
Abstract
Purpose of Review
In recent years, a family of adiponectin paralogs designated as C1q/TNF-related protein (CTRP) has attracted increasing attention. They are inflammatory adipocytokines mostly secreted from epicardial adipose tissue, which modulate the development and prognosis of coronary artery disease (CAD). This review summarizes the pathophysiological roles of individual members of the CTRP superfamily in the development of CAD.
Recent Findings
Recent studies have revealed how members of the CTRP family, CTRP1, CTRP3, CTRP5, CTRP9, CTRP12, and CTRP13, can influence both development and progression of CAD by modulating metabolic pathways, influencing immuno-inflammatory response, and regulating cardiovascular functions.
Summary
Research to date has not been sufficient to answer the specific mechanism of the CTRP family in the occurrence and development of CAD. This review explores the evidence of CTRP superfamily regulating different pathophysiology stages of CAD through the immuno-inflammation, glucose and lipid metabolism, and vascular endothelial function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Endothelial dysfunction, inflammatory response, and metabolic dysregulation are key factors involved in initiation and progression of coronary artery disease (CAD) [1, 2]. Epicardial adipose tissue (EAT) is located inside the pericardial sac, which is adjacent to the epicardium surrounding the heart. EAT is considered a metabolically active organ with endocrine activity. It can secrete a large amount of inflammatory adipocytokines, of which, adiponectin is a well-known cardiovascular protective factor [3, 4]. The C1q complement/tumor necrosis factor (TNF)–associated proteins (CTRPs) superfamily is a paralog of adiponectin, composed of CTRP1-CTRP15, which share a common structural domain with adiponectin [5]. CTRPs mRNA showed highest expression in white adipose tissue around the heart, making it the main secretory organ, though these proteins are also secreted by other viscera, such as the heart and liver [6]. Increasingly, researchers have begun to focus on the pathophysiological role of the CTRP family in cardiovascular diseases. The main members of the CTRP family related to the pathophysiological mechanisms of CAD were found to be CTRP1, CTRP3, CTRP5, CTRP9, CTRP12, and CTRP13. These proteins regulate endothelial function, inflammatory response, and metabolic dysfunction to influence CAD progression.
Regulation of Immune-Inflammation
CAD is a complex, chronic process that does not simply involve intra-arterial accumulation of cholesterol and calcium. An inflammatory response driven by both pro-inflammatory cells and cytokines also plays an important role in driving development of atherosclerosis and promoting thrombosis, leading to adverse cardiovascular events [7, 8].
CTRP1 regulates low-grade chronic inflammation in coronary atherosclerosis [9]. The inflammatory response and pro-inflammatory cytokines induce increased secretion of CTRP1, which in turn increases expression of adhesion molecules and chemokines such as TNF-α, interleukin (IL)-6, and IL-1b, by activating the p38 MAPK/NF-kB pathway [10,11,12]. These processes stimulate both in vitro and in vivo adhesion of leukocytes to endothelial cells and further promote formation of macrophages and macrophage-derived foam cells in atherosclerotic plaques, thus exerting a pro-inflammatory, pro-atherogenic effect and accelerating deterioration of CAD [10, 13]. Conversely, CTRP1 can also activate the 1P/cAMP-dependent pathway in cardiomyocytes to reduce apoptosis and inflammatory response, and thus exert a beneficial effect on the pathophysiology of ischemic heart disease [14].
CTRP3 is a potent anti-inflammatory adipokine that inhibits proinflammatory pathways in monocytes and microcells, exerting anti-inflammatory, anti-apoptotic, and cardioprotective effect during development of CAD [15, 16]. CTRP3 affects inhibitory toll-like receptors (TLRs) and nuclear factor kappa B (NF-κB) signaling pathways to reduce secretion of inflammatory adipocytokines, thus attenuating both insulin resistance and obesity-related, chronic, systemic anti-inflammatory responses [17, 18]. Furthermore, CTRP3 promotes activation of the PI3K/Akt/eNOS pathway, which inhibits endothelial inflammation induced by oxidized low-density lipoproteins (ox-LDL) by downregulating expression of CTRP, TNF-α, and IL-6, delaying atherosclerosis progression [19••].
Elevated levels of CTRP5 can promote in-stent restenosis after coronary stent implantation. CTRP5 promotes expression of matrix metalloproteinase-2, cyclin D1, and TNF-α in vascular endothelial cells, by activating Notch1, transforming growth factor (TGF)-β, and hedgehog pathways, thereby promoting the growth, migration, and inflammation of vascular smooth muscle cells (VSMC) [20•].
