
Abstract
Purpose of Review
Fabry Disease (FD) is a rare lysosomal storage disorder characterised by multiorgan accumulation of glycosphingolipid due to deficiency in the enzyme α-galactosidase A. Cardiac sphingolipid accumulation triggers various types of arrhythmias, predominantly ventricular arrhythmia, bradyarrhythmia, and atrial fibrillation. Arrhythmia is likely the primary contributor to FD mortality with sudden cardiac death, the most frequent cardiac mode of death. Traditionally FD was seen as a storage cardiomyopathy triggering left ventricular hypertrophy, diastolic dysfunction, and ultimately, systolic dysfunction in advanced disease. The purpose of this review is to outline the current evidence exploring novel mechanisms underlying the arrhythmia substrate.
Recent Findings
There is growing evidence that FD cardiomyopathy is a primary arrhythmic disease with each stage of cardiomyopathy (accumulation, hypertrophy, inflammation, and fibrosis) contributing to the arrhythmia substrate via various intracellular, extracellular, and environmental mechanisms. It is therefore important to understand how these mechanisms contribute to an individual’s risk of arrhythmia in FD.
Summary
In this review, we outline the epidemiology of arrhythmia, pathophysiology of arrhythmogenesis, risk stratification, and cardiac therapy in FD. We explore how advances in conventional cardiac investigations performed in FD patients including 12-lead electrocardiography, transthoracic echocardiography, and cardiac magnetic resonance imaging have enabled early detection of pro-arrhythmic substrate. This has allowed for appropriate risk stratification of FD patients. This paves the way for future work exploring the development of therapeutic initiatives and risk prediction models to reduce the burden of arrhythmia.
Similar content being viewed by others


Introduction
Fabry Disease (FD) in an X-linked inherited lysosomal storage disorder due to pathogenic variants in the galactosidase-α (GLA) gene, resulting in deficiency of the enzyme α-galactosidase A (α-GAL A) [1, 2]. This leads to widespread intracellular lysosomal accumulation of glycosphingolipids, namely, globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3), in multiple organs. Accumulation causes intracellular and extracellular dysfunction by direct and indirect effects that lead to predominantly cardiac, renal, and cerebrovascular manifestations with significant associated morbidity and mortality [3].
Patients exhibit a spectrum of phenotypes depending on the mutation within the GLA gene, of which there are many. Nonsense, missense variants, and premature stop codons that lead to an (almost) complete loss of function of α-GAL A enzyme are mostly associated with classic, early-onset FD characterised by severe multi-organ disease beginning usually in childhood. Missense variants that reduce α-GAL A activity are mostly associated with predominantly single-organ involvement, particularly cardiac, presenting usually later in adult life. Due to the X-linked nature of the disease, it was presumed that heterozygous females were obligate carriers. However, it is now recognised that females exhibit similar degree of cardiac involvement, albeit later in life [4].
Cardiac sphingolipid accumulation takes place in all cardiac cell types, including cardiomyocytes, conduction system cells, fibroblasts, smooth muscle, and endothelial cells [5]. The classic feature of FD cardiomyopathy is the development of left ventricular hypertrophy (LVH), which is triggered by sphingolipid accumulation, but the mechanism is not understood. Traditionally, FD has been considered a simple storage cardiomyopathy in which LVH leads to diastolic and systolic left ventricular (LV) dysfunction, presenting with breathlessness and heart failure. There is, however, increasing evidence that arrhythmia and sudden cardiac death (SCD) may be more important. This review aims to outline types of arrhythmias, epidemiology of arrhythmia, and SCD in FD; summarise the evidence on pathogenesis; identify potential methods for risk stratification; and consider therapeutic options.
Incidence and Prevalence of Arrhythmia
Tachyarrhythmia and Sudden Cardiac Death
Tachyarrhythmia is prevalent in FD and may be supraventricular or ventricular in origin. Atrial fibrillation (AF) is the most frequently reported supraventricular arrhythmia, and regular narrow complex tachycardias are less common, despite shortening of the PR interval [6]. Ventricular tachycardia (sustained and non-sustained) and ventricular fibrillation are the most frequent ventricular arrhythmias (VA), with overall prevalence between 13 and 18% [7,8,9,10,11]. Figure 1 illustrates the non-sustained ventricular tachycardia (NSVT) detected on an implanted loop recorder (ILR) in a patient with FD. The incidence of SCD varies between 0.5 and 3% (follow-up range 1.2–8 years) [7, 12, 13, 14•, 15••, 16], which is much higher than the rate in both the general population (0.03–0.1%) and patients with sarcomeric hypertrophic cardiomyopathy (0.5–1%) [17, 18]. Details of studies describing incidence and prevalence of VA and SCD are summarised in Table 1. Predictors of VA and SCD include advancing age, an annual increase in myocardial fibrosis, severity of FD measured by the Mainz Severity Score Index (MSSI), and prolongation of QRS duration on electrocardiogram (ECG) [8, 12]. Single-centre studies have demonstrated a higher prevalence of VA in males than females [7, 19]. Risk factors for VA are illustrated in Fig. 2. No change in frequency of tachyarrhythmia has been demonstrated solely according to mutation-type in FD [20].

Non-sustained ventricular tachycardia (NSVT) detected on an implantable loop recorder (ILR) in a patient with FD

Risk factors for VA
Bradyarrhythmia
Bradyarrhythmia is more common than tachyarrhythmia in FD [21••]. In a recent systematic review, weighted estimates of event rate bradyarrhythmia were 10% (pooled median follow-up time 4.5 years) [21••]. Bradyarrhythmia that have been reported in FD patients include sinus node disease (specifically, symptomatic bradycardia, sinus pauses, and chronotropic incompetence) and atrioventricular (AV) nodal disease (including first-, second-, and third-degree AV block) [22]. In a study of 29 adults with FD (82% male; mean age 43 ± 11 years), the prevalence of resting bradycardia was reported as 72% (of which 7% had shortened PR interval, 7% prolonged PR interval, and 86% normal) [23], which may be attributed to early autonomic dysfunction and chronotropic incompetence. There is even data suggesting that sinus bradycardia may ensue in childhood with a study of 26 children with FD (46% male; mean age 9.7 ± 3.8 years) demonstrating that 23% had a resting bradycardia on 12-lead ECG [24]. Bradyarrhythmia relating to clinically significant sinus node disease (severe sinus bradycardia and sinus pauses) has been reported in up to 12% [25]. Bradyarrhythmia requiring permanent pacemaker implantation (PPM) varies between 2.5 and 11% (follow-up range 1.2–8 years) [7, 9, 12, 13, 16, 26,27,28,29]. Figure 3 illustrates an asymptomatic daytime pause on ILR in a patient with FD. Details of studies describing incidence and prevalence of bradyarrhythmia are summarised in Table 2. Risk factors associated with bradyarrhythmia include increasing age, left atrial (LA) dysfunction, lower resting heart rate, prolonged PR and QRS duration, use of beta-blocker therapy, and impaired LV global longitudinal strain (GLS) [25, 26, 28]. Risk factors for bradycardia are illustrated in Fig. 4. No changes in frequency of bradycardia have been demonstrated according to gender or mutation-type in FD [20].

