols such as time of drug treatment and type of cardiomyocytes. Acute exposure with empagliflozin did not alter systolic Ca2+ transient or diastolic cytosolic Ca2+ in isolated human HF cardiomyocyte [90]. In a blinded study using healthy human-induced pluripotent stem cell cardiomyocytes long-term treatment for 2 months did also not affect Ca2+ homeostasis and EC-coupling proteins [120]. Of note, this is the only study on ion homeostasis with a treatment duration according to the time course of the clinical effects (~2 months). With respect to NHE1 inhibition, a recent study demonstrated that acute exposure with empagliflozin had no effects on the NHE1 function or the Na+ homeostasis in healthy isolated rat cardiomyocytes [121]. Differences in species and treatment protocols might limit the comparability of these studies. Thus, it is important to consider the disease/species of question when studying the effects of SGLT2i on ion-homeostasis. Finally, as Ca2+ and Na+ handling is closely intertwined with many other signalling cascades (i.e. oxidative stress, mitochondrial function), also secondary effects of SGLT2i on ion-homeostasis are conceivable.
Oxidative Stress and Inflammation
Inflammation and oxidative stress play a central role in HF development and progression. Both are associated with increasingly prevalent comorbidities such as chronic kidney disease or metabolic syndrome [122]. Particularly in HFpEF, inflammation and oxidative stress were shown to cause structural and functional diastolic dysfunction [122, 123]. Interestingly, human HFpEF myocardium treated with empagliflozin in vitro exhibited reduced markers of oxidative stress (i.e. H2O2, GSH, LPO) and inflammation (ICAM, VCAM, TNFα and IL-6) [97].
Indeed, in several studies, SGLT2i showed anti-inflammatory and anti-oxidative properties [124] as dapagliflozin reduced the inflammasome and fibrosis in mouse models of T2DM [125] and myocardial infarction [81], ipragliflozin mitigated biomarkers of oxidative stress and inflammation in diabetic mice [126, 127] and empagliflozin attenuated oxidative stress (associated with metabolic changes) in mice after myocardial infarction [128]. As increased inflammation and oxidative stress have been shown to diminish myofilament phosphorylation and therefore worsen diastolic function, improvement of inflammation and oxidative stress may serve as an explanation for the enhanced NO-sGC-PKG pathway after SGLT2i leading to a mitigated diastolic stiffness of the LV in HFpEF. In line with that, empagliflozin improved cardiac function in diabetic mice via improvement of PKG, oxidative stress and apoptosis [98]. Moreover, in a co-culture of cardiac microvascular endothelial cell with cardiomyocytes, empagliflozin reversed TNFα-mediated microvascular dysfunction leading to enhanced NO availability [129].
Autophagy and Adipokines
As recently discussed, a potential explanation for the reduction of inflammation and oxidative stress could be the effects of SGLT2i on autophagy [73]. Experimental data demonstrated restored autophagy in different tissue/cells, which could mitigate oxidative stress and inflammation [130, 131]. The improvement in autophagy has been postulated to be mediated by adenosine monophosphate-activated protein kinase (AMPK), sirtuin‐1 and hypoxia‐inducible factors [132]. Furthermore, it was reported that differences concerning AMPK activation are also associated with anti-inflammatory properties of SGLT2i in diabetic mice [125]. However, further mechanistic evidence for the effects of SGLT2i on autophagy, also with respect to cardiac function, is needed.
Another possible explanation for the favourable impact of SGLT2i on inflammation and oxidative stress could be an influence on the adipokine profile and leptin [133]. Adipokines are secreted i.e. from the epicardial adipose tissue and have been shown to contribute to obesity/metabolic syndrome-related remodelling and cardiovascular disease [134,135,136]. SGLT2i have been demonstrated to reduce epicardial adipose tissue and leptin, which was associated with reduced inflammation and oxidative stress [137, 138].
Finally, beneficial effects of erythropoietin on oxidative stress and inflammation, in particular in the metabolic syndrome, are possible [51, 139].
Conclusion
A plethora of multidirectional mechanisms of SGLT2i, which could underlie the favourable HF outcomes, has been proposed. While improvement of conventional secondary risk factors is unlikely to explain the marked benefit of these drugs on HF end points, several novel mechanisms of SGLT2i were reported, including differential volume regulation, cardiorenal mechanisms, metabolic effects, improved cardiac remodelling, direct effects on cardiac contractility and ion-homeostasis, reduction of inflammation and oxidative stress as well as an impact on autophagy and adipokines. Importantly, differences in the investigated species and disease, the treatment protocols and many other methodological varieties make it somewhat difficult to extrapolate central mechanisms of SGLT2i. So far, evidence rather points towards multifactorial effects associated with reduced systemic and myocardial inflammation and oxidative stress leading to improved cardiac function (Fig. 4). Nevertheless, while many theories about the favourable mechanisms of SGLT2i in HF have been postulated, further experimental research is clearly warranted. Translational studies i.e. utilising human samples could help to better elucidate the mechanisms of SGLT2i in HF. In any case, SGLT2i may be better termed as gliflozins at least from a cardiovascular point of view, as they seem to have multiple additional effects.

Proposed myocardial mechanisms of Gliflozins (SGLT2 inhibitors) in HF. PKG, Proteinkinase G. Image heart: ©AdobeStock/Rogatnev
Change history
04 March 2022
The original version of this paper was updated due to the insertion of Open Access funding note.
