
Abstract
ApoC-III was discovered almost 50 years ago, but for many years, it did not attract much attention. However, as epidemiological and Mendelian randomization studies have associated apoC-III with low levels of triglycerides and decreased incidence of cardiovascular disease (CVD), it has emerged as a novel and potentially powerful therapeutic approach to managing dyslipidemia and CVD risk. The atherogenicity of apoC-III has been attributed to both direct lipoprotein lipase-mediated mechanisms and indirect mechanisms, such as promoting secretion of triglyceride-rich lipoproteins (TRLs), provoking proinflammatory responses in vascular cells and impairing LPL-independent hepatic clearance of TRL remnants. Encouraging results from clinical trials using antisense oligonucleotide, which selectively inhibits apoC-III, indicate that modulating apoC-III may be a potent therapeutic approach to managing dyslipidemia and cardiovascular disease risk.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Apolipoprotein C-III (apoC-III) was first identified by Brown et al. in 1969 as a regulator of triglyceride-rich lipoproteins (TRLs) in the circulation [1]. In the blood, it is mainly present in TRLs (chylomicrons and VLDL) and, to a lesser extent, in LDL and HDL particles. Shortly after its discovery, it was identified as an inhibitor of lipoprotein lipase (LPL) activity and a key regulator of TRL concentrations in plasma [2, 3]. In addition to its potent inhibitory capacity on LPL-mediated lipolysis, apoC-III has also been shown to facilitate hepatic VLDL assembly and secretion. ApoC-III is not only a critical modulator of TRLs but also contributes to the atherogenicity of low- and high-density lipoprotein (LDL and HDL) particles [4–7]. Substantial evidence has emerged to indicate that apoC-III has several additional functions, including modulation of endothelial function and inflammation [8, 9].
The connection of apoC-III with hypertriglyceridemia and CVD risk has been confirmed in extensive animal and humans studies [10, 11]. Furthermore, given that TRL remnants are now recognized as a causal risk factor for CVD and that loss-of-function mutations of apoC-III are characterized by a robust lowering of serum triglycerides and reduced CVD risk [12, 13•, 14••, 15], apoC-III has been identified as an attractive target for the prevention of CVD. This review evaluates the role of apoC-III in the pathogenesis of CVD and summarizes its role as a novel therapeutic target.
Regulation of Apoc-III
ApoC-III is the most abundant C-apolipoprotein in humans. It is found not only in triglyceride-rich chylomicrons and very low-density lipoproteins (VLDLs) but also in HDL. In humans, apoC-III is mainly expressed in hepatocytes and enterocytes. Its expression is chiefly regulated at the transcriptional level, and the regulatory sequences of the APOC3 gene have been extensively described (reviewed in [16]). The APOC3 gene contains two regulatory elements that act as a common enhancer for a gene cluster on 11q23 that regulates the expression of three apolipoprotein genes: APOC3, APOA1, and APOA4 [16, 17].
Promoter analysis of the APOC3 gene has identified binding sites for several transcription factors including the carbohydrate response element binding protein (ChREBP) [18–20]. Thus, glucose and plasma triglyceride metabolism are linked via the regulation of APOC3 expression. Additional transcription factors that bind to the APOC3 gene include the nuclear receptors hepatocyte nuclear factor-4α (HNF-4α) [21] and liver X receptors (LXRs) [22]. Interestingly, the expression of APOC3 is upregulated by glucose and downregulated by insulin. This contrasts with most other genes in the glycolytic and lipogenic pathways, which are regulated positively both by glucose and insulin [23]. It has, therefore, been proposed that the glucose-mediated expression of APOC3 may contribute to lower lipolysis and consequently to an increase in peripheral glucose handling [20]. Substantial evidence indicates that dysregulation of the insulin signaling pathway via FoxO1 aggravates APOC3 expression as the inhibitory effect of insulin on APOC3 expression that is lost [24]. This mechanism may explain why apoC-III concentrations are increased in insulin-resistant states such as obesity, the metabolic syndrome, type 2 diabetes, and hypertriglyceridemia [10, 11, 25].
ApoC-III is a small protein (79 amino acid residues) that contains two amphipathic helices [26]. We still lack a detailed structure-function understanding of apoC-III, and it is not clear which regions are most important for attachment to lipids [26–29]. ApoC-III can undergo posttranslational modification on threonine 74, and three different glycoforms have been described with zero, one, or two sialic acids (termed apoC-III0, apoC-III1, and apoC-III2) [30]. The degree of sialylation of apoC-III has been reported to influence its function as apoC-III2 inhibits LPL-mediated hydrolysis of TRLs less efficiently than apoC-III1 [31], despite having an apparent twofold greater affinity for TRL than the other two apoC-III isoforms [32].