An asymptomatic daytime pause on ILR in a patient with FD

Risk factors for bradycardia
Atrial Fibrillation
AF is a supraventricular arrhythmia that involves irregular and disorganised atrial conduction arising due to electrophysiological disruption usually originating from the pulmonary veins. AF is prevalent in FD with reported figures between 3 and 21% [9, 25, 26]. Incidence of AF in FD from longitudinal data is reported between 3 and 31% (follow-up range 1.2–8 years) [7, 9, 12, 13, 16, 26,27,28,29]. The incidence is likely to be higher with numerous mechanisms in FD contributing to the structural, functional, and electrical substrate that is required for AF to initiate and persist. LVH raises left ventricular end-diastolic pressure which transmits to the LA, resulting in LA dilatation and impaired LA strain (see Fig. 5) [30]. LA strain impairment predicts new onset and recurrence of AF in the general population [31] and has also been found in patients with FD, with initiation of enzyme replacement therapy (ERT) improving function [32]. The same study demonstrated a higher incidence of AF and stroke in the cohort with impaired LA strain. Most of the patients had diastolic function without LVH suggesting that direct atrial sphingolipid accumulation may contribute to LA dilatation and impaired strain rather than passive effects of the LV [28].

LVH raises left ventricular end-diastolic pressure which transmits to the LA, resulting in LA dilatation and impaired LA strain
Limitations of the Data on Arrhythmia
The incidence and prevalence of both tachyarrhythmia, bradyarrhythmia, and AF are variably reported because the detection rate depends in part on the duration of monitoring. Data from small single-centre cohorts using continuous monitoring with advanced disease have detected a much higher rate of arrhythmia requiring intervention than studies using intermittent 24-h Holter monitoring [13, 33]. This is important because the annual Holter monitoring recommended by current guidelines is likely to under-estimate the frequency of arrhythmia [34]. The variation in reporting of arrhythmia is also likely to be attributable to differences in the cohorts studied, in terms of age, sex, genotype, and classical or non-classical phenotype. Studies suggest a greater number of cardiac events in males with classical FD compared with males with non-classical disease or females. However, these studies do not specify whether these relate specifically to arrhythmia [35]. Within these limitations, however, it is interesting to reflect on the possibility that FD patients seem to have a much higher risk of VA and SCD than sarcomeric hypertrophic cardiomyopathy, which is the paradigm for arrhythmogenesis and SCD [36]. Future studies using longer-term monitoring with implanted loop recorders have completed recruitment and outcomes are awaited [37].
Pathogenesis
α-GAL A is a lysosomal enzyme which catalyses the cleavage of terminal galactose from glycosphingolipids. Absence or deficiency of α-GAL A leads to a failure to catabolise galactosyl moieties [38], which contribute in turn by direct and indirect effects on generation of arrhythmia.
Direct Effects of α-GAL-A and Arrhythmia.
Cardiac arrhythmias are common and can occur before other evidence of FD cardiomyopathy. Histology of myocardial biopsies containing elements of conduction tissue clearly demonstrated sphingolipid deposition that occurs both early and consistently in most FD patients [10]. This is supported by the fact that ECG abnormalities are one of the earliest signs of cardiac involvement in children and adults. Shortening of P-wave duration and QRS width indicative of accelerated depolarisation intervals have been demonstrated in FD patients without LVH. These patients also have QTc prolongation and dispersion suggesting increased repolarisation time [39]. Studies have demonstrated crista terminalis and AV nodal glycosphingolipid accumulation which may accelerate atrial conduction and account for shortening of the P-wave duration [40, 41]. Prolongation of QTc and QTc dispersion are known to predispose to life-threatening arrhythmia, usually ventricular in origin [42]. These changes reflect the direct effect of sphingolipid accumulation in conduction tissue, resulting in altered repolarisation time and dispersion that explains the occurrence of arrhythmia before the onset of LVH, LA dilatation, or impaired LV function. Advanced techniques in digital ECG analysis are increasingly recognised as improving our ability to detect cardiac involvement in FD at an earlier stage than changes on a 12-lead ECG alone [43].
Primary Effects
Whilst intracellular sphingolipid deposition within conduction tissue may have direct pro-arrhythmic effects, accumulation can trigger other deleterious processes either within the cell or because of ‘over-spill’ into the extracellular environment. Firstly, lysosomes are directly involved in cellular autophagy (Fig. 6). A key regulator of autophagy-lysosomal fusion and mitochondrial function is the mTOR-dependent signalling pathway [44]. mTOR is located on lysosomal surfaces and upon activation through amino acid accumulation, triggers autophagy [45]. Sphingolipid accumulation has been found to disrupt mTOR activation in FD cardiac fibroblasts and thereby inhibits autophagy-lysosome fusion [46]. mTOR is also involved in mitochondrial metabolism by promoting translation of mitochondrial-related proteins, which is adversely affected by lysosomal accumulation [47]. Reduced autophagy and impaired mitochondrial metabolism may play a vital role in inducing structural and functional changes within cardiac cells that could directly trigger arrhythmia [48]. Secondly, endomyocardial biopsies of FD patients demonstrate perinuclear vacuoles of glycosphingolipid. On electron microscopy, these vacuoles had the appearance of single-membrane-bound vesicles which displaced cardiac myofibrils to the periphery of the cell and induced myofibrillolysis. Cardiomyocyte area and percentage occupied by these vacuoles were similar, indicating that increased cardiomyocyte size may be explained by these vacuoles. The same study demonstrated that in FD patients with abnormal tissue Doppler indices on transthoracic echocardiography (TTE) indicative of diastolic dysfunction, in-vitro experiments on the cardiomyocytes of these patients also demonstrated stiffening [49]. Myofibrillolysis, vacuolation, and mitochondrial dysfunction may each contribute as trophic stimuli for hypertrophy [50], which forms a substrate for arrhythmia. Thirdly, data emerging from studies modelling FD in induced pluripotent stem cells (iPSC) have identified pro-arrhythmic intracellular mechanisms. A recent study characterised the structural and electrophysiological properties of FD patient-derived IPSC-derived ventricular cardiomyocytes with iPSCs obtained from patients with FD [51]. This study demonstrated that Fabry iPSC-derived cardiomyocytes had a higher spontaneous action potential (AP) frequency, shorter AP duration with an increased peak sodium current density, and upstroke velocity, suggesting increased excitability. Calcium transients had a greater amplitude and reduction in peak width duration, which indicate increased excitability and disruption to intracellular calcium handling. It has previously been shown that ion channel expression is affected by Gb3 accumulation in neuronal and endothelial cells [52, 53]. Therefore, α-GAL A deficiency and the accumulation of glycosphingolipid may alter the expression of calcium and sodium ion channels, causing severe effects on the electrophysiological properties of the cell. These early cellular changes likely cause an increased propensity for VA during disease development [51]. There is further scope using in-vivo and ex-vivo models for evaluation of arrhythmia used in combination in FD [54].

