
Abstract
Purpose of Review
Cardiac fibroblast activation contributes to fibrosis, maladaptive remodeling and heart failure progression. This review summarizes the latest findings on cardiac fibroblast activation dynamics derived from single-cell transcriptomic analyses and discusses how this information may aid the development of new multispecific medicines.
Recent Findings
Advances in single-cell gene expression technologies have led to the discovery of distinct fibroblast subsets, some of which are more prevalent in diseased tissue and exhibit temporal changes in response to injury. In parallel to the rapid development of single-cell platforms, the advent of multispecific therapeutics is beginning to transform the biopharmaceutical landscape, paving the way for the selective targeting of diseased fibroblast subpopulations.
Summary
Insights gained from single-cell technologies reveal critical cardiac fibroblast subsets that play a pathogenic role in the progression of heart failure. Combined with the development of multispecific therapeutic agents that have enabled access to previously “undruggable” targets, we are entering a new era of precision medicine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure is the leading cause of morbidity and mortality around the world [1]. During development of chronic heart failure, excessive collagen deposition accumulates throughout the heart, resulting in interstitial fibrosis that contributes to stiffening of the heart, contractile dysfunction, and arrhythmias [2,3,4]. Interstitial fibrosis is primarily mediated by the activation of resident cardiac fibroblasts, which undergo phenotypic changes to adopt a secretory and pro-inflammatory cell state that communicates with and responds to other interstitial cells of the heart, in particular immune cells [2,3,4]. The classical activated state of a fibroblast, often referred to as a myofibroblast, represents a highly secretory cell type that produces fibrillar collagen and other extracellular matrix (ECM) proteins. Recent progress in the field has revealed that aberrant and sustained activation of fibroblasts fuels heart failure progression [4]. Consequently, targeting cardiac fibroblast activation represents a novel approach to reduce interstitial fibrosis and ameliorate heart failure.
Despite increased recognition of cardiac fibroblasts as a key driver in heart failure progression, current treatments for chronic heart failure do not specifically target fibroblasts. In this review, we will summarize recent research that highlights a pathogenic role of cardiac fibroblasts in heart failure and describe insights gained from single-cell RNA-sequencing (scRNA-seq) studies, focusing on those that have deepened our understanding of cardiac fibroblast phenotypic heterogeneity and dynamic cellular transitions in response to injury. In addition, we will discuss how these data can be leveraged for the selective targeting of disease-associated fibroblast subsets.
Cardiac Fibroblast as a Driver in Heart Failure Progression
Historically, fibroblasts have been difficult to study due to limitations in the ability to unambiguously identify these cells and manipulate them in vivo. Advances in molecular genetic tools coupled with lineage tracing have allowed for a more precise evaluation of the embryonic origin and molecular identity of resident cardiac fibroblasts (reviewed in [5,6,7,8]). In parallel, recent work has led to a greater appreciation of their causal role in heart failure progression [9, 10]. For example, in a murine model of pressure overload–induced heart failure by transverse aortic constriction (TAC), myofibroblast-specific inhibition of transforming growth factor β (TGFβ) signaling attenuated cardiac fibrosis, providing evidence that activation of tissue-resident fibroblasts by TGFβ is responsible for the fibrotic response in the heart [11•]. In addition to signaling mediated by biochemical agents such as TGFβ, maintenance of a chronic fibrotic response depends on the biophysical microenvironment and mechanical properties of the cardiac ECM, whereby matrix stiffness promotes mechano-activation of fibroblasts and perpetuates a profibrotic milieu [12,13,14].
TGFβ is considered a master regulator of myofibroblast activation [15, 16]. In addition to canonical signaling through SMAD transcription factors [11•, 17], TGFβ signals through non-canonical pathways such as p38 MAPK [18, 19]. This redistributes the transcriptional coactivator BRD4, activating profibrotic gene expression [20]. In models of genetic cardiomyopathy driven by expression of a 40-kDa fragment of cardiac myosin binding protein C (cMyBP-C) [21•] or a mutant alphaB-crystallin (CryAB R120G) [22•], conditional deletion of TGFβ signaling in myofibroblasts reduced fibrosis, improved cardiac function, and increased the probability of survival. Notably, in both of these models, the primary defect was cardiomyocyte intrinsic, and activation of fibroblasts was a secondary response to myocyte stress [21•, 22•]. The fact that inhibiting secondary fibroblast activation conferred hemodynamic benefit and arrested adverse remodeling further corroborates the notion that myofibroblasts are a major disease driver in heart failure progression.
Nevertheless, despite the critical involvement of TGFβ signaling [10, 23] and other profibrotic pathways such as angiotensin II (Ang II) and endothelin 1 [24] in the development of cardiac fibrosis, therapeutic targeting of these pathways has had limited success in fibrosis amelioration. Treatment with the renin-angiotensin-aldosterone system inhibitors, currently standard of care for heart failure, resulted in a modest reduction of cardiac fibrosis in several small clinical studies [25]. Their inability to fully abrogate the fibrotic response may be due in part to the pleiotropic and sometimes divergent roles of profibrotic mediators in different cell types. For example, SMAD3 activation in murine myofibroblasts played a protective role in cardiac injury by maintaining ECM network [17, 26], whereas SMAD3 signaling in cardiomyocytes promoted cardiomyocyte death and exacerbated systolic dysfunction after myocardial infarction (MI) [26]. Consequently, systemic inhibition of profibrotic pathways may produce both beneficial and detrimental effects.
Furthermore, cardiac fibroblasts play important homeostatic roles in the heart and contribute to the acute injury response by secreting ECM proteins in response to cardiomyocyte loss, facilitating scar formation and protecting the heart from rupture [18, 27••, 28]. Non-specific targeting of fibroblasts may therefore compromise the normal wound healing response and lead to untoward toxicities. Thus, highly specific targeting strategies in the design of therapeutic interventions against cardiac fibroblasts are needed.
Emerging evidence suggests that fibroblasts are not unidimensional cells whose sole function is to modulate ECM. Rather, they exhibit remarkable phenotypic heterogeneity [29, 30••] and mediate a diverse range of cellular functions including transducing proliferative and protective signals to cardiomyocytes [31, 32], clearing apoptotic cardiomyocytes [33], regulating electrical conduction [34, 35], and participating in inflammatory [36, 37] and angiogenic pathways [38]. This functional diversity is made possible by dynamic regulation of their gene expression profiles in response to mechanical stimuli, cytokines, or other mediators [32, 39, 40].
Taking advantage of this cellular diversity, the field has now begun to explore the feasibility of selectively targeting fibroblast subsets to reduce fibrosis. In a rodent model where up to 60% of periostin-expressing fibroblasts were ablated by diphtheria toxin, reduced cardiac fibrosis and improved heart function were observed following chronic Ang II infusion or MI [41•]. In these models, genetic recombination was driven by a tamoxifen-inducible Cre-transgene under the control of the Postn promoter [41•]. Importantly, ablation of periostin-expressing cardiac fibroblasts following MI did not compromise scar stability [41•]. In contrast, Kanisicak and colleagues generated a Postn knock-in strain containing the MerCreMer cassette and showed that all activated fibroblasts were labelled by periostin [27••]. In this model, total depletion of myofibroblasts using diphtheria toxin resulted in increased ventricular rupture and higher lethality [27••]. This inconsistency from the previous study is most likely due to differences in the genetic strategies as well as the level of cell depletion across the two approaches.