Function of Apoc-III
In addition to inhibiting LPL-mediated lipolysis of TRLs, apoC-III binds surface heparan sulfate proteoglycans (HSPGs) on the liver and cellular receptors, including the HDL receptor SR-BI [33]. However, the significance of binding to SR-BI is still unclear.
Inhibition of LPL-mediated Lipolysis
Lipolysis of TRLs is mediated by LPL, which resides on cellular surfaces. ApoC-III has been shown in vitro to inhibit lipolysis by several mechanisms, including displacing lipoproteins from the negatively charged cell surface [34], inducing conformational changes in apoE and inhibiting the activation of LPL by displacing the LPL activator apoC-II from the lipoprotein surface [35]. In addition, studies in vivo with apoC-III transgenic mice have demonstrated that overexpression of apoC-III induces marked hypertriglyceridemia due to accumulation of remnant particles with increased apoC-III and decreased apoE content compared with controls [36].
Inhibition of Lipoprotein Binding to HSPGs and the LDL Receptor Family
ApoC-III displaces apoE from lipoprotein particles [37]. This displacement can either be complete or involve only the N-terminal domain of apoE [37]. Since apoC-III impairs the clearance of TRLs and their remnants, whereas apoE mediates their clearance (by binding to hepatic HSPG and the LDL receptor), the relative proportions of apoC-III and apoE on apoB-containing lipoproteins regulate the metabolism of these lipoproteins, which is reflected in LDL and HDL subspecies. This is a very complex area of research, and Dr Sacks has made significant contributions in clarifying the crucial roles of apoE and apoC-III in apoB-lipoprotein metabolism [6].
Human Kinetic Studies
Recent evidence from kinetic studies showed that dual metabolic defects contribute to elevated TRLs in the atherogenic dyslipidemia of abdominally obese men [38•, 39, 40]. In fact, reduced catabolism of TRLs resulting from increased levels of plasma apoC-III was shown to be a more important predictor of plasma triglycerides than increased secretion rate of TRLs driven by liver fat content [38•]. Indeed, kinetic studies in humans have also provided direct evidence to show that apoB-containing lipoproteins are removed significantly more slowly from the circulation if they contain apoC-III [41]. These results may indicate that apoC-III concentration may be a good biomarker of the residual risk of CVD in subjects with atherogenic dyslipidemia; this issue remains a major clinical problem in statin-treated subjects.
The finding that removal of TRLs from the circulation is a more important determinant of plasma TG than secretion of TRL is interesting, but it remains to be clarified whether the apoC-III-induced impaired clearance of VLDL particles in subjects with atherogenic dyslipidemia is predominantly due to reduced lipolytic capacity or to the impaired removal of TRL remnants by the liver. This question is of importance when considering the potential of apoC-III inhibition as a novel treatment modality. Because the lipid oxidation capacity of the liver is limited, a potential concern is that increased lipid load from the uptake of remnant particles may lead to excess fat accumulation in hepatocytes, resulting in non-alcoholic fatty liver disease [42].
Recently, apoC-III has also been implicated in VLDL secretion [43–45]. Overexpression of apoC-III in mice and in vitro resulted in increased secretion of triglyceride-rich VLDL particles [43]. Structure-function analysis of apoC-III has identified two domains in apoC-III that seem to influence the formation and secretion of VLDL [43]. However, inhibition of apoC-III synthesis by apoC-III antisense oligonucleotides (ASO) did not reduce VLDL secretion in mice [46] so the significance of apoC-III for VLDL secretion requires further study, particularly in humans.
Measurements of ApoC-III Concentrations
Standardized clinical immunoassays for the measurements of apoC-III concentrations have not been easily available. The most common system to measure plasma apoC-III concertation has been the immunoturbidimetric assay provided by automatic autoanalyzers [47]. More recently, a sensitive sandwich ELISA to quantify apoC-III concentrations was developed for commercial use [48]. However, because apoC-III circulates in multiple glycoforms that are differentially distributed in lipoprotein fractions (VLDL, LDL, and HDL), the measurement of plasma apoC-III concentrations is not enough to understand the complexity of apoC-III in lipoprotein metabolism, particularly in the context of novel apoC-III ASO therapies. Therefore, it is critical that novel assays with high sensitivity and specificity be developed. Furthermore, to better understand the clinical importance of the different glycoforms, assays that can be easily performed in routine clinical chemistry laboratories are needed [5, 30, 49–51].