Pathophysiology of arrhythmia in Fabry Disease: Primary, secondary, and tertiary mechanisms
Secondary Effects
-
(i)
Ischaemia, LVH, and Scar
Cardiac sphingolipid accumulation only accounts for approximately 5% of the increase in ventricular mass in FD and is not associated with an increase in interstitial space [55]. Sphingolipid accumulation can be measured by detection of reduced T1 mapping times on cardiac magnetic resonance imaging (MRI) and is known to precede the development of LVH. How this triggers myocyte hypertrophy is not known. Of interest, smooth muscle cell accumulation within myocardial vasculature initiates structural and functional changes that cause microvascular and macrovascular ischaemia (Fig. 6). Stress perfusion mapping by cardiac MRI confirmed ischaemia in the absence of epicardial coronary stenosis in FD and showed this pre-dated the development of LVH in the affected myocardium [56]. FD is also characterised by the development of interstitial myocardial fibrosis, quantified by volume of extravascular, extracellular deposition of gadolinium contrast on late gadolinium enhancement (LGE) on cardiac MRI [57, 58]. LGE identifies scar, predominantly in the basal inferolateral LV wall (see Fig. 7), that can occur in women before development of hypertrophy, but seems only to develop in response to a hypertrophic stimulus in men [59]. Ischaemia, LVH, and interstitial scar are established factors in both inherited cardiomyopathies and acquired disease for arrhythmogenesis.
Fig. 7 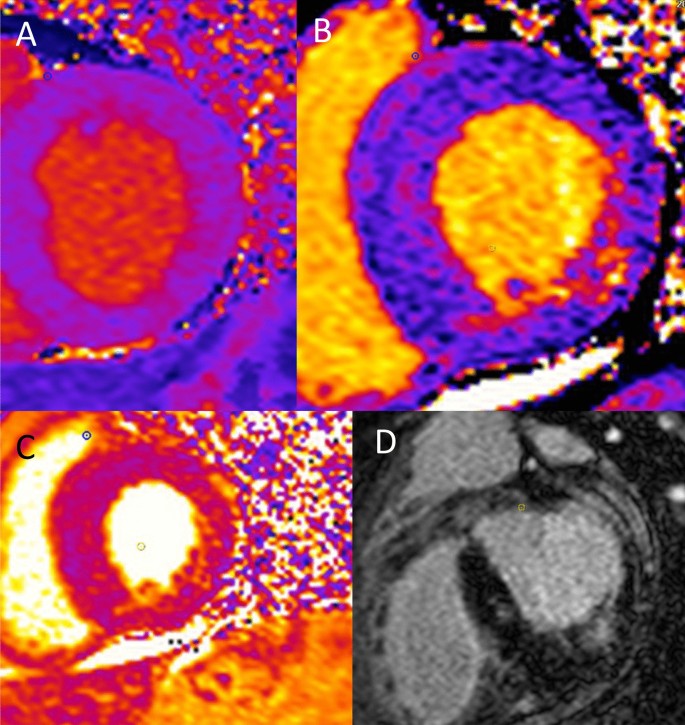
LGE identifies scar, predominantly in the basal inferolateral LV wall that can occur in women before development of hypertrophy, but seems only to develop in response to a hypertrophic stimulus in men
-
(ii)
Inflammation and Arrhythmia
Disorders of lysosomal storage are thought to have a significant effect on the immune system [60]. Lysosomal function involves maintenance of a normal immune system via antigen presentation, phagocytosis, and release of pro-inflammatory mediators [61, 62]. Specifically in FD, glycosphingolipid may cause damage-associated molecular pattern production which can induce cell apoptosis and pro-inflammatory cytokine secretion, which has been demonstrated when adding Gb3 to normal cells [63]. Leukocytes and endothelial cells in patients with FD also show signs of inflammatory activation, including generation of reactive oxygen species. A patient-specific inflammatory response may explain the significant intra-familial variability on clinical manifestations in FD. In a large study of adults with FD who underwent endomyocardial biopsy, histological evidence of myocarditis was demonstrated in 56% of patients with the presence of predominantly CD3 + T lymphocytes. Higher CD3 + T lymphocyte number correlated with increased wall thickness. In those without LVH, myocarditis was demonstrated in 38% of patients. The presence of myocarditis was associated with arrhythmia detected on Holter and ECG [64].
Multiparametric cardiac MRI studies using LGE and T1/T2 mapping have enabled non-invasive detection and quantification of myocardial inflammation, consistent with these biopsy studies. Scar in the inferolateral wall detected on LGE imaging has conventionally been considered either to be replacement or reparative fibrosis. In FD however, elevated T2 scores that reflect oedema have been found not only to co-locate to the inferolateral scar on LGE (Fig. 7) but also correlate closely to increased high-sensitivity troponin and pro-inflammatory cytokines such as interleukin 6 [65, 66]. T2 elevation in the basal inferolateral wall suggestive of focal myocardial oedema has been associated with the ECG abnormalities that may provoke arrhythmia including abnormal PR interval, bundle branch block, and QTc prolongation [67]. That this oedema and troponin release is related to inflammation has been confirmed using 18-fludeoxyglucose (18-FDG) positron emission tomography (PET). In a study of 13 adults with FD, FDG-PET/cardiac MRI confirmed that focal LGE with increased T2 values corresponded to focal, increased 18-FDG uptake and associated high-sensitivity troponin elevation [68]. Moreover, in a study of female adults without LVH who underwent 18-FDG PET/cardiac MRI, increased focal 18-FDG myocardial uptake was detected in 54% of patients of whom only 15% had LGE on cardiac MRI. Uptake of 18-FDG uptake correlated with impaired LV GLS but in this ‘early group’, high-sensitivity troponin levels were normal. A further study in females with FD demonstrated an association between focal 18-FDG uptake and pseudonormalisation of T1 values, indicating this to be another marker of inflammation at the intermediate stage before the development of fibrosis associated with cell death [69]. These findings suggest that inflammation may be an insidious process that occurs before LVH and LGE and is not inevitably associated with myocyte cell death, so may be a modifiable target for therapy [70].
-
(iii)
Multiorgan Involvement
Renal sphingolipid accumulation is prevalent and prominent in podocytes [71]. This manifests as progressive renal dysfunction including microalbuminuria, proteinuria, and progressive decline in estimated glomerular filtration rate [72]. In advanced stages, FD patients may require long-term dialysis and assessment for renal transplantation [73]. Renal dysfunction predisposes to various arrhythmias including AF, bradyarrhythmia, VA, and SCD in the general population and its high prevalence in FD is likely to be a contributory mechanism to the arrhythmia substrate (see Fig. 6). The effects of electrolyte imbalance associated with renal dysfunction (namely potassium) as well as rapid fluctuation in electrolyte levels and haemodynamic shifts during dialysis further contributes to this increase in risk in the later stages of disease [74].