References
Arnott C, Li Q, Kang A, Neuen BL, Bompoint S, Lam CSP, et al. Sodium-glucose cotransporter 2 inhibition for the prevention of cardiovascular events in patients with type 2 diabetes mellitus: A Systematic Review and Meta-Analysis. J Am Heart Assoc. 2020;9(3): e014908. https://doi.org/10.1161/JAHA.119.014908.
Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117–28. https://doi.org/10.1056/NEJMoa1504720.
Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377(7):644–57. https://doi.org/10.1056/NEJMoa1611925.
Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380(4):347–57. https://doi.org/10.1056/NEJMoa1812389.
Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393(10166):31–9. https://doi.org/10.1016/S0140-6736(18)32590-X.
Verma S, McMurray JJV, Cherney DZI. The metabolodiuretic promise of sodium-dependent glucose cotransporter 2 inhibition: the search for the sweet spot in heart failure. JAMA Cardiol. 2017;2(9):939–40. https://doi.org/10.1001/jamacardio.2017.1891.
Benham JL, Booth JE, Sigal RJ, Daskalopoulou SS, Leung AA, Rabi DM. Systematic review and meta-analysis: SGLT2 inhibitors, blood pressure and cardiovascular outcomes. Int J Cardiol Heart Vasc. 2021;33: 100725. https://doi.org/10.1016/j.ijcha.2021.100725.
Langslet G, Zinman B, Wanner C, Hantel S, Espadero RM, Fitchett D, et al. Cardiovascular outcomes and LDL-cholesterol levels in EMPA-REG OUTCOME((R)). Diab Vasc Dis Res. 2020;17(6):1479164120975256. https://doi.org/10.1177/1479164120975256.
Fitchett D, Inzucchi SE, Cannon CP, McGuire DK, Scirica BM, Johansen OE, et al. Empagliflozin reduced mortality and hospitalization for heart failure across the spectrum of cardiovascular risk in the EMPA-REG OUTCOME Trial. Circulation. 2019;139(11):1384–95. https://doi.org/10.1161/CIRCULATIONAHA.118.037778.
Fitchett D, Butler J, van de Borne P, Zinman B, Lachin JM, Wanner C, et al. Effects of empagliflozin on risk for cardiovascular death and heart failure hospitalization across the spectrum of heart failure risk in the EMPA-REG OUTCOME(R) trial. Eur Heart J. 2018;39(5):363–70. https://doi.org/10.1093/eurheartj/ehx511.
Fitchett D, Mcknight J, Lee J, George JT, Mattheus M, Woerle HJ et al., editors. Empagliflozin (EMPA) reduces heart failure irrespective of control of blood pressure (BP), low density lipoprotein cholesterol (LDL-C), and HbA1c. Diabetes; 2017: AMER DIABETES ASSOC 1701 N BEAUREGARD ST, ALEXANDRIA, VA 22311-1717 USA.
Januzzi J, Ferreira JP, Bohm M, Kaul S, Wanner C, Brueckmann M, et al. Empagliflozin reduces the risk of a broad spectrum of heart failure outcomes regardless of heart failure status at baseline. Eur J Heart Fail. 2019;21(3):386–8. https://doi.org/10.1002/ejhf.1419.
Wanner C, Lachin JM, Inzucchi SE, Fitchett D, Mattheus M, George J, et al. Empagliflozin and clinical outcomes in patients with type 2 diabetes mellitus, Established Cardiovascular Disease, and Chronic Kidney Disease. Circulation. 2018;137(2):119–29. https://doi.org/10.1161/CIRCULATIONAHA.117.028268.
McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381(21):1995–2008. https://doi.org/10.1056/NEJMoa1911303.
Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. 2020;383(15):1413–24. https://doi.org/10.1056/NEJMoa2022190.
Petrie MC, Verma S, Docherty KF, Inzucchi SE, Anand I, Belohlavek J, et al. Effect of dapagliflozin on worsening heart failure and cardiovascular death in patients with heart failure with and without diabetes. JAMA. 2020;323(14):1353–68. https://doi.org/10.1001/jama.2020.1906.
Anker SD, Butler J, Filippatos G, Khan MS, Marx N, Lam CSP, et al. Effect of empagliflozin on cardiovascular and renal outcomes in patients with heart failure by baseline diabetes status: results from the EMPEROR-Reduced trial. Circulation. 2021;143(4):337–49. https://doi.org/10.1161/CIRCULATIONAHA.120.051824.
Bhatt DL, Szarek M, Steg PG, Cannon CP, Leiter LA, McGuire DK, et al. Sotagliflozin in patients with diabetes and recent worsening heart failure. N Engl J Med. 2021;384(2):117–28. https://doi.org/10.1056/NEJMoa2030183.
Butler J, Usman MS, Khan MS, Greene SJ, Friede T, Vaduganathan M, et al. Efficacy and safety of SGLT2 inhibitors in heart failure: systematic review and meta-analysis. ESC Heart Fail. 2020;7(6):3298–309. https://doi.org/10.1002/ehf2.13169.