Role of ApoC-III in Atherogenesis and Inflammation
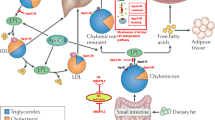
Recent epidemiological studies have clearly demonstrated that apoC-III is associated with higher risk of CVD, but what makes apoC-III atherogenic? As discussed above, apoC-III regulates lipid metabolism through multiple mechanisms with adverse effects especially on TRLs and their remnants (Fig. 1). In addition to effects on lipid metabolism, apoC-III has been shown to influence atherogenesis by increasing the affinity of LDL for artery wall proteoglycans, and thus increasing accumulation of atherogenic lipoproteins in the vessel wall [7, 52–56]. The mechanism was elusive since apoC-III by itself does not bind artery wall proteoglycans. However, Hiukka et al. identified a potential explanation by demonstrating that apoC-III influences the lipid composition not only of TRLs but also of LDL [7]. They investigated subjects with type 2 diabetes and showed that LDL with a high apoC-III/apoB molar ratio is associated with a reduction in unesterified cholesterol, sphingomyelin, and ceramide, but not phosphatidylcholine [7]. These changes in lipid composition associate with higher membrane fluidity, thus allowing apoB to acquire a conformation that is more favorable for proteoglycan binding [7, 57].

Proatherogenic action of apoC-III on lipid metabolism and atherogenicity. ApoC-III exerts strong atherogenic functions through both indirect and direct mechanisms. These include inducing oversecretion of apoC-III-containing triglyceride-rich lipoproteins (TRLs). These lipoproteins accumulate in the circulation by three mechanisms: (1) impaired lipolysis of large TRLs to smaller remnant particles, (2) impaired LPL-mediated lipolysis of TRLs on capillaries, and (3) impaired LPL-independent and LPL-dependent hepatic clearance of TRL remnants. In addition, apoC-III promotes proinflammatory responses in endothelial cells and monocytes and increases the binding affinity of LDL, leading to increased accumulation of atherogenic lipoproteins in the artery wall
Following subendothelial retention in the artery wall, LDL is exposed to several enzymes, including sphingomyelinases (SMase), that promote aggregation and fusion of retained lipoproteins [58]. Interestingly, apoC-III is a SMase activator and apoC-III-enriched LDL displays increased susceptibility to hydrolysis and aggregation by SMase [7, 59]. ApoC-III has also been linked to inflammation as apoC-III alone, or as a component of TRLs and LDL, induces activation of adhesion molecules and the proinflammatory nuclear factor-kB in monocytes and endothelial cells [60, 61].
ApoC-III and CVD Risk
Data from several observational studies show that plasma apoC-III levels associate with manifestations of CVD and the progression of coronary artery disease [5, 11]. Epidemiological studies have further demonstrated that apoC-III in plasma, VLDL, and LDL predicts CVD [10, 11, 62–65]. Importantly, in a 15-year follow-up study, age- and sex-adjusted apoC-III concentrations were shown to predict CVD mortality [65]. Recently, Qamar et al. reported that apoC-III levels in patients with type 2 diabetes patients (n = 1422) associate with elevation of triglycerides and higher coronary artery calcification (CAC) score, a measure of subclinical atherosclerosis [66•]. Notably, this relationship between apoC-III and CAC was found to be triglyceride dependent. The positive association of apoC-III levels with CVD has recently been confirmed in a meta-analysis of published data [5]. The finding of an inverse relationship between plasma apoC-III levels and incident CVD reported in a large case–control study of type 2 diabetic subjects (n = 1123) was unexpected [67]. Normally, plasma apoC-III correlates strongly with triglyceride levels. However, in this cohort of type 2 diabetics, this correlation was, at best, only modest [67]. Overall, the association of apoC-III with CVD risk has been reported to be independent of statin use, suggesting that apoC-III could become a marker for the residual risk of CVD in statin-treated patients with atherogenic dyslipidemia [63, 66•, 68].
In addition, higher apoC-III content in both VLDL and LDL shows consistent links with CVD events, highlighting the multifaceted role of apoC-III as a proatherogenic apolipoprotein (7). The available data on apoC-III in HDL particles are heterogeneous and less clear. However, two studies reported that apoC-III in HDL particles is associated with increased CHD risk [63, 69]. Remaining important questions are how apoC-III modifies HDL to become proatherogenic and if this may explain the unexpected findings in subjects with type 2 diabetes reported by Looker et al. [67].
The rapid development of molecular genetic technology has provided the tools to determine whether apoC-III has a causal role as a proatherogenic protein. Recently, a comprehensive meta-analysis of three main apoC-III polymorphisms (SstI, T-455C, and C-482T) known to associate with hypertriglyceridemia in mice [36] reported that two polymorphisms (SstI and T-455C) significantly increased the susceptibility to CHD in humans [70]. The association of the apoC-III T-455C with increased CVD risk was confirmed in another meta-analysis [71]. In contrast, some evidence emerged that rare variants of apoC-III associated with low triglycerides and high HDL cholesterol [72, 73]. In line with the lipid profile of low triglycerides and high HDL cholesterol, a lifelong deficiency of apoC-III has been shown to associate with reduced subclinical atherosclerosis, being cardioprotective [74]. Together, these data support strongly the link of polymorphisms in apoC-III with serum levels of triglycerides, as well as with either promotion or prevention of CVD.