While diphtheria toxin–mediated ablation serves as a proof-of-principle to support selective targeting of fibroblast subpopulations to reduce fibrosis, this strategy is not therapeutically viable, as it requires multiple genetic recombinations to express the toxin gene construct in a defined cell type. Alternatively, cell therapy breakthroughs in the immuno-oncology field have made it possible to selectively deplete a defined cellular subpopulation for therapeutic purposes. In particular, by expressing a chimeric antigen receptor (CAR), cytotoxic T cells can be redirected to recognize specific antigens on cancer cells and thus mediate their killing [42]. It is not unreasonable to envision that similar strategies could be adopted for the selective depletion of non-cancer cells. Proof-of-principle of such a strategy has been achieved in the study by Aghajanian and colleagues [43••], who employed T cells engineered to express a CAR against fibroblast activation protein (FAP), a cell surface protein enriched in activated fibroblasts from patients with dilated or hypertrophic cardiomyopathy [44]. Using a murine model of hypertension induced by chronic infusion of Ang II/phenylephrine (PE), selective depletion of FAP+ fibroblasts in the heart by CAR T cell transplantation resulted in reduced fibrosis and improved cardiac function in treated animals [43••]. Importantly, whether ablation of activated fibroblasts would disrupt the normal wound healing response will need to be evaluated in future studies. Together, these studies suggest that targeting specific fibroblast subpopulations involved in pathological remodeling holds promise in limiting heart failure progression. A deeper understanding of fibroblast phenotypic heterogeneity, facilitated by recent advances in single-cell technologies, will enable segregation of fibroblast subtypes for more precise targeting of subpopulations enriched in disease states.
Unraveling Fibroblast Transcriptomic Diversity Using Single-Cell Analyses
The last 10 years have seen the expansive development of single-cell transcriptomic technologies, enabling the characterization of transcriptional responses in subpopulations of cells in heterogenous tissues potentially masked by bulk RNA-seq approaches. A multitude of different scRNA-seq methods that vary at each step of the workflow have been developed, each with distinct advantages and disadvantages with regard to platform throughput, technology sensitivity, data coverage, and the per-cell cost [45, 46]. In general, all methodologies share a common workflow: single-cell isolation and capture, cell lysis, RNA reverse transcription, cDNA amplification, library preparation, sequencing, and computational analysis (reviewed in [47,48,49]). In parallel with the development of methods for single-cell isolation and RNA capture, a growing number of computational tools have also been developed (reviewed in [49, 50]), providing not only a bioinformatic pipeline for data quality control and batch normalization but also an unprecedented level of details into cellular heterogeneity and intercellular relationships.
Current high-throughput scRNA-seq technologies allow thousands of cells to be assayed simultaneously, enabling the identification of both novel and rare cell populations as well as the analysis of cell state transitions and complex intercellular communication networks [49] (Table 1). However, most high-throughput single-cell capture technologies have limits on cell size, precluding the accurate and unbiased selection of large cells like cardiomyocytes. Single-nucleus RNA-sequencing (snRNA-seq), where nuclear RNA species are sequenced instead of cytoplasmic ones, has addressed this challenge, allowing the simultaneous profiling of both cardiomyocytes and interstitial cells [51, 52••, 53••]. However, transcript detection sensitivity is reduced and isoform information is lost in snRNA-seq due to the lower abundance of RNA and enrichment of unspliced pre-mRNA in the nuclei [46]. Therefore, caution must be exercised when selecting a sequencing methodology for a particular experiment. In general, snRNA-seq may be sufficient for cell typing, whereas scRNA-seq could provide further information to facilitate target or biomarker identification beyond defining cellular heterogeneity [46].
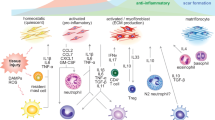
Taking advantage of single-cell transcriptomic technologies, studies in cardiac interstitial cells have revealed remarkable phenotypic plasticity of cardiac fibroblasts, whose transcriptional profile is dynamically regulated in response to external stimuli (Table 2 and Fig. 1a).

a Cardiac fibroblast dynamics in homeostasis and in response to myocardial injury revealed by scRNA-seq and snRNA-seq. Specific fibroblast subsets along with their marker genes are listed. b Examples of potential multispecific therapeutic drugs targeting cardiac fibroblasts (Fb).
Fibroblast Subtypes in Homeostasis
Fibroblast-like cells can be detected as early as 11.5 days post-coitum in the developing mouse heart, and these embryonic cells express canonical fibroblast genes such as Col1a1 and Dcn [51, 54]. This coincides with the formation of the epicardium, a mesothelial layer that migrates from the pro-epicardial organ to form a sheath of cells covering the myocardial wall [69]. From 12.5 days post-coitum, the epicardium undergoes epithelial-to-mesenchymal transition to generate interstitial fibroblasts and smooth muscle cells [69]. In both mouse and human hearts, the proportion of fibroblast-like cells was shown to increase throughout embryonic development [54, 55, 62]. Analysis of cell cycle–related genes and gene regulatory networks revealed that a major wave of fibroblast proliferation occurred during early gestation [62]. These fibroblasts gradually became more mature, accompanied by an upregulation of ECM protein expression [62]. Within the adult heart, lineage tracing suggested their continuous cellular turnover even under homeostatic conditions [59].
In the adult human heart, fibroblasts have been shown to constitute about 15–30% of all cells, with regional differences between atrial and ventricular tissues [52••, 53••]. This percentage is somewhat higher than what has been described for mouse cardiac fibroblasts, which account for less than 20% of non-myocytes under homeostatic conditions [70•]. Notably, this is not a homogeneous population of cells. In fact, single-cell analysis has provided a glimpse into the remarkable transcriptional heterogeneity of the resident fibroblast population. For example, subclustering of fibroblasts in the adult murine heart identified two transcriptionally distinct subpopulations [58•, 61, 63, 65] (Fig. 1a). Both populations expressed Col1a1, but one of them was characterized by low abundance of the canonical fibroblast markers Pdgfra and Tcf21 [58•, 61]. This distinct subpopulation instead was enriched in Wnt signaling pathway genes such as Wif1, Dkk3, Sfrp2, and Frzb [61, 63, 66•]. Their origin and function in the adult heart remain a subject of debate [61, 66•]. Unsupervised clustering of adult human cardiac fibroblasts has also identified transcriptionally distinct subpopulations. Combining scRNA-seq and snRNA-seq data from human organ donors without overt cardiovascular disease, Litviňuková and colleagues [52••] identified a subcluster of stromal cells displaying characteristics of activated fibroblasts, including expression of POSTN and FAP. Fibroblast activation in this context may result from age-related changes in cardiac physiology that leads to progressive fibrotic remodeling, or from responses to cardiac stress that were not diagnosed clinically. Similar populations were absent in an independent study using only snRNA-seq on samples collected from younger donors [53••]. This discrepancy may be attributed to differences in the health status of the donors, the more detailed clustering in Litviňuková et al., or both. Notably, in both studies, fibroblasts display chamber-specific distributions across the heart, likely related to their diverse developmental origins and specialized functions [52••, 53••]. Together, these single-cell analyses of the healthy human heart provide an information-rich reference to deepen our understanding of cardiac physiology in homeostatic conditions.