Nassif ME, Windsor SL, Tang F, Khariton Y, Husain M, Inzucchi SE, et al. Dapagliflozin effects on biomarkers, symptoms, and functional status in patients with heart failure with reduced ejection fraction: The DEFINE-HF Trial. Circulation. 2019;140(18):1463–76. https://doi.org/10.1161/CIRCULATIONAHA.119.042929.
Abraham WT, Lindenfeld J, Ponikowski P, Agostoni P, Butler J, Desai AS, et al. Effect of empagliflozin on exercise ability and symptoms in heart failure patients with reduced and preserved ejection fraction, with and without type 2 diabetes. Eur Heart J. 2021;42(6):700–10. https://doi.org/10.1093/eurheartj/ehaa943.
Petrie MC, Lee MM, Lang NN. EMPEROR-Reduced reigns while EMPERIAL whimpers. European Heart Journal. 2021;42(6):711–4. https://doi.org/10.1093/eurheartj/ehaa965.
Zannad F, Ferreira JP, Pocock SJ, Anker SD, Butler J, Filippatos G, et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet. 2020;396(10254):819–29. https://doi.org/10.1016/S0140-6736(20)31824-9.
Martinez FA, Serenelli M, Nicolau JC, Petrie MC, Chiang CE, Tereshchenko S, et al. Efficacy and safety of dapagliflozin in heart failure with reduced ejection fraction according to age: insights from DAPA-HF. Circulation. 2020;141(2):100–11. https://doi.org/10.1161/CIRCULATIONAHA.119.044133.
von Lewinski D, Rainer PP, Gasser R, Huber MS, Khafaga M, Wilhelm B, et al. Glucose-transporter-mediated positive inotropic effects in human myocardium of diabetic and nondiabetic patients. Metabolism. 2010;59(7):1020–8. https://doi.org/10.1016/j.metabol.2009.10.025.
Di Franco A, Cantini G, Tani A, Coppini R, Zecchi-Orlandini S, Raimondi L, et al. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: a new potential pharmacological target. Int J Cardiol. 2017;243:86–90. https://doi.org/10.1016/j.ijcard.2017.05.032.
Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14(1):83–90. https://doi.org/10.1111/j.1463-1326.2011.01517.x.
Kurosaki E, Ogasawara H. Ipragliflozin and other sodium-glucose cotransporter-2 (SGLT2) inhibitors in the treatment of type 2 diabetes: preclinical and clinical data. Pharmacol Ther. 2013;139(1):51–9. https://doi.org/10.1016/j.pharmthera.2013.04.003.
Lapuerta P, Zambrowicz B, Strumph P, Sands A. Development of sotagliflozin, a dual sodium-dependent glucose transporter 1/2 inhibitor. Diab Vasc Dis Res. 2015;12(2):101–10. https://doi.org/10.1177/1479164114563304.
Griffin M, Rao VS, Ivey-Miranda J, Fleming J, Mahoney D, Maulion C, et al. Empagliflozin in heart failure: diuretic and cardiorenal effects. Circulation. 2020;142(11):1028–39. https://doi.org/10.1161/CIRCULATIONAHA.120.045691.
Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab. 2013;15(9):853–62. https://doi.org/10.1111/dom.12127.
Sha S, Polidori D, Heise T, Natarajan J, Farrell K, Wang SS, et al. Effect of the sodium glucose co-transporter 2 inhibitor canagliflozin on plasma volume in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2014;16(11):1087–95. https://doi.org/10.1111/dom.12322.
Hallow KM, Helmlinger G, Greasley PJ, McMurray JJV, Boulton DW. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes Metab. 2018;20(3):479–87. https://doi.org/10.1111/dom.13126.
Verma S, McMurray JJV. SGLT2 inhibitors and mechanisms of cardiovascular benefit: a state-of-the-art review. Diabetologia. 2018;61(10):2108–17. https://doi.org/10.1007/s00125-018-4670-7.
Yanai H, Katsuyayama H. A possible mechanism for renoprotective effect of sodium-glucose cotransporter 2 inhibitor: elevation of erythropoietin production. J Clin Med Res. 2017;9(2):178–9. https://doi.org/10.14740/jocmr2857w.
Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Barsotti E, Clerico A, et al. Renal handling of ketones in response to sodium-glucose cotransporter 2 inhibition in patients with type 2 diabetes. Diabetes Care. 2017;40(6):771–6. https://doi.org/10.2337/dc16-2724.
Cohen ND, Gutman SJ, Briganti EM, Taylor AJ. Effects of empagliflozin treatment on cardiac function and structure in patients with type 2 diabetes: a cardiac magnetic resonance study. Intern Med J. 2019;49(8):1006–10. https://doi.org/10.1111/imj.14260.
Yasui A, Lee G, Hirase T, Kaneko T, Kaspers S, von Eynatten M, et al. Empagliflozin induces transient diuresis without changing long-term overall fluid balance in Japanese patients with type 2 diabetes. Diabetes Ther. 2018;9(2):863–71. https://doi.org/10.1007/s13300-018-0385-5.
Packer M, Anker SD, Butler J, Filippatos G, Ferreira JP, Pocock SJ, et al. Empagliflozin in patients with heart failure, reduced ejection fraction, and volume overload: EMPEROR-Reduced trial. J Am Coll Cardiol. 2021;77(11):1381–92. https://doi.org/10.1016/j.jacc.2021.01.033.