Two large Mendelian randomization studies have recently shown that polymorphisms in the APOC3 gene were associated with decreased incidence of CVD [13•, 14••]. In these cohorts, three loss-of-function mutations in the APOC3 gene were identified and the heterozygote carriers of these rare variants had about 39–44 % lower serum triglyceride levels, about 46 % lower apoC-III levels and a 36–40 % lower risk of CHD than non-carriers. Furthermore, the reduced risk of CVD was independent of statin use. Recently, carriers of the three loss-of-function mutations of APOC3 were shown to exhibit lower triglyceride levels together with higher HDL cholesterol and a decreased burden of coronary arterial calcification (decreased median CAC score), but no difference was observed in either carotid plaque or carotid intima-media thickness [68].
Potential Therapies to Reduce ApoC-III Plasma Levels
Currently available tools to reduce apoC-III levels and consequently TRLs and their remnants remain limited. Peroxisome proliferator activated receptor (PPAR) alfa agonists are known to downregulate the expression of apoC-III and to reduce its plasma levels in both animal and human studies [75]. Given the fact that apoC-III levels are increased in people with type 2 diabetes [76] and that fibrates reduce apoC-III levels [75, 77], it is surprising that so far no data exist on a potential link between the reduction of apoC-III and CVD outcomes from randomized clinical trials of fibrates. Statins are reported to inhibit APOC3 mRNA levels in liver cells [78] and a small reduction of apoC-III levels by statins has been reported in several relatively small studies [11, 79, 80].
The strategy to search for naturally occurring loss-of-function mutations that associate with decreased cardiovascular disease has proven successful for identifying novel therapeutic targets. For example, the translation of findings on proprotein convertase subtilisin/kexin type 9 (PCSK9) variants, from their identification in 2003 [81] to the currently ongoing phase 3 clinical outcome trials of PCSK9 inhibition with almost 40 000 persons included [82], has been performed in record time. Keys for this rapid translation from bench to clinic encompass the development of modern genetics and new techniques such as silencing RNA and novel human antibody technology [83]. ApoC-III follows the same modern pipeline for drug discovery, as ISIS Pharmaceuticals has developed an apoC-III ASO (volanesorsen) that inhibits the biosynthesis of apoC-III [46, 84]. ApoC-III ASO has been shown to robustly reduce both plasma apoC-III and triglyceride levels in multiple animal models and human volunteers in phase I studies [46]. Recent data from phase II studies confirmed that apoC-III ASO-mediated suppression of apoC-III leads to marked lowering (up to 90 %) of serum apoC-III and up to about an 80 % reduction of VLDL triglyceride levels in patients with familial chylomicron syndrome and with variable types of hypertriglyceridemia [46, 85••, 86••]. The data also demonstrated that the lowering of serum apoC-III levels and triglycerides by apoC-III ASO was dose-dependent and selective [46]. In addition, volanesorsen markedly reduced TRL remnants [85••]. These observations, together with the reduction of TRLs in the absence of LPL, support the existence of LPL-independent pathway(s) regulated by apoC-III for removal of triglycerides [86••]. Safety data from these short-term studies with apoC-III ASO therapy have not raised any major concerns; volanesorsen appears to have a good safety profile and has been well tolerated [85••, 86••]. Phase III trials with volanesorsen are ongoing (the APPROACH study and COMPASS) and will clarify the efficacy of the drug intervention in subjects with severe hypertriglyceridemia defined as triglyceride levels above 5.7 mmol/l (>500 mg/dl) [87].
Gaps in Knowledge of ApoC-III
Important contributions to our understanding of apoC-III have been made by several research groups. However, there are still important gaps in our knowledge of apoC-III (see Table 1), the closing of which will require additional research.
Conclusions
Population studies have demonstrated that plasma apoC-III is strongly associated with CVD risk. For many years, the atherogenicity of apoC-III was attributed to its LPL-mediated effects. However, recent evidence reveals that apoC-III has additional roles, for example in promoting VLDL formation and assembly and proinflammatory responses in endothelial cells and monocytes and impairing LPL-independent hepatic clearance of TRL remnants. Encouraging results from early clinical trials demonstrate that modulation of apoC-III per se is a novel and potent therapeutic approach to managing dyslipidemia and CVD risk.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Brown WV, Levy RI, Fredrickson DS. Studies of the proteins in human plasma very low density lipoproteins. J Biol Chem. 1969;244(20):5687–94.