Using available repositories of curated ligand-receptor pairs [71, 72], single-cell transcriptomic data have enabled the analysis of intercellular communication networks in heterogeneous tissues such as the heart. Employing these computational tools, scRNA-seq studies have identified dense connections between fibroblasts and multiple cardiac cell types [58•, 67]. Indeed, fibroblasts were shown to be the most trophic cell population, with multiple signaling circuits supporting the survival of other cardiac cells such as pericytes, endothelial cells, and mural cells [58•, 63]. These intercellular communication networks are disturbed in response to injury, coinciding with initiation of cardiac remodeling [64, 67]. These analyses further corroborate the diverse and essential role of the resident fibroblasts in maintaining cardiac homeostasis.
Fibroblast Subtypes in Heart Injury
In response to myocardial injury such as ischemic, mechanical, or inflammatory stress, damaged cardiomyocytes undergo apoptosis and secrete cytokines and soluble factors to trigger inflammatory infiltration and fibroblast activation [2, 4]. Activated fibroblasts are characterized by increased proliferation, ECM synthesis, expression of contractile proteins, and cytokine/growth factor secretion [2, 4]. Traditionally, detection of activated fibroblasts relied on expression of α-smooth muscle action (αSMA, ACTA2), a contractile protein whose presence defines the myofibroblast state [73]. However, emerging evidence suggests that not all activated fibroblasts express αSMA [27••, 41•, 74], and in fact, myofibroblasts are only a portion of the matrix-producing fibroblasts that underlie the fibrotic response [75••]. Hence, the field needs more reliable and definitive means to accurately profile fibroblasts after injury.
Independent studies by Fu et al. [75••] and Ivey et al. [76] using lineage tracing and reporter strains to label resident fibroblasts showed that fibroblast proliferation peaked within the first week after MI, with a 3-5-fold expansion in cell number. Cell cycle entry was confirmed at the transcriptional level by subsequent scRNA-seq analyses [61, 65, 66•, 68]. While Fu et al. [75••] observed minimal cell proliferation in regions outside of the infarct, other studies have presented contrasting evidence, demonstrating global activation of a proliferative program in the resident fibroblasts [66•, 76]. A similar temporal pattern of proliferation was observed in additional models of cardiac injury induced by TAC or isoproterenol injections, with peak proliferation occurring around the first week after injury and rapidly returning to baseline by 2 weeks [76].
Coinciding with the proliferative phase, expression of genes associated with fibroblast activation such as Acta2, Postn, and Tnc was detected 3 days after MI, suggesting that fibroblasts rapidly adopt an activated phenotype in response to injury [57, 61, 66•, 75••] (Fig. 1a). Notably, inclusion of multiple closely spaced timepoints immediately after injury allowed Forte and colleagues [66•] to uncover a novel fibroblast subpopulation appearing as early as 1 day after MI, which was termed “injury response” fibroblasts (Fig. 1a). This subcluster expressed high levels of metallothioneins Mt1-2, monocyte-macrophage chemoattractants Ccl2, Ccl7, and Csf1, and neutrophil activators Cxcl1 and Cxcl5, implicating this early fibroblast state in the initiation of the inflammatory response [66•]. A similar subcluster was also identified following MI injury in the post-natal day 8 heart, when the murine heart loses regenerative capacity [68]. By cellular trajectory analysis, these injury response cells were shown to quickly evolve into myofibroblasts by day 3, accompanied by expression of markers such as Acta2, Fn1, and Cthrc1 [66•] (Fig. 1a), highlighting the transcriptional plasticity of fibroblasts and the rapidly changing cellular landscape in the injured myocardium.
The fate of activated fibroblasts has also been a subject of extensive investigation. In models of reversible injury such as Ang II/PE, withdrawal of the injury stimuli was shown to cause dedifferentiation of myofibroblasts, with reduction in cell cycle genes and loss of αSMA expression [27••]. In humans, left ventricular assist device implantation and unloading of the heart was also associated with transcriptional normalization of the cardiac fibroblasts, which correlated with recovery of cardiac function [67]. With permanent injury such as MI, newly formed fibroblasts persisted in the infarct region and were not turned over [75••]. Nevertheless, myofibroblasts seemed to be a transient differentiated state, as αSMA expression was nearly undetectable by 2 weeks after injury [66•, 75••]. Instead, another unique differentiated state was shown to predominate the maturation phase [75••] (Fig. 1a). Termed matrifibrocytes, this fibroblast subset was closely related to myofibroblasts, but exhibited reduced contractile and secretory properties by gene expression analysis [66•]. They were enriched in genes associated with bone and cartilage remodeling, such as Comp, Chad, and Cilp2, consistent with the development of a stable, chondrogenic-like collagenous scar [66•, 75••]. A second fibroblast subset prevalent in the maturation phase was termed “late resolution” fibroblasts (Fig. 1a). They were proposed to arise from chronic pathological remodeling and expressed genes regulating TGFβ activity and ECM components, in line with continued dynamic remodeling of the myocardium during chronic injury [66•].
Presence of matrifibrocyte-like cells has been suggested in other murine models of cardiac fibrosis and heart failure such as chronic Ang II infusion [63]. However, in this study, a distinct population of Acta2+ myofibroblasts was not identified [63]. This may be explained by the choice of a single timepoint after initiation of Ang II (i.e., 2 weeks), which may have precluded the detection of early transcriptional changes in the fibroblast population. In an independent study by Ruiz-Villalba et al. [65], a distinct fibroblast subpopulation defined by markers including Cthrc1, Ddah1, Fmod, and Comp was dynamically upregulated in response to both MI and chronic Ang II infusion. Similar to matrifibrocytes, this population was characterized by gene ontology terms related to ECM assembly and collagen fibril organization [65]. However, in contrast to matrifibrocytes which persisted in the injured myocardium [66•, 75••], the relative enrichment of this subpopulation was transient, peaking at 2 weeks post-injury and then decreasing under more chronic conditions [65]. Mice deficient in Cthrc1 displayed an impaired fibrotic response and susceptibility to ventricular rupture after MI, indicating an important role for this population in the wound healing response, hence the term “reparative cardiac fibroblasts” [65]. Whether these cells represent an intermediate phenotype between myofibroblasts and matrifibrocytes remains to be investigated. Furthermore, one important question to address with regard to matrifibrocytes is the degree to which they contribute to pathological remodeling and heart failure progression, as this will shed light on whether specific targeting of this fibroblast subpopulation represents a therapeutically viable option to treat cardiac fibrosis. Another key consideration is the extent to which these observations made in murine models could translate into humans. A full spectrum of single-cell analysis of the human heart in different disease conditions, integrated with the information on the diverse subgroups of fibroblasts found in animal models, are critical for understanding the role of cardiac fibroblasts in human heart failure and for future therapeutic development.
