
Abstract
Stem cell transplant (SCT) has long been the standard of care for several hematologic, immunodeficient, and oncologic disorders. Recently, SCT has become an increasingly utilized therapy for refractory autoimmune rheumatologic disorders (ARDs). The efficacy of SCT in ARDs has been attributed to resetting an aberrant immune system either through direct immune replacement with hematopoietic stem cells or through immunomodulation with mesenchymal stem cells. Among ARDs, refractory systemic sclerosis (SSc) and systemic lupus erythematosus (SLE) are the most common indications for SCT. SCT has also been used in refractory rheumatoid arthritis, inflammatory myopathies, antiphospholipid syndrome, granulomatosis with polyangiitis, and pediatric ARDs. Complete responses have been reported in approximately 30 % of patients in all disease categories. Transplant-related mortality, however, remains a concern. Future large multi-center prospective randomized clinical trials will help to better define the specific role of SCT in the treatment of patients with ARDs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over the past 20 years, stem cell transplant (SCT) has emerged as a therapy for refractory autoimmune rheumatologic diseases (ARDs). The use of SCT in ARDs has followed on the heels of the successful application of SCT to treat a variety of hematologic, oncologic, and immunodeficient disorders including aplastic anemia, thalassemia, acute leukemias, severe combined immunodeficiency (SCID), and human immunodeficiency virus (HIV) [1–3]. Recent animal and human studies also demonstrate improvements in ARDs following SCT. SCT makes it possible to reset the immune system, shifting it from a highly pro-inflammatory disease environment to a less inflammatory one [4]. The majority of SCTs world-wide have utilized hematopoietic stem cells as opposed to mesenchymal stem cells. Data on mesenchymal stem cell transplant (MSCT) remain very limited.
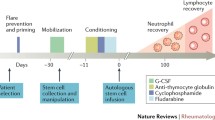
Hematopoietic stem cells have the ability to differentiate into cells belonging to both the myeloid and lymphoid lineages. For HSCTs, hematopoietic stem cells are harvested either directly from the bone marrow (bone marrow transplant) or the peripheral blood via apheresis. Collection of hematopoietic stem cells from the peripheral blood requires the use of agents such as granulocyte colony-stimulating factor (G-CSF), plerixafor, or cyclophosphamide to mobilize hematopoietic stem cells out of the bone marrow into the peripheral blood [5].
The cells used for transplantation may be obtained from either the patient (autologous) or from another person, typically an HLA-identical sibling or unrelated donor (allogeneic). The harvested cells can be manipulated to select for or deplete specific cell types. CD34+ selection is frequently used to select for hematopoietic stem cells, which also effectively eliminates T cells [6]. Prior to HSCT, patients receive a conditioning regimen comprised of chemotherapy drugs with or without radiation therapy, which may or may not result in ablation of the bone marrow (myeloablative versus non-myeloablative) depending on the doses used. Patients undergoing allogeneic transplants also receive immunosuppressive drugs to prevent graft versus host disease. Autologous transplantation is, therefore, much safer than allogeneic transplantation which carries the risk of potentially fatal graft versus host disease. However, since allogeneic transplantation replaces the immune system of the recipient with that of a healthy donor, it provides curative potential.
Mesenchymal stem cells have the potential to differentiate into various non-hematopoietic cell types including fat cells, bone, cartilage, and muscle [7, 8]. Unlike allogeneic hematopoietic stem cells, allogeneic mesenchymal stem cells lack significant immunogenicity. They reside in bone marrow, skeletal muscle, adipose tissue, synovial membranes, connective tissue, umbilical cord blood, and placental products [9]. In mesenchymal stem cell transplant (MSCT), donor cells are typically harvested from bone marrow, adipose, or umbilical cord tissue. Unlike HSCT, a conditioning regimen is not necessary prior to MSCT [10].
SCT has predominantly been investigated in patients with refractory ARDs, particularly systemic sclerosis (SSc) and systemic lupus erythematosus (SLE). Less attention has been given to other ARDs such as the inflammatory myopathies, rheumatoid arthritis, antiphospholipid syndrome, Wegener’s granulomatosis with polyangiitis, and pediatric ARDs, such as juvenile idiopathic arthritis. This article will review recent advances in SCT for the ARDs. A summary of recent clinical trials of SCT in ARDs is shown in Table 1.
Pathophysiology
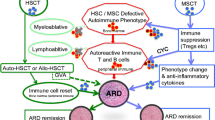
In ARDs, immune dysfunction can involve all components of the innate and humoral immune systems. Dysfunction of T and B cells, dendritic cells, and natural killer cells are common to the pathogenesis of several ARDs. Inappropriate self-antigen recognition results in both cell-mediated and humoral responses, and precipitates a pro-inflammatory cascade, resulting in end organ damage [11]. With HSCT, it is possible to eradicate the dysfunctional cells and replace them with hematopoietic stem cells that can differentiate into “healthy” B and T lymphocytes, monocytes, tissue macrophages, and dendritic cells to produce a more functional immune system. Some of the most critical cells involved in this immune rebuilding are CD34+ cells, which allow for the expansion of new T cell populations and play an important role in determining success of engraftment [12, 13]. Another critical player in immune reconstitution is CD4+CD25+FOXP3+ natural regulatory T cells which have been correlated with disease regression [12, 14, 15].