Brown WV, Baginsky ML. Inhibition of lipoprotein lipase by an apoprotein of human very low density lipoprotein. Biochem Biophys Res Commun. 1972;46(2):375–82.
Windler E, Havel RJ. Inhibitory effects of C apolipoproteins from rats and humans on the uptake of triglyceride-rich lipoproteins and their remnants by the perfused rat liver. J Lipid Res. 1985;26(5):556–65.
Mendivil CO, Rimm EB, Furtado J, Chiuve SE, Sacks FM. Low-density lipoproteins containing apolipoprotein C-III and the risk of coronary heart disease. Circulation. 2011;124(19):2065–72.
Wyler von Ballmoos MC, Haring B, Sacks FM. The risk of cardiovascular events with increased apolipoprotein CIII: a systematic review and meta-analysis. J Clin Lipidol. 2015;9(4):498–510.
Sacks FM. The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia. Curr Opin Lipidol. 2015;26(1):56–63.
Hiukka A, Stahlman M, Pettersson C, et al. ApoCIII-enriched LDL in type 2 diabetes displays altered lipid composition, increased susceptibility for sphingomyelinase, and increased binding to biglycan. Diabetes. 2009;58(9):2018–26.
Bobik A. Apolipoprotein CIII and atherosclerosis: beyond effects on lipid metabolism. Circulation. 2008;118(7):702–4.
Kawakami A, Osaka M, Aikawa M, et al. Toll-like receptor 2 mediates apolipoprotein CIII-induced monocyte activation. Circ Res. 2008;103(12):1402–9.
Norata GD, Tsimikas S, Pirillo A, Catapano AL. Apolipoprotein C-III: from pathophysiology to pharmacology. Trends Pharmacol Sci. 2015;36(10):675–87.
Ooi EM, Barrett PH, Chan DC, Watts GF. Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clin Sci (Lond). 2008;114(10):611–24.
Nordestgaard BG. Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res. 2016;118(4):547–63.
Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371(1):32–41. The study demonstrates that loss-of-function mutations in APOC3 were associated with low levels of triglycerides and a reduced risk of ischemic cardiovascular. disease.
TG, Hdl Working Group of the Exome Sequencing Project NHL, Blood I, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371(1):22–31. Altogether 18,666 genes in each of 3734 participants of European or African ancestry were sequenced in the Exome Sequencing Project. The study demonstrated that rare mutations that disrupt APOC3 function were associated with lower levels of plasma triglycerides and APOC3. Carriers of these mutations were found to have a reduced risk of coronary heart disease.
Boren J, Matikainen N, Adiels M, Taskinen MR. Postprandial hypertriglyceridemia as a coronary risk factor. Clin Chim Acta. 2014;431:131–42.
Zannis VI, Kan HY, Kritis A, Zanni EE, Kardassis D. Transcriptional regulatory mechanisms of the human apolipoprotein genes in vitro and in vivo. Curr Opin Lipidol. 2001;12(2):181–207.
Karathanasis SK, McPherson J, Zannis VI, Breslow JL. Linkage of human apolipoproteins A-I and C-III genes. Nature. 1983;304(5924):371–3.
Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101(19):7281–6.
Yamashita H, Takenoshita M, Sakurai M, et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U S A. 2001;98(16):9116–21.
Caron S, Verrijken A, Mertens I, et al. Transcriptional activation of apolipoprotein CIII expression by glucose may contribute to diabetic dyslipidemia. Arterioscler Thromb Vasc Biol. 2011;31(3):513–9.
Adamson AW, Suchankova G, Rufo C, et al. Hepatocyte nuclear factor-4alpha contributes to carbohydrate-induced transcriptional activation of hepatic fatty acid synthase. Biochem J. 2006;399(2):285–95.
Cha JY, Repa JJ. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J Biol Chem. 2007;282(1):743–51.
Towle HC. Glucose as a regulator of eukaryotic gene transcription. Trends Endocrinol Metab. 2005;16(10):489–94.
Yao Z, Wang Y. Apolipoprotein C-III and hepatic triglyceride-rich lipoprotein production. Curr Opin Lipidol. 2012;23(3):206–12.
Boren J, Taskinen MR, Olofsson SO, Levin M. Ectopic lipid storage and insulin resistance: a harmful relationship. J Intern Med. 2013;274(1):25–40.
Gangabadage CS, Zdunek J, Tessari M, Nilsson S, Olivecrona G, Wijmenga SS. Structure and dynamics of human apolipoprotein CIII. J Biol Chem. 2008;283(25):17416–27.
Liu H, Talmud PJ, Lins L, et al. Characterization of recombinant wild type and site-directed mutations of apolipoprotein C-III: lipid binding, displacement of ApoE, and inhibition of lipoprotein lipase. Biochemistry. 2000;39(31):9201–12.