Additional Applications of Single-Cell Technologies
In addition to scRNA-seq, numerous next-generation sequencing applications have made it possible to profile other biomolecules or cellular processes in a heterogeneous tissue at the single-cell level [49] (Table 1). For example, single-cell sequencing of transposase-accessible chromatin (scATAC-seq) and single-cell chromatin immune-precipitation sequencing (scChIP-seq) reveal genome-wide chromatin organization and accessibility, providing insight into gene regulatory landscapes that govern transcription [77, 78]. A recent study employing scATAC-seq identified dynamic and reversible changes in chromatin accessibility of cardiac fibroblasts that correlated with cardiac disease severity and demonstrated the plausibility of targeting transcriptional switches to reverse cardiac fibrosis [40]. Global changes to chromatin accessibility were also evident in the neonatal mouse heart after MI injury, especially in non-regenerative hearts at post-natal day 8, suggesting MI-induced transcriptional changes coinciding with cardiac remodeling [68]. Together these data demonstrated the plasticity of cardiac fibroblasts at the level of epigenetic regulation.
Another emerging technology, spatial transcriptomics, retains information on local tissue context [79, 80]. The microenvironment in which a fibroblast resides, including its surrounding ECM and neighboring cells, can have a strong influence on its phenotype and activation status [12, 39, 81]. This is an important but perhaps underappreciated aspect of fibroblast biology. Spatial transcriptomics together with the aforementioned analysis of intercellular communication networks can help depict a clearer picture of how stressed cardiomyocytes signal to recruit and activate immune cells and fibroblasts, and how fibroblasts crosstalk with other cell types to influence cardiac remodeling. Availability of spatial information will also make it possible to correlate transcriptional findings with the extent of tissue remodeling in the failing heart.
Cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) and RNA expression and protein sequencing assay (REAP-seq) are yet another variation of scRNA-seq. These techniques use oligonucleotide-barcoded antibodies to measure protein expression concurrently with cellular transcripts [82, 83]. These methodologies have been leveraged to study immune cell heterogeneity [84], and conceivably, will be helpful in providing enhanced granularity of fibroblast subsets. CITE-seq and REAP-seq are currently limited to the detection of prespecified cell surface proteins with corresponding antibodies, whereas the nascent field of single-cell proteomics with enhanced sensitivity and throughput [85] holds promise for a full integration of multimodal single-cell omics. Simultaneous interrogation of multi-level cellular regulatory mechanisms will provide a holistic view of individual cells, from upstream gene regulatory networks to downstream spatially resolved phenotypes such as protein expression.
Undoubtedly, results of these single-cell studies and associated computational analyses will need to be validated experimentally to confirm the expression profiles of genes of interest, the cellular composition of the tissue being studied, and cell state transitions in response to external stimuli. Presence of specific cellular subtypes will also need to be validated functionally, which could be achieved by genetic manipulation of one or more marker genes. Compared with bulk RNA-seq, scRNA-seq generally has lower sequencing depth, limiting its sensitivity to detect low-expressing genes and the ability to discriminate true biological signal from technical noise [86]. Notwithstanding their limitations, scRNA-seq technologies have revealed specialized fibroblast populations that are of potential therapeutic interest at an unprecedented level of resolution. This rich information will inform development of novel treatments targeting cardiac fibroblasts.
Novel Therapeutic Strategies for Targeting Cardiac Fibroblasts
The success of targeting pathogenic fibroblasts to achieve disease amelioration in preclinical models has pointed to a novel treatment paradigm for heart failure. Nevertheless, given the wide distribution of fibroblasts and lack of specific markers to label them [4, 6], how to achieve therapeutic specificity without inadvertent targeting of other cardiac cell types and/or other tissue fibroblasts becomes a major hurdle in drug development. Innovations in the biopharmaceutical industry over the last two decades have provided a potential solution, with the development and commercialization of multispecific drugs. In contrast to the classical “one target and one drug” approach where a molecule binds to a specific target and modulates its function, multispecific therapeutics work by engaging two or more entities (reviewed in [87]) (Fig. 1b). As a result of this engagement, a therapeutic agent can be localized to the desired site of action; alternatively, a target can be brought into close proximity of an endogenous effector [87]. These powerful capabilities have the potential to transform medicine with extraordinary precision, enabling access to biological entities previously considered intractable.
Currently, most commercialized multispecific agents are approved for oncology or hematology indications [87], but their utilization in other therapeutic areas is rapidly emerging. For example, a proof-of-concept study illustrated the feasibility of targeting cardiac fibroblast subsets using T cells engineered to express a CAR against FAP [43••]. A CAR is comprised of an extracellular antigen-binding domain, typically a single-chain variable fragment derived from an antibody, and an intracellular signaling domain derived from the T cell receptor ζ chain and costimulatory domains such as CD28, 4-1BB, or OX40 [42] (Fig. 1b). This construction programs T cells to recognize a specific protein expressed on the surface of target cells, leading to their cytotoxic killing [42]. FAP, expressed specifically by stromal cells, is a cell surface protein induced at sites of active ECM turnover [44], making it a desirable target for depleting activated fibroblasts without affecting other cardiac cells and quiescent fibroblasts. However, what percentage of FAP+ cells was depleted by CAR T cells and to what extent FAP+ cell depletion was required for amelioration of fibrosis was not reported in this study. In addition, whether non-specific actions of the modified T cells could contribute to the observed phenotype remains unclear. Nevertheless, this study represents an important milestone in the advancement of cardiovascular medicine beyond traditional small molecules and biologics. Subsequent attempts at utilizing CAR T cells to reduce tissue fibrosis targeted the cell surface protein urokinase-type plasminogen activator receptor (uPAR), whose expression is induced in senescent cells [88]. uPAR-specific CAR T cells improved senescence-associated liver fibrosis induced by carbon tetrachloride or a high-fat diet by targeting both macrophages and hepatic stellate cells [88].
CAR T cell immunotherapy is one example of multispecific “matchmakers” that work by inducing proximity, in this case, between a target cell type and effector T cells. Bispecific CD3 engagers represent another strategy that redirects T cell activity towards a defined cell population. As the name implies, these are bispecific antibody–based formats with one domain that binds a surface antigen on target cells and a second domain that binds the CD3 subunit of T cell receptor, leading to T cell activation and lysis of the target cells [89] (Fig. 1b). Recently, bispecific antibody–based biologics that redirect natural killer cells have also been developed [90]. These matchmakers have great therapeutic potential because they harness an endogenous biological mechanism for effector function rather than having to directly modulate the target [87]. As a result, most biological entities such as macromolecules, organelles, and even cells can be targeted with high specificity. Nevertheless, in addition to the inherent challenges faced by multispecific therapeutics (reviewed in [87]), successful targeting of cardiac fibroblasts requires knowledge of the precise expression patterns of the target cell surface antigens, as any on-target killing of cells other than the intended fibroblast subpopulation(s) poses safety risks that are unlikely to be acceptable for a cardiovascular disease indication.