Risk Stratification
Given the susceptibility to arrhythmia in patients with FD across all spectrums of the disease, risk stratification is imperative to establish those who would benefit from therapy. In the case of sarcomeric hypertrophic cardiomyopathy (HCM) there is a validated and universally accepted prediction model to evaluate individualised risk of SCD [75]. Those in a high-risk category are offered a primary-prevention implantable cardioverter defibrillator (ICD) to reduce the risk of VA and SCD. No such tool is available in FD on which to base decisions on therapy. To date, there have been single-centre studies and systematic reviews confirming risk factors for arrhythmia discussed so far, including advancing age, male gender, prior arrhythmia, LVH, and scar on LGE [21••, 76]. One important note is that, once patients have a risk of one type of arrhythmia, the change in substrate that takes place in FD seems to place the individual at increased risk of any arrythmia. For example, in a study evaluating device implantation in FD, one patient with a PPM died from sustained VF detected on device interrogation [16]. This suggests that, given the progressive nature of FD cardiomyopathy, considerable caution should be taken on implanting cardiac devices without a defibrillator function even in the absence of VA. In the absence of an individual risk calculator, there have been two recent large studies that used deep cardiac phenotyping to develop risk models in FD, with arrhythmia a major component of cardiovascular events recorded [14•, 15••].
A longitudinal prospective cohort study of 200 adults (average age 46 years; 61% female) with FD undergoing cardiac MRI developed a prognostic model to generate risk estimates for major adverse cardiac events (MACE) over a median follow-up of 4.5 years [14•]. MACE outcomes, based on time to first event, included hospitalisation for heart failure, myocardial infarction, coronary revascularisation, VT, new onset AF, bradycardia requiring implantation of a PPM, aborted SCD, implantation of an ICD, or cardiovascular death. A new parameter was developed from the cardiac MRI data, T1 dispersion, calculated as a standard deviation of T1 times extracted from all voxels consisting of the basal and mid-ventricular short-axis T1 maps in the mid-wall of the myocardium. In this study, the composite outcome occurred in 43 participants (21% total), at an annualised rate of 4.8% per year, and the most frequent component of the composite outcome was NSVT. The rate of VA was 0.44% per year, AF 1.67% per year, and bradycardia requiring pacemaker implantation 1.11% per year. Pooled univariable Cox regression for time to the composite outcome was performed, and variables with the highest statistic were age, maximum wall thickness (MWT), indexed LV mass, LV GLS, QRS duration, and MSSI. An internally validated model was developed which accurately predicted the 5-year risk of MACE using age, indexed LV mass, and native non-contrast T1 dispersion in all adults with FD.
In a recently published retrospective observational multi-centre study of 314 adults with FD, patients were staged on the degree of FD cardiomyopathy using TTE-based parameters [15••]. Study endpoints were used to test the prognostic value of the staging classification. Endpoints included all-cause mortality, cardiac mortality, major arrhythmia (bradyarrhythmia requiring PPM or tachyarrhythmia cardioversion/ICD therapy), new onset AF, heart failure hospitalisation, and ischaemic stroke. Patients were classified as stage 0 (no cardiac involvement), stage 1 (LVH with MWT > 12 mm), stage 2 (LA enlargement with indexed LAl volume > 34 ml/m2), or stage 3 (ventricular impairment). Older patients had more advanced disease and the majority with AF or cardiac device implantation were in stages 2 and 3. In total, 18% of patients met a study endpoint. Occurrence of cardiac events increased with stage and arrhythmic events accounted for 45% of these over the 8-year follow-up, with event rates for bradyarrhythmia 7.2%, VA 3.3%, and new onset AF 7.6% with a higher prevalence in more advanced stages. Interestingly, the stage with the highest frequency of PR duration abnormalities was stage 0, consistent with the known early electrical changes. Importantly, the role of LA dilatation and dysfunction as a parameter for prognosticating is highlighted here. LA dilatation may reflect elevated left ventricular end-diastolic pressure from LVH but dilatation may pre-date LVH secondary to direct atrial myopathy [77]. A greater proportion of female patients were in stage 2 (atrial enlargement) compared with males. Conversely, a greater proportion of male patients were in stage 1 (LVH), highlighting the varied pathophysiology and arrhythmic risk based on gender observed in FD [15•]. This study highlights how a greater burden of cardiac involvement correlates with cardiac events but with the large number of endpoints forming the composite (with arrhythmia included); this precludes accurate arrhythmic prediction.
Various risk-prediction data provide conclusive evidence of arrhythmic risk in other cardiomyopathies including sarcomeric HCM, lamin A/C, dilated cardiomyopathy, cardiac sarcoid, and cardiac amyloid [75, 78,79,80,81]. These enable clinicians to assess an individual’s risk of arrhythmia using conventional investigations including 12-lead ECG, Holter, TTE, and CMR. The existing risk prediction studies in FD provide more detailed information confirming that arrythmia is common but that other major adverse cardiovascular events traditionally included in cardiovascular outcome trials, including hospitalisation for heart failure, myocardial infarction, and cardiovascular death, are less frequent and follow-up time needs to be over longer periods. For arrhythmia, which clearly represent a significant burden to the patient in terms of quality of life, further work is needed to define individual risk in the way that the HCM-SCD risk calculator performs [75]. A prospective multi-centre international randomised control trial is currently underway in adults with FD comparing the rate of significant arrhythmia using ILRs [37]. This trial will also collect data from a wide variety of investigations with the aim to develop a robust risk stratification tool in FD.
Therapy
Conventional Risk Modification
Prevalence of conventional risk factors for atherosclerosis in FD is higher than in the general population suggesting accelerated atherosclerosis [82]. Interestingly, novel mechanisms of atherosclerotic disease have also been shown specifically in FD suggesting accelerated atherosclerosis via non-conventional mechanisms [82]. Although there is no randomised evidence of benefit specifically in FD, these data highlight the importance of aggressive risk factor modification including lipid-lowering therapy, antihypertensive therapy, and strict glycaemic control. Lifestyle modification including smoking cessation and regular physical exercise is also recommended [83]. Elevated systolic blood pressure has an incremental impact on progression of cardiomyopathy and subsequent arrhythmic risk due to relation with myocardial hypertrophy and geometry [84]. The benefits of angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blocker (ARB) therapy in reducing cardiovascular risk have been demonstrated in sarcomeric HCM [85].