CTRP9 is the closest paralog of adiponectin, and its anti-inflammatory and anti-atherosclerosis features allow it to play a cardioprotective role in the CAD process [21]. CTRP9 stimulates adenosine monophosphate (AMP)–activated protein kinase pathway to inhibit expression of adhesion molecules such as intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 in endothelial cells, decreases secretion of proinflammatory cytokines such as TNF-α and monocyte chemoattractant protein (MCP)-1 within atherosclerotic plaques, reduces proliferation VSMC, promotes vasodilation (further inhibiting inflammatory responses in macrophages), and thus increases stability of atherosclerotic plaques [22, 23•, 24].
CTRP12 is associated with inflammatory cytokines and plays a potential role in atherosclerosis. CTRP12 is known to reduce the expression of pro-inflammatory cytokines and decrease macrophage accumulation within adipose tissue in obese mice, and also was found to inhibit the secretion of inflammatory cytokines IL-6 and TNF-α in CAD patients [25, 26]. Therefore, overexpression of CTRP12 exerts an anti-inflammatory effect during both development and deterioration of CAD.
CTRP13 inhibits macrophage activation and infiltration of vessel walls, reduces plaque formation, and thereby inhibits development of atherosclerosis. Furthermore, it prevents proliferation and migration of macrophages by downregulating lipid uptake, delays local and systemic inflammatory responses during atherosclerosis by promoting autophagy (in macrophages), and accelerates CD36-dependent degradation of autophagolysosomal pathways, thus reducing number of macrophages in lesions [27, 28]. A case-control study found that CTRP13 led to decreases in obesity and inflammation and that it showed negative correlation with proinflammatory cytokines such as TNF-α and IL-6, while TNF-α and body mass index (BMI) were its independent negative predictors [16].
Effects on Glucose and Lipid Metabolism
Glucose and lipid metabolism are the two major processes involved in increasing the risk and severity of CAD. Abnormal metabolism affects activity of regulatory pathways, composition of the final product, degree of inflammation, and coronary-plaque formation, thus contributing to the development of CAD and accelerating occurrence of adverse cardiovascular events [29, 30].
CTRP1 is involved in regulation of obesity-related, metabolic, and cardiovascular diseases, and affects cardiac metabolism by primarily regulating blood glucose and lipid metabolism [31]. Increased expression of CTRP1 could improve insulin sensitivity and glucose tolerance, which in turn may increase glucose metabolism and reduce adiposity in an overnutritional state [32]. CTRP1 increases fatty acid oxidation and energy expenditure. It inhibits acetyl-CoA carboxylase (ACC) via the AMP protein kinase (AMPK) pathway to attenuate obesity [33].
CTRP3 is a cardioprotective, anti-inflammatory cytokine. It improves insulin sensitivity, enhances insulin-mediated glucose uptake, and reduces hepatocyte gluconeogenesis (and subsequent glucose output), thereby slowing down development of CAD [34, 35]. A cross-sectional study found low levels of CTRP3 in association with CAD, though its levels in correlation to obesity and diabetes showed sex-specific differences [36].
CTRP5 upregulates 12/-15-lipoxygenase (LOX) expression via the signal transducer and activator of transcription (STAT)-6 signaling pathway. Inhibition of the STAT6-12/15-lipoxygenase-dependent pathway attenuates CTRP5-induced transcytosis and oxidative modification of the LDL transendothelial monolayers, thereby retarding development and progression of early-stage atherosclerosis [37].
CTRP9 regulates lipid metabolism and enhances the AMPK/mTOR autophagy signaling pathway to enhance acid-lipid-mediated cholesterol efflux, increases the level of expression of the cholesterol-transporting receptors like ATP-binding membrane cassette transporter (ABC) A1, and G1 (ABCG1), which accelerates cholesterol efflux from foam cells, thereby preventing THP-1 macrophages from forming foam cells and slowing progression of early atherosclerosis [24, 38]. An in vivo study of mice showed that overexpression of CTRP9 reduced fasting insulin levels and fasting blood glucose, increasing insulin sensitivity [39, 40]. CTRP9 correlated positively with parameters of glucose metabolism by activating Akt, AMPK, and p42/44 MAPK pathways, and further increasing glucose uptake [41].