Joshi SS, Singh T, Newby DE, Singh J. Sodium-glucose co-transporter 2 inhibitor therapy: mechanisms of action in heart failure. Heart. 2021:heartjnl-2020-318060. doi:https://doi.org/10.1136/heartjnl-2020-318060.
Patel AR, Kuvin JT, Pandian NG, Smith JJ, Udelson JE, Mendelsohn ME, et al. Heart failure etiology affects peripheral vascular endothelial function after cardiac transplantation. J Am Coll Cardiol. 2001;37(1):195–200. https://doi.org/10.1016/s0735-1097(00)01057-3.
Chilton R, Tikkanen I, Cannon CP, Crowe S, Woerle HJ, Broedl UC, et al. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes Metab. 2015;17(12):1180–93. https://doi.org/10.1111/dom.12572.
Solini A, Giannini L, Seghieri M, Vitolo E, Taddei S, Ghiadoni L, et al. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: a pilot study. Cardiovasc Diabetol. 2017;16(1):138. https://doi.org/10.1186/s12933-017-0621-8.
Li H, Shin SE, Seo MS, An JR, Choi IW, Jung WK, et al. The anti-diabetic drug dapagliflozin induces vasodilation via activation of PKG and Kv channels. Life Sci. 2018;197:46–55. https://doi.org/10.1016/j.lfs.2018.01.032.
Scheen AJ. Effects of reducing blood pressure on cardiovascular outcomes and mortality in patients with type 2 diabetes: focus on SGLT2 inhibitors and EMPA-REG OUTCOME. Diabetes Res Clin Pract. 2016;121:204–14. https://doi.org/10.1016/j.diabres.2016.09.016.
Guo M, Ding J, Li J, Wang J, Zhang T, Liu C, et al. SGLT2 inhibitors and risk of stroke in patients with type 2 diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2018;20(8):1977–82. https://doi.org/10.1111/dom.13295.
Rangaswami J, Bhalla V, Blair JEA, Chang TI, Costa S, Lentine KL, et al. Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American Heart Association. Circulation. 2019;139(16):e840–78. https://doi.org/10.1161/cir.0000000000000664.
Heerspink HJL, Stefansson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. 2020;383(15):1436–46. https://doi.org/10.1056/NEJMoa2024816.
Wheeler DC, Stefansson BV, Batiushin M, Bilchenko O, Cherney DZI, Chertow GM, et al. The dapagliflozin and prevention of adverse outcomes in chronic kidney disease (DAPA-CKD) trial: baseline characteristics. Nephrol Dial Transplant. 2020;35(10):1700–11. https://doi.org/10.1093/ndt/gfaa234.
Cherney DZI, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. 2014;129(5):587–97. https://doi.org/10.1161/CIRCULATIONAHA.113.005081.
Mazer CD, Hare GMT, Connelly PW, Gilbert RE, Shehata N, Quan A, et al. Effect of empagliflozin on erythropoietin levels, iron stores, and red blood cell morphology in patients with type 2 diabetes mellitus and coronary artery disease. Circulation. 2020;141(8):704–7. https://doi.org/10.1161/CIRCULATIONAHA.119.044235.
Maruyama T, Takashima H, Oguma H, Nakamura Y, Ohno M, Utsunomiya K, et al. Canagliflozin improves erythropoiesis in diabetes patients with anemia of chronic kidney disease. Diabetes Technol Ther. 2019;21(12):713–20. https://doi.org/10.1089/dia.2019.0212.
Sano M, Goto S. Possible mechanism of hematocrit elevation by sodium glucose cotransporter 2 inhibitors and associated beneficial renal and cardiovascular effects. Circulation. 2019;139(17):1985–7. https://doi.org/10.1161/CIRCULATIONAHA.118.038881.
Inzucchi SE, Zinman B, Fitchett D, Wanner C, Ferrannini E, Schumacher M, et al. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA-REG OUTCOME trial. Diabetes Care. 2018;41(2):356–63. https://doi.org/10.2337/dc17-1096.
Zelniker TA, Braunwald E. Mechanisms of cardiorenal effects of sodium-glucose cotransporter 2 inhibitors: JACC state-of-the-art review. J Am Coll Cardiol. 2020;75(4):422–34. https://doi.org/10.1016/j.jacc.2019.11.031.
Bailey CJ. Uric acid and the cardio-renal effects of SGLT2 inhibitors. Diabetes Obes Metab. 2019;21(6):1291–8. https://doi.org/10.1111/dom.13670.
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2008;359(17):1811–21. https://doi.org/10.1056/NEJMra0800885.
Lytvyn Y, Perkins BA, Cherney DZ. Uric acid as a biomarker and a therapeutic target in diabetes. Can J Diabetes. 2015;39(3):239–46. https://doi.org/10.1016/j.jcjd.2014.10.013.
Zhao Y, Xu L, Tian D, Xia P, Zheng H, Wang L, et al. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: a meta-analysis of randomized controlled trials. Diabetes Obes Metab. 2018;20(2):458–62. https://doi.org/10.1111/dom.13101.
Devenny JJ, Godonis HE, Harvey SJ, Rooney S, Cullen MJ, Pelleymounter MA. Weight loss induced by chronic dapagliflozin treatment is attenuated by compensatory hyperphagia in diet-induced obese (DIO) rats. Obesity (Silver Spring). 2012;20(8):1645–52. https://doi.org/10.1038/oby.2012.59.
Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, Heise T, et al. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest. 2014;124(2):499–508. https://doi.org/10.1172/JCI72227.
Brown AJM, Gandy S, McCrimmon R, Houston JG, Struthers AD, Lang CC. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: the DAPA-LVH trial. Eur Heart J. 2020;41(36):3421–32. https://doi.org/10.1093/eurheartj/ehaa419.
Yamada K, Nakayama H, Yoshinobu S, Kawano S, Tsuruta M, Nohara M, et al. Effects of a sodium glucose co-transporter 2 selective inhibitor, ipragliflozin, on the diurnal profile of plasma glucose in patients with type 2 diabetes: a study using continuous glucose monitoring. J Diabetes Investig. 2015;6(6):699–707. https://doi.org/10.1111/jdi.12370.
Daniele G, Xiong J, Solis-Herrera C, Merovci A, Eldor R, Tripathy D, et al. Dapagliflozin enhances fat oxidation and ketone production in patients with type 2 diabetes. Diabetes Care. 2016;39(11):2036–41. https://doi.org/10.2337/dc15-2688.
Ferrannini E, Mark M, Mayoux E. CV Protection in the EMPA-REG OUTCOME trial: a “Thrifty Substrate” hypothesis. Diabetes Care. 2016;39(7):1108–14. https://doi.org/10.2337/dc16-0330.
Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME study? A unifying hypothesis J Diabetes Care. 2016;39(7):1115–22. https://doi.org/10.2337/dc16-0542.
Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human Heart failure. Circulation. 2016;133(8):706–16. https://doi.org/10.1161/CIRCULATIONAHA.115.017545.
Nielsen R, Moller N, Gormsen LC, Tolbod LP, Hansson NH, Sorensen J, et al. Cardiovascular effects of treatment with the ketone body 3-hydroxybutyrate in chronic heart failure patients. Circulation. 2019;139(18):2129–41. https://doi.org/10.1161/CIRCULATIONAHA.118.036459.
Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, et al. Catabolic Defect of branched-chain amino acids promotes heart failure. Circulation. 2016;133(21):2038–49. https://doi.org/10.1161/CIRCULATIONAHA.115.020226.
Kappel BA, Lehrke M, Schutt K, Artati A, Adamski J, Lebherz C, et al. Effect of empagliflozin on the metabolic signature of patients with type 2 diabetes mellitus and cardiovascular disease. Circulation. 2017;136(10):969–72. https://doi.org/10.1161/CIRCULATIONAHA.117.029166.
Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. 2019;73(15):1931–44. https://doi.org/10.1016/j.jacc.2019.01.056.
Lopaschuk GD, Verma S. Empagliflozin’s fuel hypothesis: not so soon. Cell Metab. 2016;24(2):200–2. https://doi.org/10.1016/j.cmet.2016.07.018.
Packer M. Molecular, cellular, and clinical evidence that sodium-glucose cotransporter 2 inhibitors act as neurohormonal antagonists when used for the treatment of chronic heart failure. J Am Heart Assoc. 2020;9(16): e016270. https://doi.org/10.1161/JAHA.120.016270.
Trum M, Wagner S, Maier LS, Mustroph J. CaMKII and GLUT1 in heart failure and the role of gliflozins. Biochim Biophys Acta Mol Basis Dis. 2020;1866(6): 165729. https://doi.org/10.1016/j.bbadis.2020.165729.
Avogaro A, Fadini GP, Del Prato S. Reinterpreting cardiorenal protection of renal sodium-glucose cotransporter 2 inhibitors via cellular life history programming. Diabetes Care. 2020;43(3):501–7. https://doi.org/10.2337/dc19-1410.
Verma S, Mazer CD, Yan AT, Mason T, Garg V, Teoh H, et al. Effect of empagliflozin on left ventricular mass in patients with type 2 diabetes mellitus and coronary artery disease: the EMPA-HEART CardioLink-6 randomized clinical trial. Circulation. 2019;140(21):1693–702. https://doi.org/10.1161/CIRCULATIONAHA.119.042375.
Verma S, Garg A, Yan AT, Gupta AK, Al-Omran M, Sabongui A, et al. Effect of empagliflozin on left ventricular mass and diastolic function in individuals with diabetes: an important clue to the EMPA-REG OUTCOME trial? Diabetes Care. 2016;39(12):e212–3. https://doi.org/10.2337/dc16-1312.
Hwang IC, Cho GY, Yoon YE, Park JJ, Park JB, Lee SP, et al. Different effects of SGLT2 inhibitors according to the presence and types of heart failure in type 2 diabetic patients. Cardiovasc Diabetol. 2020;19(1):69. https://doi.org/10.1186/s12933-020-01042-3.
Singh JSS, Mordi IR, Vickneson K, Fathi A, Donnan PT, Mohan M, et al. Dapagliflozin versus placebo on left ventricular remodeling in patients with diabetes and heart failure: the REFORM trial. Diabetes Care. 2020;43(6):1356–9. https://doi.org/10.2337/dc19-2187.
Shi L, Zhu D, Wang S, Jiang A, Li F. Dapagliflozin attenuates cardiac remodeling in mice model of cardiac pressure overload. Am J Hypertens. 2019;32(5):452–9. https://doi.org/10.1093/ajh/hpz016.