Arrhythmia
Patients with FD tolerate arrhythmia supraventricular tachycardia and AF poorly due to impaired diastolic filling. Therefore, rhythm control is the favoured strategy with either pharmacological intervention, direct current cardioversion (DCCV), or ablation, although long-term success is compromised by progressive atrial dilatation, impaired atrial function, and change in atrial structure. Although there is now general support for early ablation in all patients presenting with AF, there remain concerns regarding long-term efficacy in FD [86]. In those with refractory AF not amenable to cardioversion, careful rate control is important, with the additional concern regarding co-existing risk of bradyarrhythmia and conduction disease. Beta blockers or calcium channel antagonists are generally favoured as first line for rate-limiting strategies, although amiodarone is generally avoided long term in FD. Amiodarone may alter lysosomal pH, subsequent enzyme activity, and trigger lysosomal dysfunction. It has also been shown to induce phospholipidosis via inhibition of lysosomal phospholipase activity, triggering phospholipid accumulation and development of lamellar bodies [87]. Amiodarone may induce acute heart failure decompensation with features of amiodarone toxicity confirmed on endomyocardial biopsy in a case study of amiodarone initiation in FD [88]. Frequent Holter or wearable ECG monitoring is recommended.
Although anticoagulation is recommended for life in all FD patients with a history of AF due to the high incidence of stroke, none of the current scoring systems for assessing risk of stroke, such as the CHA2DS2VASc scoring system [89], have been validated for use in FD. There is little systematic data on choice of anticoagulation (direct oral anticoagulant versus warfarin). However, direct oral anticoagulation may be advantageous due to lower risks of intracranial bleeding, as FD may be associated with cerebral micro-bleeding [90].
As described, cardiac device implantation with PPM or ICD is high due to increased risk of bradyarrhythmia and VA, particularly in advanced disease [21, 76]. However, with no specific guide for device implantation in FD currently available and as FD is specifically excluded from conventional risk calculators for HCM for primary prevention, device implantation is generally for secondary prevention after a significant arrhythmic event or aborted SCD [83].
Enzyme Replacement Therapy
The mainstay of systemic FD-related therapy is ERT (recombinant α-GAL A) to replace deficient α-GAL A. Current recommendations are that in adult males with classical variants and enzyme activity < 5%, FD therapy is to be considered at diagnosis. In adult females and males with non-classical variants, FD therapy is recommended in those with an LV wall thickness > 13 mm in males and > 12 mm in females, indexed LV mass on TTE/CMR above normal for age and sex or the presence of LGE on CMR [34]. As with all FD therapy, better outcomes are reported with early initiation. There is limited efficacy if commenced > 50 years of age in terms of stabilising LV mass compared to if started < 30 years of age [72]. Those without LGE have seen improvements in LV mass, strain, and exercise capacity with ERT, which is not observed in those with LGE [91]. Efficacy in FD cardiomyopathy is mixed. Once many of the cardinal features of FD cardiomyopathy have developed, which increase arrhythmic risk, the benefits of ERT initiation are significantly limited. There is currently no definitive evidence that ERT reduces the burden of arrhythmia in FD [72, 92].
Oral Chaperone Therapy
Oral chaperone therapy (OCT) is a pharmacological chaperone licensed for use in adults with FD with residual enzyme and an amenable GLA mutation. The mechanism is to stabilise the mutant enzyme, increase bioavailability, and traffic this to the lysosome whereby metabolism of sphingolipid can take place [93]. As with ERT, efficacy of OCT in established cardiomyopathy is limited and there is no evidence demonstrating the benefit of OCT on reducing arrhythmia burden. OCT may slow organ damage with studies showing a stabilisation and, in some cases, mild reduction in LVMi [94, 95]. However, in those with evidence of LGE, cardiomyopathy still progresses even with OCT [96].
Transplantation
In patients with significant symptomatic heart failure, intractable arrhythmia despite all optimal therapy (medical and cardiac devices including synchronisation), transplantation may be a viable option. The major barrier to cardiac transplantation in FD is the progressive and multi-organ nature of the disease. Patients often have co-existing end-organ renal disease and so combined cardiac and renal transplantation may need to be considered. The involvement of multiple organs and increased disease burden has a significant impact on exercise capacity and mental health which affects transplant candidacy [97]. Finally, in patients with non-classical disease including cardiac variants, these are often late-onset in age which presents another barrier to transplantation [98].
Conclusion
Mechanisms underpinning susceptibility for arrhythmia in FD are vast. Electrical instability begins early in the disease. Inflammation, previously presumed a more advanced process, also begins early and is a significant contributor to the arrhythmia substrate. Understanding the molecular mechanisms will enable better targeted therapy to reduce the burden of arrhythmia and SCD in this at-risk cohort. The development of a robust FD-specific risk-stratification tool will enable at-risk patients with FD to be identified and offered primary prevention cardiac device therapy. Further work is essential in these domains to improve cardiac outcomes in FD.
Availability of Data and Materials
Not applicable as this review did not involve the collection of data.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Desnick RJ, Blieden LC, Sharp HL, Hofschire PJ, Moller JH. Cardiac valvular anomalies in Fabry disease. Clinical, morphologic, and biochemical studies. Circulation. 1976;54(5):818–25.
Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138(4):338–46.
Mehta A, Beck M, Eyskens F, Feliciani C, Kantola I, Ramaswami U, et al. Fabry disease: a review of current management strategies. QJM. 2010;103(9):641–59.
Niemann M, Herrmann S, Hu K, Breunig F, Strotmann J, Beer M, et al. Differences in Fabry cardiomyopathy between female and male patients: consequences for diagnostic assessment. JACC Cardiovasc Imaging. 2011;4(6):592–601.
Nair V, Belanger EC, Veinot JP. Lysosomal storage disorders affecting the heart: a review. Cardiovasc Pathol. 2019;39:12–24.
Namdar M. Electrocardiographic changes and arrhythmia in Fabry disease. Front Cardiovasc Med. 2016;3:7.
Shah JS, Hughes DA, Sachdev B, Tome M, Ward D, Lee P, et al. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol. 2005;96(6):842–6.
Krämer J, Niemann M, Störk S, Frantz S, Beer M, Ertl G, et al. Relation of burden of myocardial fibrosis to malignant ventricular arrhythmias and outcomes in Fabry disease. Am J Cardiol. 2014;114(6):895–900.
Acharya D, Robertson P, Kay GN, Jackson L, Warnock DG, Plumb VJ, et al. Arrhythmias in Fabry cardiomyopathy. Clin Cardiol. 2012;35(12):738–40.
Frustaci A, Morgante E, Russo MA, Scopelliti F, Grande C, Verardo R, et al. Pathology and function of conduction tissue in Fabry disease cardiomyopathy. Circ Arrhythm Electrophysiol. 2015;8(4):799–805.
Deva DP, Hanneman K, Li Q, Ng MY, Wasim S, Morel C, et al. Cardiovascular magnetic resonance demonstration of the spectrum of morphological phenotypes and patterns of myocardial scarring in Anderson-Fabry disease. J Cardiovasc Magn Reson. 2016;18:14.
Patel V, O’Mahony C, Hughes D, Rahman MS, Coats C, Murphy E, et al. Clinical and genetic predictors of major cardiac events in patients with Anderson-Fabry disease. Heart. 2015;101(12):961–6.