CTRP12 inhibits gluconeogenesis and increases glucose uptake in hepatocytes and adipocytes by activating the PI3K-Akt signaling pathway and improving insulin sensitivity [42]. CTRP12 exerts a beneficial effect on glucose and insulin metabolism and plays a potential detrimental role in atherosclerosis via its association with insulin resistance, decreased high-density lipoprotein cholesterol, and increased BMI [43, 44].
CTRP13 exerts a beneficial effect during insulin-mediated glucose uptake, which can reduce glucose output in hepatocytes by inhibiting the expression of gluconeogenic enzymes. CTRP13 reduces phosphorylation of AMPK in adipocytes, muscle cells, and hepatocytes; promotes AMPK signaling pathway activation to increase glucose uptake in adipocytes; and inhibits stress-activated protein kinase/JNK stress signaling, to decrease lipid-induced, insulin resistance in hepatocytes, thereby reducing hepatocytic gluconeogenesis and decreasing insulin resistance [34]. In addition, CTRP13 hydrolyzes cholesterol droplets stored in macrophages, inhibits intracellular influx of cholesterol, and promotes cholesterol efflux, thus inhibiting the formation of foam cells and decelerating progression of atherosclerosis [45, 46].
Mechanisms of Vascular and Myocardial Injury
Coronary artery endothelial injury is an early event in the pathological process of atherosclerosis, mediated via immune-inflammation, oxidative stress, and biochemicals [47, 48]. Endothelial cells are constantly exposed to these stimulating factors and strive to maintain antithrombotic, anti-inflammatory, and anti-proliferative homeostasis through compensatory responses [49]. This normal homeostasis, when impaired, can aggravate subsequently the inflammatory response, leading to vasomotor dysfunction and ventricular remodeling, as seen after myocardial infarction [50, 51].
CTRP1 may serve as a vasculoprotective adipokine, with similar effects on vascular injury as seen with adiponectin. Increased expression of CTRP1 reduces neointimal hyperplasia and cell proliferation in damaged arteries after vascular injury, through inhibition of VSMC growth via cAMP-dependent pathways. In addition, it attenuates accumulation of macrophages in damaged blood vessels, while leaving the degree of both apoptosis and reendothelialization unaffected [52, 53]. CTRP1 prevents pathological vascular remodeling, inhibits formation of collateral circulation by inhibiting in vitro angiogenesis of endothelial progenitor cells, and prevents vascular stenosis after injury [54]. CTRP1 is an acute phase reactant of inflammation and is positively associated with fibrinogen, which can cause cross-linking and aggregation of platelets, leading to thrombosis, thereby indicating an association with adverse cardiovascular events [55].
CTRP3 reduces myocardial damage after ischemia and plays a cardioprotective role. CTRP3 attenuates pathological myocardial remodeling after an acute infarction through inhibition of myocardial fibrosis and enhances the survival and regeneration of ischemic cardiomyocytes [56, 57]. In addition, CTRP3 can possibly increase Akt phosphorylation and induce expression of hypoxia-inducible factor 1-α, vascular endothelial growth factor, and promote secretion of angiogenic factors from endothelial cells, which can contribute to angiogenesis [56].
CTRP9 has a higher vasoactive potency than adiponectin and plays an important role in the regulation of vascular stiffness [58]. It can promote vasodilation, inhibit both neointimal hyperplasia and endothelium-dependent VSMC proliferation, attenuate atherosclerosis, and exert a protective effect on cardiac remodeling after acute myocardial infarction [59,60,61]. Overexpression of CTRP9 in circulation and in EAT was found to significantly attenuate VSMC proliferation and neointimal formation [62].