Lee TM, Chang NC, Lin SZ. Dapagliflozin, a selective SGLT2 inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic Biol Med. 2017;104:298–310. https://doi.org/10.1016/j.freeradbiomed.2017.01.035.
Lee HC, Shiou YL, Jhuo SJ, Chang CY, Liu PL, Jhuang WJ, et al. The sodium-glucose co-transporter 2 inhibitor empagliflozin attenuates cardiac fibrosis and improves ventricular hemodynamics in hypertensive heart failure rats. Cardiovasc Diabetol. 2019;18(1):45. https://doi.org/10.1186/s12933-019-0849-6.
Li C, Zhang J, Xue M, Li X, Han F, Liu X, et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol. 2019;18(1):15. https://doi.org/10.1186/s12933-019-0816-2.
Hsu JC, Wang CY, Su MM, Lin LY, Yang WS. Effect of empagliflozin on cardiac function, adiposity, and diffuse fibrosis in patients with type 2 diabetes mellitus. Sci Rep. 2019;9(1):15348. https://doi.org/10.1038/s41598-019-51949-5.
Matsutani D, Sakamoto M, Kayama Y, Takeda N, Horiuchi R, Utsunomiya K. Effect of canagliflozin on left ventricular diastolic function in patients with type 2 diabetes. Cardiovasc Diabetol. 2018;17(1):73. https://doi.org/10.1186/s12933-018-0717-9.
Soga F, Tanaka H, Tatsumi K, Mochizuki Y, Sano H, Toki H, et al. Impact of dapagliflozin on left ventricular diastolic function of patients with type 2 diabetic mellitus with chronic heart failure. Cardiovasc Diabetol. 2018;17(1):132. https://doi.org/10.1186/s12933-018-0775-z.
Shim CY, Seo J, Cho I, Lee CJ, Cho IJ, Lhagvasuren P, et al. Randomized, controlled trial to evaluate the effect of dapagliflozin on left ventricular diastolic function in patients with type 2 diabetes mellitus: the IDDIA trial. Circulation. 2021;143(5):510–2. https://doi.org/10.1161/CIRCULATIONAHA.120.051992.
Santos-Gallego CG, Vargas-Delgado AP, Requena-Ibanez JA, Garcia-Ropero A, Mancini D, Pinney S, et al. Randomized trial of empagliflozin in nondiabetic patients with heart failure and reduced ejection fraction. J Am Coll Cardiol. 2021;77(3):243–55. https://doi.org/10.1016/j.jacc.2020.11.008.
Lee MMY, Brooksbank KJM, Wetherall K, Mangion K, Roditi G, Campbell RT, et al. Effect of empagliflozin on left ventricular volumes in patients with type 2 diabetes, or prediabetes, and heart failure with reduced ejection fraction (SUGAR-DM-HF). Circulation. 2021;143(6):516–25. https://doi.org/10.1161/CIRCULATIONAHA.120.052186.
Pabel S, Wagner S, Bollenberg H, Bengel P, Kovacs A, Schach C, et al. Empagliflozin directly improves diastolic function in human heart failure. Eur J Heart Fail. 2018;20(12):1690–700. https://doi.org/10.1002/ejhf.1328.
Connelly KA, Zhang Y, Visram A, Advani A, Batchu SN, Desjardins JF, et al. Empagliflozin improves diastolic function in a nondiabetic rodent model of heart failure with preserved ejection fraction. JACC Basic Transl Sci. 2019;4(1):27–37. https://doi.org/10.1016/j.jacbts.2018.11.010.
Habibi J, Aroor AR, Sowers JR, Jia G, Hayden MR, Garro M, et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc Diabetol. 2017;16(1):9. https://doi.org/10.1186/s12933-016-0489-z.
Hammoudi N, Jeong D, Singh R, Farhat A, Komajda M, Mayoux E, et al. Empagliflozin improves left ventricular diastolic dysfunction in a genetic model of type 2 diabetes. Cardiovasc Drugs Ther. 2017;31(3):233–46. https://doi.org/10.1007/s10557-017-6734-1.
Moellmann J, Klinkhammer BM, Droste P, Kappel B, Haj-Yehia E, Maxeiner S, et al. Empagliflozin improves left ventricular diastolic function of db/db mice. Biochim Biophys Acta Mol Basis Dis. 2020;1866(8): 165807. https://doi.org/10.1016/j.bbadis.2020.165807.
Byrne NJ, Parajuli N, Levasseur JL, Boisvenue J, Beker DL, Masson G, et al. Empagliflozin prevents worsening of cardiac function in an experimental model of pressure overload-induced heart failure. JACC Basic Transl Sci. 2017;2(4):347–54. https://doi.org/10.1016/j.jacbts.2017.07.003.
Rau M, Thiele K, Hartmann NK, Schuh A, Altiok E, Mollmann J, et al. Empagliflozin does not change cardiac index nor systemic vascular resistance but rapidly improves left ventricular filling pressure in patients with type 2 diabetes: a randomized controlled study. Cardiovasc Diabetol. 2021;20(1):6. https://doi.org/10.1186/s12933-020-01175-5.
Kolijn D, Pabel S, Tian Y, Lodi M, Herwig M, Carrizzo A, et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Galpha oxidation. Cardiovasc Res. 2021;117(2):495–507. https://doi.org/10.1093/cvr/cvaa123.