Weidemann F, Maier SK, Störk S, Brunner T, Liu D, Hu K, et al. Usefulness of an implantable loop recorder to detect clinically relevant arrhythmias in patients with advanced fabry cardiomyopathy. Am J Cardiol. 2016;118(2):264–74.
• Orsborne C, Bradley J, Bonnett LJ, Pleva LA, Naish JH, Clark DG, et al. Validated model for prediction of adverse cardiac outcome in patients with Fabry disease. J Am Coll Cardiol. 2022;80(10):982–94. This important studyhighlights how risk-prediction for arrhythmia can be accurately modelled in Fabry disease using advance cardiac magnetic resonance techniques but requires further external validation.
•• Meucci MC, Lillo R, Del Franco A, Monda E, Iannaccone G, Baldassarre R, et al. Prognostic implications of the extent of cardiac damage in patients with Fabry disease. J Am Coll Cardiol. 2023;82(15):1524–34. This study highlights how risk prediction formajor adverse cardiac events including arrhythmia can be evaluated using conventional transthoracic echocardiography which patients with FD will undergo routinely as part of their clinical care.
Vijapurapu R, Geberhiwot T, Jovanovic A, Baig S, Nordin S, Kozor R, et al. Study of indications for cardiac device implantation and utilisation in Fabry cardiomyopathy. Heart. 2019;105(23):1825–31.
Pelliccia F, Pasceri V, Limongelli G, Autore C, Basso C, Corrado D, et al. Long-term outcome of nonobstructive versus obstructive hypertrophic cardiomyopathy: a systematic review and meta-analysis. Int J Cardiol. 2017;243:379–84.
Elliott PM, Gimeno JR, Thaman R, Shah J, Ward D, Dickie S, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart. 2006;92(6):785–91.
Weidemann F, Niemann M, Störk S, Breunig F, Beer M, Sommer C, et al. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med. 2013;274(4):331-41.
Germain DP, Brand E, Burlina A, Cecchi F, Garman SC, Kempf J, et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: a multicenter Fabry Registry study. Mol Genet Genomic Med. 2018.
•• Vijapurapu R, Roy A, Demetriades P, Warfield A, Hughes DA, Moon J, et al. Systematic review of the incidence and clinical risk predictors of atrial fibrillation and permanent pacemaker implantation for bradycardia in Fabry disease. Open Heart. 2023;10(2). This review assessing atrial fibrillation and bradycardia requiring device implantation concludes that whilst reporting of both are limited to single-centre studies and have variable reporting dependent on diagnostic modality, the burden of brady-arrhythmia and atrial fibrillation requiring therapy are likely to be much higher than reported. Factors such as left ventricular hypertrophy, age and atrial volume dilatation may contribute to the burden of arrhythmia.
Hagège A, Réant P, Habib G, Damy T, Barone-Rochette G, Soulat G, et al. Fabry disease in cardiology practice: literature review and expert point of view. Arch Cardiovasc Dis. 2019;112(4):278–87.
Lobo T, Morgan J, Bjorksten A, Nicholls K, Grigg L, Centra E, et al. Cardiovascular testing in Fabry disease: exercise capacity reduction, chronotropic incompetence and improved anaerobic threshold after enzyme replacement. Intern Med J. 2008;38(6):407–14.
Wilson HC, Hopkin RJ, Madueme PC, Czosek RJ, Bailey LA, Taylor MD, et al. Arrhythmia and clinical cardiac findings in children with Anderson-Fabry disease. Am J Cardiol. 2017;120(2):251–5.
Di LZ, Pichette M, Nadeau R, Bichet DG, Poulin F. Severe bradyarrhythmia linked to left atrial dysfunction in Fabry disease—a cross-sectional study. Clin Cardiol. 2018;41(9):1207–13.
O’Mahony C, Coats C, Cardona M, Garcia A, Calcagnino M, Murphy E, et al. Incidence and predictors of anti-bradycardia pacing in patients with Anderson-Fabry disease. Europace. 2011;13(12):1781–8.
Sené T, Lidove O, Sebbah J, Darondel JM, Picard H, Aaron L, et al. Cardiac device implantation in Fabry disease: a retrospective monocentric study. Medicine (Baltimore). 2016;95(40): e4996.
Pichette M, Serri K, Pagé M, Di LZ, Bichet DG, Poulin F. Impaired left atrial function in Fabry disease: a longitudinal speckle-tracking echocardiography study. J Am Soc Echocardiogr. 2017;30(2):170-9.e2.
Talbot AS, Lewis NT, Nicholls KM. Cardiovascular outcomes in Fabry disease are linked to severity of chronic kidney disease. Heart. 2015;101(4):287–93.
Fan JL, Su B, Zhao X, Zhou BY, Ma CS, Wang HP, et al. Correlation of left atrial strain with left ventricular end-diastolic pressure in patients with normal left ventricular ejection fraction. Int J Cardiovasc Imaging. 2020;36(9):1659–66.
Hauser R, Nielsen AB, Skaarup KG, Lassen MCH, Duus LS, Johansen ND, et al. Left atrial strain predicts incident atrial fibrillation in the general population: the Copenhagen City Heart Study. Eur Heart J Cardiovasc Imaging. 2021;23(1):52–60.
Fukuda Y, Onishi T, Suzuki A, Tanaka H, Fukuzawa K, Yoshida A, et al. Follow-up of cardiac fabry disease treated by cardiac resynchronization therapy. CASE (Phila). 2017;1(4):134–7.
Roy A, Vijapurapu R, Kurdi H, Orsborne C, Woolfson P, Kalla M, et al. Clinical utilisation of implantable loop recorders in adults with Fabry disease—a multi-centre snapshot study. Front Cardiovasc Med. 2023;10:1323214.
Hiwot T, Hughes D, Ramaswami U. Guidelines for the treatment of Fabry disease. British Inherited Metabolic Diseases Group2020.
Arends M, Wanner C, Hughes D, Mehta A, Oder D, Watkinson OT, et al. Characterization of classical and nonclassical fabry disease: a multicenter study. J Am Soc Nephrol. 2017;28(5):1631–41.
Vijapurapu R, Bradlow W, Leyva F, Moon JC, Zegard A, Lewis N, et al. Cardiac device implantation and device usage in Fabry and hypertrophic cardiomyopathy. Orphanet J Rare Dis. 2022;17(1):6.
Vijapurapu R, Kozor R, Hughes DA, Woolfson P, Jovanovic A, Deegan P, et al. A randomised controlled trial evaluating arrhythmia burden, risk of sudden cardiac death and stroke in patients with Fabry disease: the role of implantable loop recorders (RaILRoAD) compared with current standard practice. Trials. 2019;20(1):314.
Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency N Engl J Med. 1967;276(21):1163–7.
Namdar M, Steffel J, Vidovic M, Brunckhorst CB, Holzmeister J, Lüscher TF, et al. Electrocardiographic changes in early recognition of Fabry disease. Heart. 2011;97(6):485–90.