Conclusion
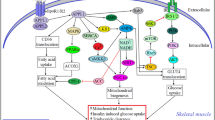
The CTRP family plays an important role in all stages of CAD by regulating immuno-inflammation, glucose and lipid metabolism, and vascular endothelial function. (Table 1) CTRP1 represents as pro-inflammatory and pro-atherosclerotic markers by contributing toward the secretion of inflammatory cytokines and adhesion molecules and promoting the formation of foam cells from macrophages. CTRP5 promotes VSMC growth, migration, and inflammation. In contrast, CTRP3, CTRP9, CTRP12, and CTRP13 activate anti-inflammatory and anti-atherosclerotic mechanisms of CAD, by inhibiting endothelial inflammation and reducing plaque formation (mediated via inhibition of both inflammatory cytokine secretion and expression of adhesion molecules). Also, these four family members reduce macrophage accumulation and foam-cell formation. CTRP family members regulate vascular endothelial inflammation and plaque formation by regulating glucose and lipid metabolism. This protein superfamily could improve insulin sensitivity, decrease insulin resistance, increase glucose tolerance, enhance glucose uptake, and reduce gluconeogenesis. Furthermore, they also enhance expression of cholesterol transport receptors, promote cholesterol efflux, and increase fatty acid oxidation. CTRP1, CTRP3, and CTRP9 increase expression of HIF1α and vascular endothelial growth factor, promote secretion of endothelial cell angiogenic factors, inhibit neointimal hyperplasia and VSMC proliferation, and inhibit myocardial fibrosis, thus supporting the survival and regeneration of ischemic cardiomyocytes (Fig. 1).

Macrophages derive from monocytes, which form foam cells following phagocytosis of lipids. The accumulation of macrophages and foam cells promotes the formation of atherosclerotic plaques. Vascular injury promotes secretion of inflammatory cytokines, adhesion molecules, and chemokines, which aggravates the inflammatory response of the vascular endothelium and promotes plaque formation. Endothelial cell angiogenic factors promote endothelial cell proliferation and enhance the survival and regeneration of ischemic cardiomyocytes. Vascular remodeling due to long-term chronic inflammation stimulation manifests as the thickened blood vessel wall and the narrowed lumen
CTRP1 and CTRP5, as possible risk factors for CAD, elevate in patients with CAD and associate with the severity of coronary stenosis. On the contrary, CTRP3, CTRP9, CTRP12, and CTRP13, as protective factors for CAD, decrease in patients with CAD. Thereby, this review on CTRP superfamily may take unique insight into the development and progression of CAD. Positive results from such research and further understanding of their molecular mechanisms will promote adding these biomarkers to CAD diagnostic guidelines.
Abbreviations
- CAD:
-
Coronary artery disease
- EAT:
-
Epicardial adipose tissue
- TNF:
-
Tumor necrosis factor
- CTRPs:
-
C1q complement/tumor necrosis factor (TNF)–associated proteins
- TLR:
-
Toll-like receptor
- NF-κB:
-
Nuclear factor kappa B
- Ox-LDL:
-
Oxidized low-density lipoproteins
- IL:
-
Interleukin
- MMP:
-
Matrix metalloproteinase
- TGF:
-
Transforming growth factor
- AMP:
-
Adenosine monophosphate
- ICAM:
-
Intercellular adhesion molecule
- VCAM:
-
Vascular cell adhesion molecule
- MCP:
-
Monocyte chemoattractant protein
- VSMC:
-
Vascular smooth muscle cells
- BMI:
-
Body mass index
- AMPK:
-
AMP protein kinase
- ACC:
-
Acetyl-CoA carboxylase
- LOX:
-
Lipoxygenase
- STAT:
-
Signal transducer and activator of transcription
- ABC:
-
ATP-binding membrane cassette transporter
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213(3):337–54.
Xu S, Xu Y, Liu P, et al. The novel coronary artery disease risk gene JCAD/KIAA1462 promotes endothelial dysfunction and atherosclerosis. Eur Heart J. 2019;40(29):2398–408.
Aprigliano G, Scuteri L, Iafelice I, Li Volsi L, Cuko B, Palloshi A, et al. Epicardial adipose tissue thickness and acute coronary syndrome: a matter of how much or how? Int J Cardiol. 2015;199:8–9.
Hou H, Ge S, Zhao L, Wang C, Wang W, Zhao X, et al. An updated systematic review and meta-analysis of association between adiponectin gene polymorphisms and coronary artery disease. Omics. 2017;21(6):340–51.
Wong GW, Wang J, Hug C, Tsao TS, Lodish HF. A family of Acrp30/adiponectin structural and functional paralogs. Proc Natl Acad Sci U S A. 2004;101(28):10302–7.
Schaffler A, Buechler C. CTRP family: linking immunity to metabolism. Trends Endocrinol Metab. 2012;23(4):194–204.
Nguyen MT, Fernando S, Schwarz N, et al. Inflammation as a therapeutic target in atherosclerosis. J Clin Med. 2019;8(8):1109.
Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci. 2018;132(12):1243–52.