Sparrow JT, Pownall HJ, Hsu FJ, Blumenthal LD, Culwell AR, Gotto AM. Lipid binding by fragments of apolipoprotein C-III-1 obtained by thrombin cleavage. Biochemistry. 1977;16(25):5427–31.
Lins L, Flore C, Chapelle L, Talmud PJ, Thomas A, Brasseur R. Lipid-interacting properties of the N-terminal domain of human apolipoprotein C-III. Protein Eng. 2002;15(6):513–20.
Trenchevska O, Schaab MR, Nelson RW, Nedelkov D. Development of multiplex mass spectrometric immunoassay for detection and quantification of apolipoproteins C-I, C-II, C-III and their proteoforms. Methods. 2015;81:86–92.
Holdsworth G, Stocks J, Dodson P, Galton DJ. An abnormal triglyceride-rich lipoprotein containing excess sialylated apolipoprotein C-III. J Clin Invest. 1982;69(4):932–9.
Mann CJ, Troussard AA, Yen FT, et al. Inhibitory effects of specific apolipoprotein C-III isoforms on the binding of triglyceride-rich lipoproteins to the lipolysis-stimulated receptor. J Biol Chem. 1997;272(50):31348–54.
Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med (Berl). 2006;84(4):276–94.
Ebara T, Ramakrishnan R, Steiner G, Shachter NS. Chylomicronemia due to apolipoprotein CIII overexpression in apolipoprotein E-null mice. Apolipoprotein CIII-induced hypertriglyceridemia is not mediated by effects on apolipoprotein E. J Clin Invest. 1997;99(11):2672–81.
Lambert DA, Smith LC, Pownall H, Sparrow JT, Nicolas JP, Gotto Jr AM. Hydrolysis of phospholipids by purified milk lipoprotein lipase. Effect of apoprotein CII, CIII, A and E, and synthetic fragments. Clin Chim Acta. 2000;291(1):19–33.
Ito Y, Azrolan N, O’Connell A, Walsh A, Breslow JL. Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science. 1990;249(4970):790–3.
Narayanaswami V, Ryan RO. Molecular basis of exchangeable apolipoprotein function. Biochim Biophys Acta. 2000;1483(1):15–36.
Boren J, Watts GF, Adiels M, et al. Kinetic and related determinants of plasma triglyceride concentration in abdominal obesity: multicenter tracer kinetic study. Arterioscler Thromb Vasc Biol. 2015;35(10):2218–24. Factors that predict the kinetics of very-low density lipoprotein 1 (VLDL1) triglycerides were studied using stable isotopes. Results show that plasma triglyceride concentrations in abdominal obesity are determined by the kinetics of VLDL1 subspecies, catabolism being mainly dependent on apoC-III concentration and secretion on liver fat content.
Taskinen MR, Adiels M, Westerbacka J, et al. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler Thromb Vasc Biol. 2011;31(9):2144–50.
Carpentier AC. Hypertriglyceridemia associated with abdominal obesity: getting contributing factors into perspective. Arterioscler Thromb Vasc Biol. 2015;35(10):2076–8.
Zheng C, Khoo C, Furtado J, Sacks FM. Apolipoprotein C-III and the metabolic basis for hypertriglyceridemia and the dense low-density lipoprotein phenotype. Circulation. 2010;121(15):1722–34.
Huff MW, Assini JM, Hegele RA. Gene therapy for hypercholesterolemia: sweet dreams and flying machines. Circ Res. 2014;115(6):542–5.
Yao Z. Human apolipoprotein C-III—a new intrahepatic protein factor promoting assembly and secretion of very low density lipoproteins. Cardiovasc Hematol Disord Drug Targets. 2012;12(2):133–40.
Taskinen MR, Boren J. New insights into the pathophysiology of dyslipidemia in type 2 diabetes. Atherosclerosis. 2015;239(2):483–95.
Adiels M, Olofsson SO, Taskinen MR, Boren J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28(7):1225–36.
Graham MJ, Lee RG, Bell 3rd TA, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112(11):1479–90.
Rifai N, Silverman LM. Immunoturbidimetric techniques for quantifying apolipoproteins CII and CIII. Clin Chem. 1986;32(10):1969–72.
Wang Y, Song Z, Wagner JD, Pachuk C, Subramanian RR. Development of a sensitive ELISA to quantify apolipoprotein CIII in nonhuman primate serum. J Lipid Res. 2011;52(6):1265–71.
Yassine HN, Trenchevska O, Ramrakhiani A, et al. The association of human apolipoprotein C-III sialylation proteoforms with plasma triglycerides. PLoS One. 2015;10(12), e0144138.
van den Broek I, Romijn FP, Nouta J, et al. Automated multiplex LC-MS/MS assay for quantifying serum apolipoproteins A-I, B, C-I, C-II, C-III, and E with qualitative apolipoprotein E phenotyping. Clin Chem. 2016;62(1):188–97.