Xue M, Li T, Wang Y, Chang Y, Cheng Y, Lu Y, et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin Sci (Lond). 2019;133(15):1705–20. https://doi.org/10.1042/CS20190585.
Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263–71. https://doi.org/10.1016/j.jacc.2013.02.092.
Hamdani N, Bishu KG, von Frieling-Salewsky M, Redfield MM, Linke WA. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res. 2013;97(3):464–71. https://doi.org/10.1093/cvr/cvs353.
Pabel S, Hamdani N, Sossalla S. A mechanistic rationale for the investigation of sodium-glucose co-transporter 2 inhibitors in heart failure with preserved ejection fraction. Letter regarding the article ‘Baseline characteristics of patients with heart failure with preserved ejection fraction in the EMPEROR-Preserved trial’. Eur J Heart Fail. 2020. doi:https://doi.org/10.1002/ejhf.2091.
Murphy SP, Ibrahim NE, Januzzi JL Jr. Heart failure with reduced ejection fraction: a review. JAMA. 2020;324(5):488–504. https://doi.org/10.1001/jama.2020.10262.
Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198–205. https://doi.org/10.1038/415198a.
Wagner S, Maier LS, Bers DM. Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ Res. 2015;116(12):1956–70. https://doi.org/10.1161/circresaha.116.304678.
Fischer TH, Maier LS, Sossalla S. The ryanodine receptor leak: how a tattered receptor plunges the failing heart into crisis. Heart Fail Rev. 2013;18(4):475–83. https://doi.org/10.1007/s10741-012-9339-6.
Baartscheer A, Schumacher CA, Wust RC, Fiolet JW, Stienen GJ, Coronel R, et al. Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia. 2017;60(3):568–73. https://doi.org/10.1007/s00125-016-4134-x.
Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R, Fiolet JW. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57(4):1015–24.
Trum M, Riechel J, Lebek S, Pabel S, Sossalla ST, Hirt S, et al. Empagliflozin inhibits Na(+) /H(+) exchanger activity in human atrial cardiomyocytes. ESC Heart Fail. 2020;7(6):4429–37. https://doi.org/10.1002/ehf2.13024.
Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A, et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia. 2018;61(3):722–6. https://doi.org/10.1007/s00125-017-4509-7.
Sossalla S, Wagner S, Rasenack EC, Ruff H, Weber SL, Schondube FA, et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts–role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;45(1):32–43. https://doi.org/10.1016/j.yjmcc.2008.03.006.
Sossalla S, Maurer U, Schotola H, Hartmann N, Didie M, Zimmermann WH, et al. Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na(+) current. Basic Res Cardiol. 2011;106(2):263–72. https://doi.org/10.1007/s00395-010-0136-x.
Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010;107(9):1150–61. https://doi.org/10.1161/CIRCRESAHA.110.220418.
Bertero E, Prates Roma L, Ameri P, Maack C. Cardiac effects of SGLT2 inhibitors: the sodium hypothesis. Cardiovasc Res. 2018;114(1):12–8. https://doi.org/10.1093/cvr/cvx149.
Philippaert K, Kalyaanamoorthy S, Fatehi M, Long W, Soni S, Byrne NJ, et al. Cardiac late sodium channel current is a molecular target for the sodium/glucose cotransporter 2 inhibitor empagliflozin. Circulation. 2021;143(22):2188–204. https://doi.org/10.1161/CIRCULATIONAHA.121.053350.
Dybkova N, Ahmad S, Pabel S, Tirilomis P, Hartmann N, Fischer TH, et al. Differential regulation of sodium channels as a novel proarrhythmic mechanism in the human failing heart. Cardiovasc Res. 2018;114(13):1728–37. https://doi.org/10.1093/cvr/cvy152.
Mustroph J, Wagemann O, Lucht CM, Trum M, Hammer KP, Sag CM, et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018;5(4):642–8. https://doi.org/10.1002/ehf2.12336.
Pabel S, Mustroph J, Stehle T, Lebek S, Dybkova N, Keyser A, et al. Dantrolene reduces CaMKIIdeltaC-mediated atrial arrhythmias. Europace. 2020;22(7):1111–8. https://doi.org/10.1093/europace/euaa079.
Mustroph J, Neef S, Maier LS. CaMKII as a target for arrhythmia suppression. Pharmacol Ther. 2017;176:22–31. https://doi.org/10.1016/j.pharmthera.2016.10.006.
Mustroph J, Sag CM, Bahr F, Schmidtmann AL, Gupta SN, Dietz A, et al. Loss of CASK accelerates heart failure development. Circ Res. 2021. https://doi.org/10.1161/CIRCRESAHA.120.318170.
Pabel S, Reetz F, Dybkova N, Shomroni O, Salinas G, Mustroph J, et al. Long-term effects of empagliflozin on excitation-contraction-coupling in human induced pluripotent stem cell cardiomyocytes. J Mol Med (Berl). 2020. https://doi.org/10.1007/s00109-020-01989-6.
Chung YJ, Park KC, Tokar S, Eykyn TR, Fuller W, Pavlovic D, et al. Off-target effects of SGLT2 blockers: empagliflozin does not inhibit Na+/H+ exchanger-1 or lower [Na+]i in the heart. Cardiovasc Res. 2020. https://doi.org/10.1093/cvr/cvaa323.