Wang XQ, Liu ZH, Xue L, Lu L, Gao J, Shen Y, et al. C1q/TNF-related protein 1 links macrophage lipid metabolism to inflammation and atherosclerosis. Atherosclerosis. 2016;250:38–45.
Lu L, Zhang RY, Wang XQ, et al. C1q/TNF-related protein-1: an adipokine marking and promoting atherosclerosis. Eur Heart J. 2016;37(22):1762–71.
Muendlein A, Leiherer A, Saely C, Ebner J, Geiger K, Brandtner EM, et al. The novel adipokine CTRP1 is significantly associated with the incidence of major adverse cardiovascular events. Atherosclerosis. 2019;286:1–6.
Kim D, Park SY. C1q and TNF related protein 1 regulates expression of inflammatory genes in vascular smooth muscle cells. Genes Genomics. 2019;41(4):397–406.
Xin Y, Lyu X, Wang C, Fu Y, Zhang S, Tian C, et al. Elevated circulating levels of CTRP1, a novel adipokine, in diabetic patients. Endocr J. 2014;61(9):841–7.
Yuasa D, Ohashi K, Shibata R, et al. C1q/TNF-related protein-1 functions to protect against acute ischemic injury in the heart. FASEB J. 2016;30(3):1065–75.
Hofmann C, Chen N, Obermeier F, Paul G, Büchler C, Kopp A, et al. C1q/TNF-related protein-3 (CTRP-3) is secreted by visceral adipose tissue and exerts antiinflammatory and antifibrotic effects in primary human colonic fibroblasts. Inflamm Bowel Dis. 2011;17(12):2462–71.
Fadaei R, Moradi N, Baratchian M, Aghajani H, Malek M, Fazaeli AA, et al. Association of C1q/TNF-related protein-3 (CTRP3) and CTRP13 serum levels with coronary artery disease in subjects with and without type 2 diabetes mellitus. PLoS One. 2016;11(12):e0168773.
Petersen PS, Wolf RM, Lei X, et al. Immunomodulatory roles of CTRP3 in endotoxemia and metabolic stress. Phys Rep. 2016;4(5):e12735.
Schmid A, Kopp A, Hanses F, Karrasch T, Schäffler A. C1q/TNF-related protein-3 (CTRP-3) attenuates lipopolysaccharide (LPS)-induced systemic inflammation and adipose tissue Erk-1/-2 phosphorylation in mice in vivo. Biochem Biophys Res Commun. 2014;452(1):8–13.
•• Chen L, Qin L, Liu X, et al. CTRP3 alleviates ox-LDL-induced inflammatory response and endothelial dysfunction in mouse aortic endothelial cells by activating the PI3K/Akt/eNOS pathway. Inflammation. 2019;42(4):1350–9 Suggests the pathological pathway of CTRP3 affecting metabolism and inflammation.
• Shen Y, Li C, Zhang RY, et al. Association of increased serum CTRP5 levels with in-stent restenosis after coronary drug-eluting stent implantation: CTRP5 promoting inflammation, migration and proliferation in vascular smooth muscle cells. Int J Cardiol. 2017;228:129–36 This is the first study to demonstrate the role of CTRP member in patients after PCI.
Wang J, Hang T, Cheng XM, et al. Associations of C1q/TNF-related protein-9 levels in serum and epicardial adipose tissue with coronary atherosclerosis in humans. Biomed Res Int. 2015;971683:17.
Li J, Zhang P, Li T, Liu Y, Zhu Q, Chen T, et al. CTRP9 enhances carotid plaque stability by reducing pro-inflammatory cytokines in macrophages. Biochem Biophys Res Commun. 2015;458(4):890–5.
• Jung CH, Lee MJ, Kang YM, et al. C1q/TNF-related protein-9 inhibits cytokine-induced vascular inflammation and leukocyte adhesiveness via AMP-activated protein kinase activation in endothelial cells. Mol Cell Endocrinol. 2016;419:235–43 This is the first study to demonstrate that CTRP9 attenuates cytokine-induced vascular inflammation in endothelial cells mediated by AMPK activation.
Zhang L, Liu Q, Zhang H, Wang XD, Chen SY, Yang Y, et al. C1q/TNF-related protein 9 inhibits THP-1 macrophage foam cell formation by enhancing autophagy. J Cardiovasc Pharmacol. 2018;72(4):167–75.