Jastrzebski M, Bacior B, Dimitrow PP, Kawecka-Jaszcz K. Electrophysiological study in a patient with Fabry disease and a short PQ interval. Europace. 2006;8(12):1045–7.
Aryana A, Fifer MA, Ruskin JN, Mela T. Short PR interval in the absence of preexcitation: a characteristic finding in a patient with Fabry disease. Pacing Clin Electrophysiol. 2008;31(6):782–3.
Straus SM, Kors JA, De Bruin ML, van der Hooft CS, Hofman A, Heeringa J, et al. Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J Am Coll Cardiol. 2006;47(2):362–7.
Vijapurapu R, Maanja M, Schlegel T, Augusto J, Kurdi H, Moon JC, et al. Advanced electrocardiography predicts early cardiac involvement and incident arrhythmias in Fabry disease. EP Europace. 2022;24(1):euac053.30.
Maruyama H, Taguchi A, Nishikawa Y, Guili C, Mikame M, Nameta M, et al. Medullary thick ascending limb impairment in the Gla. FASEB J. 2018;32(8):4544–59.
Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020;21(2):101–18.
Waldek S, Feriozzi S. Fabry nephropathy: a review — how can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014;15:72.
Puertollano R, Ferguson SM, Brugarolas J, Ballabio A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018;37(11).
Ivanova M. Altered sphingolipids metabolism damaged mitochondrial functions: lessons learned from Gaucher and Fabry diseases. J Clin Med. 2020;9(4).
Chimenti C, Hamdani N, Boontje NM, DeCobelli F, Esposito A, Bronzwaer JG, et al. Myofilament degradation and dysfunction of human cardiomyocytes in Fabry disease. Am J Pathol. 2008;172(6):1482–90.
Barbey F, Brakch N, Linhart A, Rosenblatt-Velin N, Jeanrenaud X, Qanadli S, et al. Cardiac and vascular hypertrophy in Fabry disease: evidence for a new mechanism independent of blood pressure and glycosphingolipid deposition. Arterioscler Thromb Vasc Biol. 2006;26(4):839–44.
Birket MJ, Raibaud S, Lettieri M, Adamson AD, Letang V, Cervello P, et al. A human stem cell model of Fabry disease implicates LIMP-2 accumulation in cardiomyocyte pathology. Stem Cell Reports. 2019;13(2):380–93.
Choi L, Vernon J, Kopach O, Minett MS, Mills K, Clayton PT, et al. The Fabry disease-associated lipid Lyso-Gb3 enhances voltage-gated calcium currents in sensory neurons and causes pain. Neurosci Lett. 2015;594:163–8.
Choi S, Kim JA, Na HY, Cho SE, Park S, Jung SC, et al. Globotriaosylceramide induces lysosomal degradation of endothelial KCa3.1 in fabry disease. Arterioscler Thromb Vasc Biol. 2014;34(1):81–9.
Cumberland MJ, Riebel LL, Roy A, O’Shea C, Holmes AP, Denning C, et al. Basic research approaches to evaluate cardiac arrhythmia in heart failure and beyond. Front Physiol. 2022;13: 806366.
Nordin S, Kozor R, Baig S, Abdel-Gadir A, Medina-Menacho K, Rosmini S, et al. Cardiac phenotype of prehypertrophic Fabry disease. Circ Cardiovasc Imaging. 2018;11(6): e007168.
Knott KD, Augusto JB, Nordin S, Kozor R, Camaioni C, Xue H, et al. Quantitative myocardial perfusion in Fabry disease. Circ Cardiovasc Imaging. 2019;12(7): e008872.
Moon JC, Sachdev B, Elkington AG, McKenna WJ, Mehta A, Pennell DJ, et al. Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J. 2003;24(23):2151–5.
Seydelmann N, Liu D, Krämer J, Drechsler C, Hu K, Nordbeck P, et al. High-sensitivity troponin: a clinical blood biomarker for staging cardiomyopathy in Fabry disease. J Am Heart Assoc. 2016;5(6).
Pieroni M, Moon JC, Arbustini E, Barriales-Villa R, Camporeale A, Vujkovac AC, et al. Cardiac involvement in Fabry disease: JACC review topic of the week. J Am Coll Cardiol. 2021;77(7):922–36.
Vitner EB, Platt FM, Futerman AH. Common and uncommon pathogenic cascades in lysosomal storage diseases. J Biol Chem. 2010;285(27):20423–7.
Schmid D, Dengjel J, Schoor O, Stevanovic S, Münz C. Autophagy in innate and adaptive immunity against intracellular pathogens. J Mol Med (Berl). 2006;84(3):194–202.
Hsing LC, Rudensky AY. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol Rev. 2005;207:229–41.
De Francesco PN, Mucci JM, Ceci R, Fossati CA, Rozenfeld PA. Fabry disease peripheral blood immune cells release inflammatory cytokines: role of globotriaosylceramide. Mol Genet Metab. 2013;109(1):93–9.
Frustaci A, Verardo R, Grande C, Galea N, Piselli P, Carbone I, et al. Immune-mediated myocarditis in Fabry disease cardiomyopathy. J Am Heart Assoc. 2018;7(17): e009052.
Nordin S, Kozor R, Bulluck H, Castelletti S, Rosmini S, Abdel-Gadir A, et al. Cardiac Fabry disease with late gadolinium enhancement is a chronic inflammatory cardiomyopathy. J Am Coll Cardiol. 2016;68(15):1707–8.
Yogasundaram H, Nikhanj A, Putko BN, Boutin M, Jain-Ghai S, Khan A, et al. Elevated inflammatory plasma biomarkers in patients with Fabry disease: a critical link to heart failure with preserved ejection fraction. J Am Heart Assoc. 2018;7(21): e009098.
Augusto JB, Nordin S, Vijapurapu R, Baig S, Bulluck H, Castelletti S, et al. Myocardial edema, myocyte injury, and disease severity in Fabry disease. Circ Cardiovasc Imaging. 2020;13(3): e010171.
Nappi C, Altiero M, Imbriaco M, Nicolai E, Giudice CA, Aiello M, et al. First experience of simultaneous PET/MRI for the early detection of cardiac involvement in patients with Anderson-Fabry disease. Eur J Nucl Med Mol Imaging. 2015;42(7):1025–31.
Imbriaco M, Nappi C, Ponsiglione A, Pisani A, Dell’Aversana S, Nicolai E, et al. Hybrid positron emission tomography-magnetic resonance imaging for assessing different stages of cardiac impairment in patients with Anderson-Fabry disease: AFFINITY study group. Eur Heart J Cardiovasc Imaging. 2019;20(9):1004–11.
Spinelli L, Imbriaco M, Nappi C, Nicolai E, Giugliano G, Ponsiglione A, et al. Early cardiac involvement affects left ventricular longitudinal function in females carrying α-galactosidase A mutation: role of hybrid positron emission tomography and magnetic resonance imaging and speckle-tracking echocardiography. Circ Cardiovasc Imaging. 2018;11(4): e007019.