An exciting class of matchmakers mobilizes endogenous molecular machinery to achieve targeted degradation of proteins, nucleic acids, and even organelles (reviewed in [91]). Examples include the heterobifunctional proteolysis-targeting chimeras (PROTACs), which are chimeric small molecules with one module binding the target protein and the other binding a ubiquitin ligase (Fig. 1b). The induced proximity between the two leads to ubiquitylation of the target protein followed by its degradation in the proteasome [92]. Though still in its infancy, targeted degradation as a therapeutic modality has garnered considerable momentum in the industry due to its versatility, effectiveness in abolishing all functions of a target, and potentially long-lasting effect [91].
In addition to matchmakers, another class of multispecific therapeutics termed “tetherbodies” enrich a drug at the desired site of action, thus reducing side effects in off-target tissues [87]. Examples include antibody-drug conjugates and RNA-based therapeutics targeted for a specific tissue. Current antibody-drug conjugates in development are designed for oncology indications, taking advantage of tumor-specific cell surface antigens that concentrate a cytotoxic drug at tumor cells (reviewed in [93]) (Fig. 1b). A similar strategy targeting cardiac fibroblasts for cytotoxic killing may be employed. Alternatively, cell-specific inhibition of profibrotic pathways such as TGFβ signaling could be realized with a bispecific format, using a TGFβ inhibitor as one arm and an antibody against a fibroblast-specific antigen as the other arm. Development of tetherbodies for enrichment of therapeutics at the site of action has been hampered by the paucity of information on tissue-restricted cell surface receptors. Knowledge gained from multi-omics studies at the single-cell level may provide a breakthrough and greatly expand the utility of these agents.
RNA-based therapeutics such as small-interfering RNA (siRNA), antisense oligonucleotide (ASO), modified mRNA and microRNA mimetics greatly expand the domain of druggable targets while offering the advantages of ease of sequence-based design and high specificity (reviewed in [94, 95]). They offer vast opportunities for novel forms of therapeutic mechanisms that are difficult to achieve with traditional small molecules and biologics, such as upregulating or repressing gene expression, altering mRNA splicing, and targeting non-coding RNAs [96]. Currently, the most successful targeting approach for oligonucleotide therapies involves chemical conjugation to ligands for the hepatocyte-specific asialoglycoprotein receptor, resulting in uptake by the liver [97, 98]. Additional carriers are actively being explored to enhance transmembrane delivery into extrahepatic tissues, although none has reached clinical development. Recently, using fatty acids such as docosanoic acid and myristic acid as carriers, researchers were able to achieve 30–45% gene knockdown in the heart [99, 100]. However, to mediate uptake by defined cell types such as the cardiac fibroblast, a highly specific receptor-targeting moiety such as a ligand, aptamer, or antibody would be more desirable as a carrier (Fig. 1b). Proof-of-concept studies in animals have demonstrated this potential, revealing efficient gene knockdown with conjugated siRNA in defined cell types including activated lymphocytes [101], macrophages [102], or tumor cells [102, 103].
Multispecific therapeutic agents rely on target tissue-enriched proteins to achieve specificity; however, it is unlikely that a unique marker is only expressed specifically in cardiac fibroblasts [6]. Inadvertent targeting of other tissues would narrow the therapeutic window. To address this challenge, inspiration could be drawn from recent innovations in the oncology field. For example, dual-receptor CAR T cells have been designed where a synthetic Notch receptor for one antigen induces the expression of a CAR for a second antigen [104, 105]. These T cells are only activated in the presence of both antigens, thus enhancing discrimination between target cells and “bystander” cells [104, 105]. Another strategy involves masking the antigen-binding site of an antibody therapeutic with a peptide, which gets cleaved by proteases specifically found in the tumor microenvironment [106].
Conclusion
Thanks to advances in genetic manipulation and scRNA-seq technologies, the field of cardiac fibroblast biology is entering a new era, with ever more recognition for the diverse roles these cells play in cardiac physiology. Our current understanding of cardiac fibroblasts is based primarily on genetic manipulation within animal models of disease, and it is critical that findings from these studies be validated in humans. Recent scRNA-seq studies in the healthy human heart are encouraging and will serve as valuable resources in this regard [52••, 53••, 67]. Further leveraging of single-cell technologies to identify and characterize pathological states of fibroblasts in the setting of human heart failure will be critical to a better understanding of their contribution to disease progression and to development of novel treatment strategies.
With multispecific drugs significantly expanding the therapeutic toolbox, specific targeting of tissue fibroblasts may soon become a reality. However, multispecific therapeutics are inherently more complex than traditional small molecules or biologics. For them to realize their full potential, challenges specific to multispecific formats must be considered early in their development [87]. Taken together, technological advances are revolutionizing the way we study human diseases. In addition, we anticipate that they will aid in the development of novel and effective therapeutic strategies to target cardiac fibroblasts, a key player in cardiovascular disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139–596. https://doi.org/10.1161/CIR.0000000000000757.
Frangogiannis NG. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Asp Med. 2019;65:70–99. https://doi.org/10.1016/j.mam.2018.07.001.
Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. 2014;70:74–82. https://doi.org/10.1016/j.yjmcc.2013.11.015.
Humeres C, Frangogiannis NG. Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl Sci. 2019;4(3):449–67. https://doi.org/10.1016/j.jacbts.2019.02.006.
Tallquist MD. Cardiac fibroblasts: from origin to injury. Curr Opin Physiol. 2018;1:75–9. https://doi.org/10.1016/j.cophys.2017.08.002.
Tallquist MD, Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol. 2017;14(8):484–91. https://doi.org/10.1038/nrcardio.2017.57.
Furtado MB, Costa MW, Rosenthal NA. The cardiac fibroblast: origin, identity and role in homeostasis and disease. Differentiation. 2016;92(3):93–101. https://doi.org/10.1016/j.diff.2016.06.004.
Swonger JM, Liu JS, Ivey MJ, Tallquist MD. Genetic tools for identifying and manipulating fibroblasts in the mouse. Differentiation. 2016;92(3):66–83. https://doi.org/10.1016/j.diff.2016.05.009.
Gonzalez A, Schelbert EB, Diez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol. 2018;71(15):1696–706. https://doi.org/10.1016/j.jacc.2018.02.021.
Gourdie RG, Dimmeler S, Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat Rev Drug Discov. 2016;15(9):620–38. https://doi.org/10.1038/nrd.2016.89.
. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127(10):3770–83. https://doi.org/10.1172/JCI94753This key study demonstrated that activation of canonical TGFβ signaling in tissue-resident cardiac fibroblasts was the major mediator of the fibrotic response in the heart.
Yokota T, McCourt J, Ma F, Ren S, Li S, Kim TH, et al. Type V collagen in scar tissue regulates the size of scar after heart injury. Cell. 2020;182(3):545–62 e23. https://doi.org/10.1016/j.cell.2020.06.030.