Fadaei R, Moradi N, Kazemi T, Chamani E, Azdaki N, Moezibady SA, et al. Decreased serum levels of CTRP12/adipolin in patients with coronary artery disease in relation to inflammatory cytokines and insulin resistance. Cytokine. 2019;113:326–31.
Enomoto T, Ohashi K, Shibata R, et al. Adipolin/C1qdc2/CTRP12 protein functions as an adipokine that improves glucose metabolism. J Biol Chem. 2011;286(40):34552–8.
Wang C, Xu W, Liang M, Huang D, Huang K. CTRP13 inhibits atherosclerosis via autophagy-lysosome-dependent degradation of CD36. FASEB J. 2019;33(2):2290–300.
Mendez-Barbero N, Esteban V, Villahoz S, et al. A major role for RCAN1 in atherosclerosis progression. EMBO Mol Med. 2013;5(12):1901–17.
Widecka K, Safranow K, Lewandowski M, Przybycień K, Gorący J, Kornacewicz-Jach Z. Angiographic severity of coronary artery disease and cardiovascular risk in acute coronary syndrome in patients with metabolic syndrome. Kardiol Pol. 2018;76(3):662–8.
Adeva-Andany MM, Martinez-Rodriguez J, Gonzalez-Lucan M, et al. Insulin resistance is a cardiovascular risk factor in humans. Diabetes Metab Syndr. 2019;13(2):1449–55.
Ouchi N, Walsh K. Cardiovascular and metabolic regulation by the adiponectin/C1q/tumor necrosis factor-related protein family of proteins. Circulation. 2012;125(25):3066–8.
Peterson JM, Aja S, Wei Z, Wong GW. CTRP1 protein enhances fatty acid oxidation via AMP-activated protein kinase (AMPK) activation and acetyl-CoA carboxylase (ACC) inhibition. J Biol Chem. 2012;287(2):1576–87.
Shen L, Wang S, Ling Y, Liang W. Association of C1q/TNF-related protein-1 (CTRP1) serum levels with coronary artery disease. J Int Med Res. 2019;47(6):2571–9.
Wei Z, Peterson JM, Wong GW. Metabolic regulation by C1q/TNF-related protein-13 (CTRP13): activation OF AMP-activated protein kinase and suppression of fatty acid-induced JNK signaling. J Biol Chem. 2011;286(18):15652–65.
Peterson JM, Wei Z, Wong GW. C1q/TNF-related protein-3 (CTRP3), a novel adipokine that regulates hepatic glucose output. J Biol Chem. 2010;285(51):39691–701.
Wagner RM, Sivagnanam K, Clark WA, et al. Divergent relationship of circulating CTRP3 levels between obesity and gender: a cross-sectional study. PeerJ. 2016;4:e2573.
Li C, Chen JW, Liu ZH, Shen Y, Ding FH, Gu G, et al. CTRP5 promotes transcytosis and oxidative modification of low-density lipoprotein and the development of atherosclerosis. Atherosclerosis. 2018;278:197–209.
Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328(5986):1689–93.
Peterson JM, Wei Z, Seldin MM, et al. CTRP9 transgenic mice are protected from diet-induced obesity and metabolic dysfunction. Am J Phys Regul Integr Comp Phys. 2013;305(5):R522–33.
Wei Z, Lei X, Petersen PS, Aja S, Wong GW. Targeted deletion of C1q/TNF-related protein 9 increases food intake, decreases insulin sensitivity, and promotes hepatic steatosis in mice. Am J Physiol Endocrinol Metab. 2014;306(7):E779–90.
Moradi N, Fadaei R, Emamgholipour S, Kazemian E, Panahi G, Vahedi S, et al. Association of circulating CTRP9 with soluble adhesion molecules and inflammatory markers in patients with type 2 diabetes mellitus and coronary artery disease. PLoS One. 2018;13(1):e0192159.
Wei Z, Peterson JM, Lei X, et al. C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J Biol Chem. 2012;287(13):10301–15.
Tan BK, Lewandowski KC, O’Hare JP, Randeva HS. Insulin regulates the novel adipokine adipolin/CTRP12: in vivo and ex vivo effects. J Endocrinol. 2014;221(1):111–9.
Tan BK, Chen J, Hu J, Amar O, Mattu HS, Ramanjaneya M, et al. Circulatory changes of the novel adipokine adipolin/CTRP12 in response to metformin treatment and an oral glucose challenge in humans. Clin Endocrinol. 2014;81(6):841–6.
Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–21.
Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13(6):655–67.
He L, Huang X, Kanisicak O, et al. Preexisting endothelial cells mediate cardiac neovascularization after injury. J Clin Invest. 2017;127(8):2968–81.
Vieceli Dalla Sega F, Mastrocola R, Aquila G, et al. KRIT1 deficiency promotes aortic endothelial dysfunction. Int J Mol Sci. 2019;20(19):4930.
Sari CI, Eikelis N, Head GA, et al. Android fat deposition and its association with cardiovascular risk factors in overweight young males. Front Physiol. 2019;10:1162.
Obata JE, Horikoshi T, Nakamura T, et al. Sustained endothelial dysfunction in the infarct-related coronary artery is associated with left ventricular adverse remodeling in survivors of ST-segment elevation myocardial infarction. J Cardiol. 2019;75(3):219–338 [Online ahead of print].
Horikoshi T, Obata JE, Nakamura T, Fujioka D, Watanabe Y, Nakamura K, et al. Persistent dysfunction of coronary endothelial vasomotor responses is related to atheroma plaque progression in the infarct-related coronary artery of AMI survivors. J Atheroscler Thromb. 2019;26(12):1062–74.
Kanemura N, Shibata R, Ohashi K, Ogawa H, Hiramatsu-Ito M, Enomoto T, et al. C1q/TNF-related protein 1 prevents neointimal formation after arterial injury. Atherosclerosis. 2017;257:138–45.
Wu YJ, Bond M, Sala-Newby GB, Newby AC. Altered S-phase kinase-associated protein-2 levels are a major mediator of cyclic nucleotide-induced inhibition of vascular smooth muscle cell proliferation. Circ Res. 2006;98(9):1141–50.
Shen Y, Lu L, Liu ZH, Wu F, Zhu JZ, Sun Z, et al. Increased serum level of CTRP1 is associated with low coronary collateralization in stable angina patients with chronic total occlusion. Int J Cardiol. 2014;174(1):203–6.
Ang L, Mahmud E. Monitoring oral antiplatelet therapy: is it justified? Ther Adv Cardiovasc Dis. 2008;2(6):485–96.
Yi W, Sun Y, Yuan Y, Lau WB, Zheng Q, Wang X, et al. C1q/tumor necrosis factor-related protein-3, a newly identified adipokine, is a novel antiapoptotic, proangiogenic, and cardioprotective molecule in the ischemic mouse heart. Circulation. 2012;125(25):3159–69.
Wu D, Lei H, Wang JY, Zhang CL, Feng H, Fu FY, et al. CTRP3 attenuates post-infarct cardiac fibrosis by targeting Smad3 activation and inhibiting myofibroblast differentiation. J Mol Med. 2015;93(12):1311–25.
Jung CH, Lee MJ, Kang YM, Jang JE, Leem J, Lee YL, et al. Association of serum C1q/TNF-related protein-9 concentration with arterial stiffness in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2014;99(12):E2477–84.
Zheng Q, Yuan Y, Yi W, et al. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler Thromb Vasc Biol. 2011;31(11):2616–23.
Kambara T, Ohashi K, Shibata R, Ogura Y, Maruyama S, Enomoto T, et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J Biol Chem. 2012;287(23):18965–73.
Sun Y, Yi W, Yuan Y, Lau WB, Yi D, Wang X, et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation. 2013;128(11 Suppl 1):S113–20.
Uemura Y, Shibata R, Ohashi K, Enomoto T, Kambara T, Yamamoto T, et al. Adipose-derived factor CTRP9 attenuates vascular smooth muscle cell proliferation and neointimal formation. FASEB J. 2013;27(1):25–33.
Funding
Lixian Sun was supported by grants from Hebei Province Government Science and Technology Agency (Grant No. 17277769D).
Author information
Authors and Affiliations
Contributions
Wenjun Fan did critical review of the manuscript. Lixian Sun drafted and did critical revision of manuscript. Yueqiao Si drafted and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Vascular Biology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Si, Y., Fan, W. & Sun, L. A Review of the Relationship Between CTRP Family and Coronary Artery Disease. Curr Atheroscler Rep 22, 22 (2020). https://doi.org/10.1007/s11883-020-00840-0
Published:
DOI: https://doi.org/10.1007/s11883-020-00840-0