Zhazykbayeva S, Pabel S, Mugge A, Sossalla S, Hamdani N. The molecular mechanisms associated with the physiological responses to inflammation and oxidative stress in cardiovascular diseases. Biophys Rev. 2020;12(4):947–68. https://doi.org/10.1007/s12551-020-00742-0.
Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 2016;4(4):312–24. https://doi.org/10.1016/j.jchf.2015.10.007.
Yaribeygi H, Atkin SL, Butler AE, Sahebkar A. Sodium-glucose cotransporter inhibitors and oxidative stress: an update. J Cell Physiol. 2019;234(4):3231–7. https://doi.org/10.1002/jcp.26760.
Ye Y, Bajaj M, Yang HC, Perez-Polo JR, Birnbaum Y. SGLT-2 Inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. Further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc Drugs Ther. 2017;31(2):119–32. https://doi.org/10.1007/s10557-017-6725-2.
Tahara A, Kurosaki E, Yokono M, Yamajuku D, Kihara R, Hayashizaki Y, et al. Effects of sodium-glucose cotransporter 2 selective inhibitor ipragliflozin on hyperglycaemia, oxidative stress, inflammation and liver injury in streptozotocin-induced type 1 diabetic rats. J Pharm Pharmacol. 2014;66(7):975–87. https://doi.org/10.1111/jphp.12223.
Tahara A, Kurosaki E, Yokono M, Yamajuku D, Kihara R, Hayashizaki Y, et al. Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur J Pharmacol. 2013;715(1–3):246–55. https://doi.org/10.1016/j.ejphar.2013.05.014.
Yurista SR, Sillje HHW, Oberdorf-Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, et al. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. 2019;21(7):862–73. https://doi.org/10.1002/ejhf.1473.
Juni RP, Kuster DWD, Goebel M, Helmes M, Musters RJP, van der Velden J, et al. Cardiac microvascular endothelial enhancement of cardiomyocyte function is impaired by inflammation and restored by empagliflozin. JACC Basic Transl Sci. 2019;4(5):575–91. https://doi.org/10.1016/j.jacbts.2019.04.003.
Mizuno M, Kuno A, Yano T, Miki T, Oshima H, Sato T, et al. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol Rep. 2018;6(12): e13741. https://doi.org/10.14814/phy2.13741.
Xu C, Wang W, Zhong J, Lei F, Xu N, Zhang Y, et al. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem Pharmacol. 2018;152:45–59. https://doi.org/10.1016/j.bcp.2018.03.013.
Packer M. Autophagy stimulation and intracellular sodium reduction as mediators of the cardioprotective effect of sodium-glucose cotransporter 2 inhibitors. Eur J Heart Fail. 2020;22(4):618–28. https://doi.org/10.1002/ejhf.1732.
Packer M. Do sodium-glucose co-transporter-2 inhibitors prevent heart failure with a preserved ejection fraction by counterbalancing the effects of leptin? A novel hypothesis Diabetes Obes Metab. 2018;20(6):1361–6. https://doi.org/10.1111/dom.13229.
Lau DC, Dhillon B, Yan H, Szmitko PE, Verma S. Adipokines: molecular links between obesity and atheroslcerosis. Am J Physiol Heart Circ Physiol. 2005;288(5):H2031-41. https://doi.org/10.1152/ajpheart.01058.2004.
Patel VB, Shah S, Verma S, Oudit GY. Epicardial adipose tissue as a metabolic transducer: role in heart failure and coronary artery disease. Heart Fail Rev. 2017;22(6):889–902. https://doi.org/10.1007/s10741-017-9644-1.
Sattar N, McLaren J, Kristensen SL, Preiss D, McMurray JJ. SGLT2 Inhibition and cardiovascular events: why did EMPA-REG outcomes surprise and what were the likely mechanisms? Diabetologia. 2016;59(7):1333–9. https://doi.org/10.1007/s00125-016-3956-x.
Garvey WT, Van Gaal L, Leiter LA, Vijapurkar U, List J, Cuddihy R, et al. Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism. 2018;85:32–7. https://doi.org/10.1016/j.metabol.2018.02.002.
Sato T, Aizawa Y, Yuasa S, Kishi S, Fuse K, Fujita S, et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc Diabetol. 2018;17(1):6. https://doi.org/10.1186/s12933-017-0658-8.
Maiese K, Chong ZZ, Hou J, Shang YC. Erythropoietin and oxidative stress. Curr Neurovasc Res. 2008;5(2):125–42. https://doi.org/10.2174/156720208784310231.
Funding
Open Access funding enabled and organized by Projekt DEAL. S. Pabel and S. Sossalla are funded by the Else-Kröner-Fresenius Stiftung. S. Pabel is funded by the German Society of Internal Medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
N. Hamdani has nothing to report. S. Pabel receive speakers/consultancy honoraria from AstraZeneca. S. Sossalla and M. Luedde receive speaker’s/consultancy honoraria from Boehringer Ingelheim Pharma GmbH and AstraZeneca.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Translational Research in Heart Failure
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pabel, S., Hamdani, N., Luedde, M. et al. SGLT2 Inhibitors and Their Mode of Action in Heart Failure—Has the Mystery Been Unravelled?. Curr Heart Fail Rep 18, 315–328 (2021). https://doi.org/10.1007/s11897-021-00529-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-021-00529-8