Zhang J, Fan G, Zhao H, Wang Z, Li F, Zhang P, et al. Targeted inhibition of focal adhesion kinase attenuates cardiac fibrosis and preserves heart function in adverse cardiac remodeling. Sci Rep. 2017;7:43146. https://doi.org/10.1038/srep43146.
Yang J, Savvatis K, Kang JS, Fan P, Zhong H, Schwartz K, et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat Commun. 2016;7:13710. https://doi.org/10.1038/ncomms13710.
Frangogiannis N. Transforming growth factor-beta in tissue fibrosis. J Exp Med. 2020;217(3):e20190103. https://doi.org/10.1084/jem.20190103.
Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–38. https://doi.org/10.1038/nrneph.2016.48.
Russo I, Cavalera M, Huang S, Su Y, Hanna A, Chen B, et al. Protective effects of activated myofibroblasts in the pressure-overloaded myocardium are mediated through Smad-dependent activation of a matrix-preserving program. Circ Res. 2019;124(8):1214–27. https://doi.org/10.1161/CIRCRESAHA.118.314438.
Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, et al. Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation. 2017;136(6):549–61. https://doi.org/10.1161/CIRCULATIONAHA.116.026238.
Li Y, Li Z, Zhang C, Li P, Wu Y, Wang C, et al. Cardiac fibroblast-specific activating transcription factor 3 protects against heart failure by suppressing MAP2K3-p38 signaling. Circulation. 2017;135(21):2041–57. https://doi.org/10.1161/CIRCULATIONAHA.116.024599.
Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, et al. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circ Res. 2019;125(7):662–77. https://doi.org/10.1161/CIRCRESAHA.119.315125.
. Meng Q, Bhandary B, Bhuiyan MS, James J, Osinska H, Valiente-Alandi I, et al. Myofibroblast-specific TGFbeta receptor II signaling in the fibrotic response to cardiac myosin binding protein C-induced cardiomyopathy. Circ Res. 2018;123(12):1285–97. https://doi.org/10.1161/CIRCRESAHA.118.313089.
. Bhandary B, Meng Q, James J, Osinska H, Gulick J, Valiente-Alandi I, et al. Cardiac fibrosis in proteotoxic cardiac disease is dependent upon myofibroblast TGF -beta signaling. J Am Heart Assoc. 2018;7(20):e010013. https://doi.org/10.1161/JAHA.118.010013Using murine models of genetic cardiomyopathy, these two papers demonstrated that aberrant activation of cardiac fibroblasts is a bona fide disease driver in heart failure progression, even when the primary insult was cardiomyocyte specific.
Walton KL, Johnson KE, Harrison CA. Targeting TGF-beta mediated SMAD signaling for the prevention of fibrosis. Front Pharmacol. 2017;8:461. https://doi.org/10.3389/fphar.2017.00461.
Rodriguez-Pascual F, Busnadiego O, Gonzalez-Santamaria J. The profibrotic role of endothelin-1: is the door still open for the treatment of fibrotic diseases? Life Sci. 2014;118(2):156–64. https://doi.org/10.1016/j.lfs.2013.12.024.
Fang L, Murphy AJ, Dart AM. A clinical perspective of anti-fibrotic therapies for cardiovascular disease. Front Pharmacol. 2017;8:186. https://doi.org/10.3389/fphar.2017.00186.
Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, et al. Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation. 2018;137(7):707–24. https://doi.org/10.1161/CIRCULATIONAHA.117.029622.
. Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. https://doi.org/10.1038/ncomms12260This seminal paper utilized genetic lineage tracing in the mouse to demonstrate that in the setting of cardiac injury, activated fibroblasts derive from tissue-resident fibroblasts of the Tcf21 lineage. It further showed that periostin+ myofibroblasts are necessary for the fibrotic response in the heart.
Maruyama S, Nakamura K, Papanicolaou KN, Sano S, Shimizu I, Asaumi Y, et al. Follistatin-like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Mol Med. 2016;8(8):949–66. https://doi.org/10.15252/emmm.201506151.
Hookway TA, Matthys OB, Mendoza-Camacho FN, Rains S, Sepulveda JE, Joy DA, et al. Phenotypic variation between stromal cells differentially impacts engineered cardiac tissue function. Tissue Eng Part A. 2019;25(9-10):773–85. https://doi.org/10.1089/ten.TEA.2018.0362.
. Doll S, Dressen M, Geyer PE, Itzhak DN, Braun C, Doppler SA, et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat Commun. 2017;8(1):1469. https://doi.org/10.1038/s41467-017-01747-2This key paper established a region and cell type–resolved proteome map of the healthy human heart. It provides crucial information to facilitate the understanding of the regional differences in cardiac physiology and will serve as a reference for future studies assessing protein expression changes in disease states.
Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong TT, Shaw RM, et al. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell. 2009;16(2):233–44. https://doi.org/10.1016/j.devcel.2008.12.007.
Lighthouse JK, Burke RM, Velasquez LS, Dirkx RA Jr, Aiezza A 2nd, Moravec CS, et al. Exercise promotes a cardioprotective gene program in resident cardiac fibroblasts. JCI Insight. 2019;4(1). https://doi.org/10.1172/jci.insight.92098.
Nakaya M, Watari K, Tajima M, Nakaya T, Matsuda S, Ohara H, et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J Clin Invest. 2017;127(1):383–401. https://doi.org/10.1172/JCI83822.
Kohl P, Camelliti P, Burton FL, Smith GL. Electrical coupling of fibroblasts and myocytes: relevance for cardiac propagation. J Electrocardiol. 2005;38(4 Suppl):45–50. https://doi.org/10.1016/j.jelectrocard.2005.06.096.
Nagaraju CK, Dries E, Gilbert G, Abdesselem M, Wang N, Amoni M, et al. Myofibroblast modulation of cardiac myocyte structure and function. Sci Rep. 2019;9(1):8879. https://doi.org/10.1038/s41598-019-45078-2.
Humeres C, Vivar R, Boza P, Munoz C, Bolivar S, Anfossi R, et al. Cardiac fibroblast cytokine profiles induced by proinflammatory or profibrotic stimuli promote monocyte recruitment and modulate macrophage M1/M2 balance in vitro. J Mol Cell Cardiol. 2016. https://doi.org/10.1016/j.yjmcc.2016.10.014.
Anzai A, Choi JL, He S, Fenn AM, Nairz M, Rattik S, et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J Exp Med. 2017;214(11):3293–310. https://doi.org/10.1084/jem.20170689.
Mouton AJ, Ma Y, Rivera Gonzalez OJ, Daseke MJ 2nd, Flynn ER, Freeman TC, et al. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res Cardiol. 2019;114(2):6. https://doi.org/10.1007/s00395-019-0715-4.
Yu J, Seldin MM, Fu K, Li S, Lam L, Wang P, et al. Topological arrangement of cardiac fibroblasts regulates cellular plasticity. Circ Res. 2018;123(1):73–85. https://doi.org/10.1161/CIRCRESAHA.118.312589.