Koska J, Yassine H, Trenchevska O, et al. Disialylated apolipoprotein C-III proteoform is associated with improved lipids in prediabetes and type 2 diabetes. J Lipid Res. 2016;57(5):894–905.
Olin-Lewis K, Krauss RM, La Belle M, et al. ApoC-III content of apoB-containing lipoproteins is associated with binding to the vascular proteoglycan biglycan. J Lipid Res. 2002;43(11):1969–77.
Davidsson P, Hulthe J, Fagerberg B, et al. A proteomic study of the apolipoproteins in LDL subclasses in patients with the metabolic syndrome and type 2 diabetes. J Lipid Res. 2005;46(9):1999–2006.
Boren J, Gustafsson M, Skalen K, Flood C, Innerarity TL. Role of extracellular retention of low density lipoproteins in atherosclerosis. Curr Opin Lipidol. 2000;11(5):451–6.
Gustafsson M, Boren J. Mechanism of lipoprotein retention by the extracellular matrix. Curr Opin Lipidol. 2004;15(5):505–14.
Gustafsson M, Flood C, Jirholt P, Boren J. Retention of atherogenic lipoproteins in atherogenesis. Cell Mol Life Sci. 2004;61(1):4–9.
Segrest J, Jones M, Mishra V, et al. Apolipoprotein B-100: conservation of lipid-associating amphipathic secondary structural motifs in nine species of vertebrates. J Lipid Res. 1998;3:85–102.
Schissel SL, Jiang X, Tweedie-Hardman J, et al. Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral pH. Implications for atherosclerotic lesion development. J Biol Chem. 1998;273(5):2738–46.
Ahmad TY, Beaudet AL, Sparrow JT, Morrisett JD. Human lysosomal sphingomyelinase: substrate efficacy of apolipoprotein/sphingomyelin complexes. Biochemistry. 1986;25(15):4415–20.
Kawakami A, Aikawa M, Alcaide P, Luscinskas FW, Libby P, Sacks FM. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation. 2006;114(7):681–7.
Kawakami A, Aikawa M, Nitta N, Yoshida M, Libby P, Sacks FM. Apolipoprotein CIII-induced THP-1 cell adhesion to endothelial cells involves pertussis toxin-sensitive G protein- and protein kinase C alpha-mediated nuclear factor-kappaB activation. Arterioscler Thromb Vasc Biol. 2007;27(1):219–25.
Luc G, Fievet C, Arveiler D, et al. Apolipoproteins C-III and E in apoB- and non-apoB-containing lipoproteins in two populations at contrasting risk for myocardial infarction: the ECTIM study. Etude Cas Temoins sur ’Infarctus du Myocarde. J Lipid Res. 1996;37(3):508–17.
Sacks FM, Alaupovic P, Moye LA, et al. VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the Cholesterol and Recurrent Events (CARE) trial. Circulation. 2000;102(16):1886–92.
Lee SJ, Campos H, Moye LA, Sacks FM. LDL containing apolipoprotein CIII is an independent risk factor for coronary events in diabetic patients. Arterioscler Thromb Vasc Biol. 2003;23(5):853–8.
Scheffer PG, Teerlink T, Dekker JM, et al. Increased plasma apolipoprotein C-III concentration independently predicts cardiovascular mortality: the Hoorn Study. Clin Chem. 2008;54(8):1325–30.
Qamar A, Khetarpal SA, Khera AV, Qasim A, Rader DJ, Reilly MP. Plasma apolipoprotein C-III levels, triglycerides, and coronary artery calcification in type 2 diabetics. Arterioscler Thromb Vasc Biol. 2015;35(8):1880–8. An important study showing that in persons with T2DM, increased plasma apoC-III is associated with higher triglycerides, less favorable cardiometabolic phenotypes, and higher coronary artery calcification, a measure of subclinical atherosclerosis.
Looker HC, Colombo M, Agakov F, et al. Protein biomarkers for the prediction of cardiovascular disease in type 2 diabetes. Diabetologia. 2015;58(6):1363–71.
Natarajan P, Kohli P, Baber U, et al. Association of APOC3 loss-of-function mutations with plasma lipids and subclinical atherosclerosis: the multi-ethnic BioImage study. J Am Coll Cardiol. 2015;66(18):2053–5.
Jensen MK, Rimm EB, Furtado JD, Sacks FM. Apolipoprotein C-III as a potential modulator of the association between HDL-cholesterol and incident coronary heart disease. J Am Heart Assoc. 2012;1:jah3-e000232.