Fall B, Scott CR, Mauer M, Shankland S, Pippin J, Jefferson JA, et al. Urinary podocyte loss is increased in patients with Fabry disease and correlates with clinical severity of Fabry nephropathy. PLoS ONE. 2016;11(12): e0168346.
Germain DP, Charrow J, Desnick RJ, Guffon N, Kempf J, Lachmann RH, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015;52(5):353–8.
Silva CAB, Moura-Neto JA, Dos Reis MA, Vieira Neto OM, Barreto FC. Renal manifestations of Fabry disease: a narrative review. Can J Kidney Health Dis. 2021;8:2054358120985627.
Mc Causland FR, Tumlin JA, Roy-Chaudhury P, Koplan BA, Costea AI, Kher V, et al. Intradialytic hypotension and cardiac arrhythmias in patients undergoing maintenance hemodialysis: results from the monitoring in dialysis study. Clin J Am Soc Nephrol. 2020;15(6):805–12.
O’Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. 2014;35(30):2010–20.
Baig S, Edward NC, Kotecha D, Liu B, Nordin S, Kozor R, et al. Ventricular arrhythmia and sudden cardiac death in Fabry disease: a systematic review of risk factors in clinical practice. Europace. 2018;20(FI2):f153–61.
Boyd AC, Lo Q, Devine K, Tchan MC, Sillence DO, Sadick N, et al. Left atrial enlargement and reduced atrial compliance occurs early in Fabry cardiomyopathy. J Am Soc Echocardiogr. 2013;26(12):1415–23.
Hasselberg NE, Edvardsen T, Petri H, Berge KE, Leren TP, Bundgaard H, et al. Risk prediction of ventricular arrhythmias and myocardial function in Lamin A/C mutation positive subjects. Europace. 2014;16(4):563–71.
Okada DR, Smith J, Derakhshan A, Gowani Z, Misra S, Berger RD, et al. Ventricular arrhythmias in cardiac sarcoidosis. Circulation. 2018;138(12):1253–64.
Laptseva N, Rossi VA, Sudano I, Schwotzer R, Ruschitzka F, Flammer AJ, et al. Arrhythmic manifestations of cardiac amyloidosis: challenges in risk stratification and clinical management. J Clin Med. 2023;12(7).
Sammani A, Kayvanpour E, Bosman LP, Sedaghat-Hamedani F, Proctor T, Gi WT, et al. Predicting sustained ventricular arrhythmias in dilated cardiomyopathy: a meta-analysis and systematic review. ESC Heart Fail. 2020;7(4):1430–41.
Roy A, Umar H, Ochoa-Ferraro A, Warfield A, Lewis N, Geberhiwot T, et al. Atherosclerosis in Fabry disease—a contemporary review. J Clin Med. 2021;10(19).
Baig S, Vijapurapu R, Alharbi F, Nordin S, Kozor R, Moon J, et al. Diagnosis and treatment of the cardiovascular consequences of Fabry disease. QJM. 2019;112(1):3–9.
Krämer J, Bijnens B, Störk S, Ritter CO, Liu D, Ertl G, et al. Left ventricular geometry and blood pressure as predictors of adverse progression of Fabry cardiomyopathy. PLoS ONE. 2015;10(11): e0140627.
Ammirati E, Contri R, Coppini R, Cecchi F, Frigerio M, Olivotto I. Pharmacological treatment of hypertrophic cardiomyopathy: current practice and novel perspectives. Eur J Heart Fail. 2016;18(9):1106–18.
Kirchhof P, Camm AJ, Goette A, Brandes A, Eckardt L, Elvan A, et al. Early rhythm-control therapy in patients with atrial fibrillation. N Engl J Med. 2020;383(14):1305–16.
Reasor MJ, Kacew S. Drug-induced phospholipidosis: are there functional consequences? Exp Biol Med (Maywood). 2001;226(9):825–30.
Fine NM, Wang Y, Khan A. Acute decompensated heart failure after initiation of amiodarone in a patient with Anderson-Fabry disease. Can J Cardiol. 2019;35(1):104.e5-.e7;35(1):104.e5-e7.
Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the euro heart survey on atrial fibrillation. Chest. 2010;137(2):263–72.
Reisin RC, Romero C, Marchesoni C, Nápoli G, Kisinovsky I, Cáceres G, et al. Brain MRI findings in patients with Fabry disease. J Neurol Sci. 2011;305(1–2):41–4.
Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, Störk S, et al. Long-term effects of enzyme replacement therapy on fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation. 2009;119(4):524–9.
Ortiz A, Abiose A, Bichet DG, Cabrera G, Charrow J, Germain DP, et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: data from the Fabry Registry. J Med Genet. 2016;53(7):495–502.
Benjamin ER, Della Valle MC, Wu X, Katz E, Pruthi F, Bond S, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 2017;19(4):430–8.
Hughes DA, Nicholls K, Shankar SP, Sunder-Plassmann G, Koeller D, Nedd K, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017;54(4):288–96.
Lenders M, Nordbeck P, Kurschat C, Karabul N, Kaufeld J, Hennermann JB, et al. Treatment of Fabry’s disease with migalastat: outcome from a prospective observational multicenter study (FAMOUS). Clin Pharmacol Ther. 2020;108(2):326–37.
Gatterer C, Beitzke D, Graf S, Lenz M, Sunder-Plassmann G, Mann C, et al. Long-term monitoring of cardiac involvement under migalastat treatment using magnetic resonance tomography in Fabry disease. Life (Basel). 2023;13(5).
Kyem G, Okorozo A, Hamdan H, Tuffaha AM. A case report of kidney after heart transplant in patient with Fabry disease. Transplant Proc. 2023;55(8):1975–7.
Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, Burlina A, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416–27.
Funding
K. G. is supported by grants from the Medical Research Council (MR/V009540/1) and The National Centre for the 3Rs and Industry (NC/T001747/1). The Institute of Cardiovascular Sciences, University of Birmingham, has received an Accelerator Award by the British Heart Foundation (AA/18/2/34218). M. C. is funded by a National Centre for the 3Rs/British Heart Foundation (BHF) studentship (NC/T001747/1) and an NC3Rs Early Career Engagement Award.
Author information
Authors and Affiliations
Contributions
R. P. S. and T. G. — idea and conceptualisation. A. R. and M. C. — writing up of manuscript. R. P. S., T. G., A. H., M. K., and K. G. — review and editing of manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical Approval and Consent to Participate
Not applicable as this is a review of published literature.
Consent for Publication
Not applicable as this is a review of published literature.
Conflict of Interest
The authors declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roy, A., Cumberland, M.J., O’Shea, C. et al. Arrhythmogenesis in Fabry Disease. Curr Cardiol Rep (2024). https://doi.org/10.1007/s11886-024-02053-2
Accepted:
Published:
DOI: https://doi.org/10.1007/s11886-024-02053-2