Alexanian M, Przytycki PF, Micheletti R, Padmanabhan A, Ye L, Travers JG, et al. A transcriptional switch governing fibroblast plasticity underlies reversibility of chronic heart disease. bioRxiv. 2020:2020.07.21.214874. https://doi.org/10.1101/2020.07.21.214874.
. Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, et al. Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ Res. 2016;118(12):1906–17. https://doi.org/10.1161/CIRCRESAHA.116.308643Using a killer gene approach mediated by diphtheria toxin to deplete periostin-expressing cardiac fibroblasts, this work highlighted the potential of selective ablation of a fibroblast subpopulation as a new treatment paradigm for heart failure.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5. https://doi.org/10.1126/science.aar6711.
. Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573(7774):430–3. https://doi.org/10.1038/s41586-019-1546-zThis seminal study demonstrated proof-of-principle for the selective therapeutic depletion of a defined fibroblast subpopulation to improve cardiac performance in a mouse model of heart failure. These results represent the first step towards using immunotherapy for the treatment of chronic cardiac diseases.
Tillmanns J, Hoffmann D, Habbaba Y, Schmitto JD, Sedding D, Fraccarollo D, et al. Fibroblast activation protein alpha expression identifies activated fibroblasts after myocardial infarction. J Mol Cell Cardiol. 2015;87:194–203. https://doi.org/10.1016/j.yjmcc.2015.08.016.
Yamawaki TM, Lu DR, Ellwanger DC, Bhatt D, Manzanillo P, Arias V, et al. Systematic comparison of high-throughput single-cell RNA-seq methods for immune cell profiling. BMC Genomics. 2021;22(1):66. https://doi.org/10.1186/s12864-020-07358-4.
Ding J, Adiconis X, Simmons SK, Kowalczyk MS, Hession CC, Marjanovic ND, et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat Biotechnol. 2020;38(6):737–46. https://doi.org/10.1038/s41587-020-0465-8.
Massaia A, Chaves P, Samari S, Miragaia RJ, Meyer K, Teichmann SA, et al. Single cell gene expression to understand the dynamic architecture of the heart. Front Cardiovasc Med. 2018;5:167. https://doi.org/10.3389/fcvm.2018.00167.
Haque A, Engel J, Teichmann SA, Lonnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9(1):75. https://doi.org/10.1186/s13073-017-0467-4.
Paik DT, Cho S, Tian L, Chang HY, Wu JC. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat Rev Cardiol. 2020;17(8):457–73. https://doi.org/10.1038/s41569-020-0359-y.
Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol. 2019;15(6):e8746. https://doi.org/10.15252/msb.20188746.
Hu P, Liu J, Zhao J, Wilkins BJ, Lupino K, Wu H, et al. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018;32(19-20):1344–57. https://doi.org/10.1101/gad.316802.118.
. Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, et al. Cells of the adult human heart. Nature. 2020. https://doi.org/10.1038/s41586-020-2797-4. These two landmark papers provided a region-resolved cellular map of the healthy human heart using large-scale scRNA-seq and snRNA-seq analysis. These information-rich resources will help understanding the cellular and transcriptional diversity of the human heart and will facilitate identification of cell type–specific therapeutic targets.
. Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC Jr, et al. Transcriptional and cellular diversity of the human heart. Circulation. 2020. https://doi.org/10.1161/CIRCULATIONAHA.119.045401These two landmark papers provided a region-resolved cellular map of the healthy human heart using large-scale scRNA-seq and snRNA-seq analysis. These information-rich resources will help understanding the cellular and transcriptional diversity of the human heart and will facilitate identification of cell type–specific therapeutic targets.
DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, et al. Single-cell resolution of temporal gene expression during heart development. Dev Cell. 2016;39(4):480–90. https://doi.org/10.1016/j.devcel.2016.10.001.
Li G, Xu A, Sim S, Priest JR, Tian X, Khan T, et al. Transcriptomic profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev Cell. 2016;39(4):491–507. https://doi.org/10.1016/j.devcel.2016.10.014.
Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552(7683):110–5. https://doi.org/10.1038/nature24676.
Gladka MM, Molenaar B, de Ruiter H, van der Elst S, Tsui H, Versteeg D, et al. Single-cell sequencing of the healthy and diseased heart reveals cytoskeleton-associated protein 4 as a new modulator of fibroblasts activation. Circulation. 2018;138(2):166–80. https://doi.org/10.1161/CIRCULATIONAHA.117.030742.
. Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, et al. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 2018;22(3):600–10. https://doi.org/10.1016/j.celrep.2017.12.072This key paper presented a detailed analysis of non-myocyte population in the healthy mouse heart by scRNA-seq.
Kretzschmar K, Post Y, Bannier-Helaouet M, Mattiotti A, Drost J, Basak O, et al. Profiling proliferative cells and their progeny in damaged murine hearts. Proc Natl Acad Sci U S A. 2018;115(52):E12245–E54. https://doi.org/10.1073/pnas.1805829115.
Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J, et al. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019;33(21-22):1491–505. https://doi.org/10.1101/gad.329763.119.
Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. 2019;8. https://doi.org/10.7554/eLife.43882.
Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019;26(7):1934–50 e5. https://doi.org/10.1016/j.celrep.2019.01.079.
McLellan MA, Skelly DA, Dona MSI, Squiers GT, Farrugia GE, Gaynor TL, et al. High-resolution transcriptomic profiling of the heart during chronic stress reveals cellular drivers of cardiac fibrosis and hypertrophy. Circulation. 2020. https://doi.org/10.1161/CIRCULATIONAHA.119.045115.
Ren Z, Yu P, Li D, Li Z, Liao Y, Wang Y, et al. Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation. 2020;141(21):1704–19. https://doi.org/10.1161/CIRCULATIONAHA.119.043053.
Ruiz-Villalba A, Romero JP, Hernandez SC, Vilas-Zornoza A, Fortelny N, Castro-Labrador L, et al. Single-cell RNA sequencing analysis reveals a crucial role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) cardiac fibroblasts after myocardial infarction. Circulation. 2020;142(19):1831–47. https://doi.org/10.1161/CIRCULATIONAHA.119.044557.
. Forte E, Skelly DA, Chen M, Daigle S, Morelli KA, Hon O, et al. Dynamic interstitial cell response during myocardial infarction predicts resilience to rupture in genetically diverse mice. Cell Rep. 2020;30(9):3149–63 e6. https://doi.org/10.1016/j.celrep.2020.02.008This work presented a comprehensive analysis of time-dependent changes to cardiac interstitial cell transcriptional program, covering both acute and chronic phases of remodeling post-MI injury.
Wang L, Yu P, Zhou B, Song J, Li Z, Zhang M, et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat Cell Biol. 2020;22(1):108–19. https://doi.org/10.1038/s41556-019-0446-7.
Wang Z, Cui M, Shah AM, Tan W, Liu N, Bassel-Duby R, et al. Cell-type-specific gene regulatory networks underlying murine neonatal heart regeneration at single-cell resolution. Cell Rep. 2020;33(10):108472. https://doi.org/10.1016/j.celrep.2020.108472.