Zhang JZ, Xie X, Ma YT, et al. Association between apolipoprotein C-III gene polymorphisms and coronary heart disease: a meta-analysis. Aging Dis. 2016;7(1):36–44.
Sun Y, Zhou RB, Chen DM. APOA5 -1131T>C and APOC3 -455T>C polymorphisms are associated with an increased risk of coronary heart disease. Genet Mol Res. 2015;14(4):18218–28.
Tachmazidou I, Dedoussis G, Southam L, et al. A rare functional cardioprotective APOC3 variant has risen in frequency in distinct population isolates. Nat Commun. 2013;4:2872.
Timpson NJ, Walter K, Min JL, et al. A rare variant in APOC3 is associated with plasma triglyceride and VLDL levels in Europeans. Nat Commun. 2014;5:4871.
Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322(5908):1702–5.
Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98(19):2088–93.
Hiukka A, Fruchart-Najib J, Leinonen E, Hilden H, Fruchart JC, Taskinen MR. Alterations of lipids and apolipoprotein CIII in very low density lipoprotein subspecies in type 2 diabetes. Diabetologia. 2005;48(6):1207–15.
Malmendier CL, Lontie JF, Delcroix C, Dubois DY, Magot T, De Roy L. Apolipoproteins C-II and C-III metabolism in hypertriglyceridemic patients. Effect of a drastic triglyceride reduction by combined diet restriction and fenofibrate administration. Atherosclerosis. 1989;77(2–3):139–49.
Schoonjans K, Peinado-Onsurbe J, Fruchart JC, Tailleux A, Fievet C, Auwerx J. 3-Hydroxy-3-methylglutaryl CoA reductase inhibitors reduce serum triglyceride levels through modulation of apolipoprotein C-III and lipoprotein lipase. FEBS Lett. 1999;452(3):160–4.
Dallinga-Thie GM, Berk II P, Bootsma AH, Jansen H. Diabetes Atorvastatin Lipid intervention Study G: atorvastatin decreases apolipoprotein C-III in apolipoprotein B-containing lipoprotein and HDL in type 2 diabetes: a potential mechanism to lower plasma triglycerides. Diabetes Care. 2004;27(6):1358–64.
Karalis IK, Bergheanu SC, Wolterbeek R, et al. Effect of increasing doses of Rosuvastatin and Atorvastatin on apolipoproteins, enzymes and lipid transfer proteins involved in lipoprotein metabolism and inflammatory parameters. Curr Med Res Opin. 2010;26(10):2301–13.
Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–6.
Stein EA, Swergold GD. Potential of proprotein convertase subtilisin/kexin type 9 based therapeutics. Curr Atheroscler Rep. 2013;15(3):310.
Bernelot Moens SJ, van Capelleveen JC, Stroes ES. Inhibition of ApoCIII: the next PCSK9? Curr Opin Lipidol. 2014;25(6):418–22.
Huff MW, Hegele RA. Apolipoprotein C-III: going back to the future for a lipid drug target. Circ Res. 2013;112(11):1405–8.
Gaudet D, Alexander VJ, Baker BF, et al. Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med. 2015;373(5):438–47. A total of 57 patients were treated with antisense inhibition of apoC-III. No safety concerns were identified in this short-term study. Results demonstrate that the intervention was associated with significant lowering of triglyceride levels, among patients with a broad range of baseline levels, through selective antisense inhibition of APOC3 synthesis.
Gaudet D, Brisson D, Tremblay K, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371(23):2200–6. The inhibitor of APOC3 messenger RNA (mRNA) was administered to three patients with the familial chylomicronemia syndrome. After 13 weeks of study-drug administration, plasma APOC3 levels were reduced by 71 to 90% and triglyceride levels by 56 to 86%. These data support the role of APOC3 as a key regulator of LPL-independent pathways of triglyceride metabolism.
Gaudet D. Novel therapies for severe dyslipidemia originating from human genetics. Curr Opin Lipidol. 2016;27(2):112–24.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Marja-Riitta Taskinen declares personal fees from Amgen, AstraZeneca, and Chiesi Pharma, grant support and personal fees from Sanofi Aventis and Novo Nordisk, and grant support from Merck Sharp & Dohme.
Jan Borén declares grant support and personal fees from AstraZeneca, Sanofi-Aventis, NovoNordisk, and Amgen and personal fees from Merck Sharp & Dome.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Clinical Trials and Their Interpretations
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Taskinen, MR., Borén, J. Why Is Apolipoprotein CIII Emerging as a Novel Therapeutic Target to Reduce the Burden of Cardiovascular Disease?. Curr Atheroscler Rep 18, 59 (2016). https://doi.org/10.1007/s11883-016-0614-1
Published:
DOI: https://doi.org/10.1007/s11883-016-0614-1