Meilhac SM, Buckingham ME. The deployment of cell lineages that form the mammalian heart. Nat Rev Cardiol. 2018;15(11):705–24. https://doi.org/10.1038/s41569-018-0086-9.
. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, et al. Revisiting cardiac cellular composition. Circ Res. 2016;118(3):400–9. https://doi.org/10.1161/CIRCRESAHA.115.307778Using a combination of cellular markers and genetic tools, this key study provided the first comprehensive analysis of the cellular composition of the mouse and human heart.
Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods. 2020;17(2):159–62. https://doi.org/10.1038/s41592-019-0667-5.
Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. 2020;15(4):1484–506. https://doi.org/10.1038/s41596-020-0292-x.
Shinde AV, Humeres C, Frangogiannis NG. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta Mol basis Dis. 2017;1863(1):298–309. https://doi.org/10.1016/j.bbadis.2016.11.006.
Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124(7):2921–34. https://doi.org/10.1172/JCI74783.
. Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. 2018;128(5):2127–43. https://doi.org/10.1172/JCI98215Using genetic lineage tracing and transcriptional profiling, Fu and colleagues defined the dynamic phenotypic changes of fibroblasts populations in the remodeling heart after MI injury and revealed a specialized fibroblast subpopulation in the mature scar termed “matrifibrocytes,” a long-lasting chronic state of fibroblasts.
Ivey MJ, Kuwabara JT, Pai JT, Moore RE, Sun Z, Tallquist MD. Resident fibroblast expansion during cardiac growth and remodeling. J Mol Cell Cardiol. 2018;114:161–74. https://doi.org/10.1016/j.yjmcc.2017.11.012.
Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–90. https://doi.org/10.1038/nature14590.
Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015;33(11):1165–72. https://doi.org/10.1038/nbt.3383.
Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 2019;568(7751):235–9. https://doi.org/10.1038/s41586-019-1049-y.
Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363(6434):1463–7. https://doi.org/10.1126/science.aaw1219.
Ugolini GS, Pavesi A, Rasponi M, Fiore GB, Kamm R, Soncini M. Human cardiac fibroblasts adaptive responses to controlled combined mechanical strain and oxygen changes in vitro. Elife. 2017;6. https://doi.org/10.7554/eLife.22847.
Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–8. https://doi.org/10.1038/nmeth.4380.
Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. 2017;35(10):936–9. https://doi.org/10.1038/nbt.3973.
Dominguez Conde C, Teichmann SA. Deciphering immunity at high plexity and resolution. Nat Rev Immunol. 2020;20(2):77–8. https://doi.org/10.1038/s41577-019-0254-0.
Swaminathan J, Boulgakov AA, Hernandez ET, Bardo AM, Bachman JL, Marotta J, et al. Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat Biotechnol. 2018. https://doi.org/10.1038/nbt.4278.
Zhang MJ, Ntranos V, Tse D. Determining sequencing depth in a single-cell RNA-seq experiment. Nat Commun. 2020;11(1):774. https://doi.org/10.1038/s41467-020-14482-y.
Deshaies RJ. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature. 2020;580(7803):329–38. https://doi.org/10.1038/s41586-020-2168-1.
Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583(7814):127–32. https://doi.org/10.1038/s41586-020-2403-9.
Viardot A, Bargou R. Bispecific antibodies in haematological malignancies. Cancer Treat Rev. 2018;65:87–95. https://doi.org/10.1016/j.ctrv.2018.04.002.
Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. 2019;177(7):1701–13 e16. https://doi.org/10.1016/j.cell.2019.04.041.
Verma R, Mohl D, Deshaies RJ. Harnessing the power of proteolysis for targeted protein inactivation. Mol Cell. 2020;77(3):446–60. https://doi.org/10.1016/j.molcel.2020.01.010.
Pettersson M, Crews CM. PROteolysis TArgeting Chimeras (PROTACs) - past, present and future. Drug Discov Today Technol. 2019;31:15–27. https://doi.org/10.1016/j.ddtec.2019.01.002.
Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–37. https://doi.org/10.1038/nrd.2016.268.
Lu D, Thum T. RNA-based diagnostic and therapeutic strategies for cardiovascular disease. Nat Rev Cardiol. 2019;16(11):661–74. https://doi.org/10.1038/s41569-019-0218-x.
Lieberman J. Tapping the RNA world for therapeutics. Nat Struct Mol Biol. 2018;25(5):357–64. https://doi.org/10.1038/s41594-018-0054-4.
Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35(3):222–9. https://doi.org/10.1038/nbt.3802.
Nair JK, Willoughby JL, Chan A, Charisse K, Alam MR, Wang Q, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136(49):16958–61. https://doi.org/10.1021/ja505986a.
Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N Engl J Med. 2017;376(1):41–51. https://doi.org/10.1056/NEJMoa1609243.
Biscans A, Coles A, Haraszti R, Echeverria D, Hassler M, Osborn M, et al. Diverse lipid conjugates for functional extra-hepatic siRNA delivery in vivo. Nucleic Acids Res. 2019;47(3):1082–96. https://doi.org/10.1093/nar/gky1239.
Biscans A, Coles A, Echeverria D, Khvorova A. The valency of fatty acid conjugates impacts siRNA pharmacokinetics, distribution, and efficacy in vivo. J Control Release. 2019;302:116–25. https://doi.org/10.1016/j.jconrel.2019.03.028.
Peer D, Zhu P, Carman CV, Lieberman J, Shimaoka M. Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function-associated antigen-1. Proc Natl Acad Sci U S A. 2007;104(10):4095–100. https://doi.org/10.1073/pnas.0608491104.
Kedmi R, Veiga N, Ramishetti S, Goldsmith M, Rosenblum D, Dammes N, et al. A modular platform for targeted RNAi therapeutics. Nat Nanotechnol. 2018;13(3):214–9. https://doi.org/10.1038/s41565-017-0043-5.
Weinstein S, Toker IA, Emmanuel R, Ramishetti S, Hazan-Halevy I, Rosenblum D, et al. Harnessing RNAi-based nanomedicines for therapeutic gene silencing in B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113(1):E16–22. https://doi.org/10.1073/pnas.1519273113.
Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. 2016;164(4):770–9. https://doi.org/10.1016/j.cell.2016.01.011.
Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell. 2019;35(3):489–503 e8. https://doi.org/10.1016/j.ccell.2019.02.003.
Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl Med. 2013;5(207):207ra144. https://doi.org/10.1126/scitranslmed.3006682.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
All authors are employees of Amgen Inc.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Regenerative Medicine
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shi, S.Y., Luo, X., Yamawaki, T.M. et al. Recent Advances in Single-Cell Profiling and Multispecific Therapeutics: Paving the Way for a New Era of Precision Medicine Targeting Cardiac Fibroblasts. Curr Cardiol Rep 23, 82 (2021). https://doi.org/10.1007/s11886-021-01517-z
Accepted:
Published:
DOI: https://doi.org/10.1007/s11886-021-01517-z