In MSCT, the approach is immunomodulatory rather than immunoablative [10, 16]. Mesenchymal stem cells have little immunogenicity due to low levels of major histocompatibility complex-I (MHC-I) and the absence of HC-II or co-stimulatory molecules such as B7-1(CD-80), B7-2 (CD86), or CD40. Mesenchymal stem cells down-regulate immune responses by blocking pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α and interferon-γ, and by secreting inhibitory soluble factors such as interleukin (IL) 6 and macrophage colony-stimulating factor [4, 17–19].
The physiology of immune reconstitution remains incompletely understood. A recent study by Szodoray et al. of 12 patients with a variety of refractory ARDs (SSc, rheumatoid arthritis, SLE, and overlap syndrome with features of myositis and rheumatoid arthritis) has provided initial insights into the chronology of immune resetting following autologous HSCT in ARDs. Using serial flow cytometry, this group showed that the first “immune” cells to appear after transplantation were CD56+ natural killer cells. Naive B cells regenerated within 2 months, whereas repopulation of naive T cells took longer. Immune reconstitution was complete at approximately 5–6 months [20]. These findings mirror the chronology of immune reconstitution reported in patients transplanted for hematologic, immunodeficient, and oncologic diseases [21].
Systemic Sclerosis
SSc is characterized by CD4+ and CD8+ T cell activation, autoantibody production (e.g., anti-Scl-70), and cytokine secretion (e.g., IL-13, IL-6) [22, 23]. In SSc, the target organs are primarily the lungs, skin, gastrointestinal tract, and pulmonary vasculature, which undergo extensive fibrosis [24–26]. SSc is classified as either limited or diffuse based upon the extent of skin involvement. Severe manifestations include digital ulceration, pulmonary arterial hypertension, interstitial lung disease, and scleroderma renal crisis. Treatment is symptom-targeted with no proven disease-modifying agent or cure. Due to the lack of effective therapies, SCT has been investigated as an alternative treatment for severe and refractory SSc. Notably, SSc is now the most common rheumatologic disease for which HSCT is performed, with more than 250 reports in the published literature to date [27••].
Reported results with HSCT have been promising in SSc, with patients achieving a complete remission or reduction in disease severity [28, 29] .The European Group for Blood and Marrow Transplantation (EBMT) registry reported a 50 % progression-free survival of 50 % at 5 years and 39 % at 7 years for SSc patients treated with either autologous or allogeneic HSCT. [30]. Overall survival at 7 years in the transplanted patient group was 70 % [30].
More recent studies have focused on specific endpoints unique to SSc, particularly skin fibrosis and interstitial lung disease. Results of the American Scleroderma Stem Cell versus Immune Suppression Trial (ASSIST) were published in 2011 [31•]. In this randomized controlled phase II trial, 19 patients were randomized to either autologous HSCT with cyclophosphamide and anti-thymocyte globulin or to treatment with monthly intravenous cyclophosphamide. The trial was restricted to patients less than 60 years of age who had interstitial lung disease or modified Rodnan skin scores >14 with other internal organ involvement. At 12 months, all 10 patients randomized to the HSCT arm achieved the primary endpoints defined as an improvement of >25 % in the modified Rodnan skin score or >10 % improvement in their forced vital capacity. None of the 9 patients on the monthly cyclophosphamide treatment arm showed improvement [31•]. In a separate study of 26 patients by Henes et al., improvements in modified Rodnan skin score were reported as early as 6 months following a radiation-free conditioning regimen and autologous HSCT with CD34+ selected cells. At 4.4 years, the overall response rate was 91 %, and the progression free survival was 74 % [32].
Two large ongoing randomized prospective clinical trials evaluating HSCT in SSc, the Autologous Stem Cell Transplantation International Scleroderma (ASTIS) trial and the Scleroderma Cyclophosphamide or Transplant (SCOT) trial, are expected to better define the role of SCT in SSc [31•]. ASTIS is a 2-year multicenter randomized controlled phase III trial comparing autologous HSCT following high dose cyclophosphamide and anti-thymocyte globulin for conditioning with CD34+ HSC selection versus high dose monthly IV cyclophosphamide [34, 35]. The primary endpoint of the ASTIS trial is event-free survival defined as either the time from randomization until death or persistent major organ failure [35]. Preliminary data on the 156 patients enrolled on the study that were presented at the June 2012 European Union League Against Rheumatism meeting show fewer deaths in the transplant arm, although the 100-day transplant-related mortality was 10 % [36]. The SCOT trial is a phase II/III randomized study comparing myeloablative (total body irradiation and cyclophosphamide) autologous HSCT versus monthly cyclophosphamide with similar endpoints as the ASTIS trial [37]. The study designs of the ASSIST, ASTIS, and SCOT trials are shown in Table 2 for comparison.
Results from these trials may provide some insight into the efficacy of the different conditioning regimens used for autologous HSCT for SSc. Recent reports suggesting genomic instability in patients with SSc have fueled concerns regarding the use of total body irradiation in myeloablative regimens in these patients and the potential for increased susceptibility to second malignancies [27••]. Secondary analyses of the ongoing SSc trials should help to clarify this concern.
Systemic Lupus Erythematosus
SLE is a multisystem immune disorder characterized by autoantibody production (e.g., anti-double-stranded DNA antibody) against cellular components. Clinical features include arthritis, malar rash, vasculitis, immune-mediated cytopenias, serositis, glomerulonephritis, and central nervous system disease. Lupus nephritis is a particularly severe complication associated with increased mortality. Standard treatment involves cyclophosphamide, azathioprine, mycophenolate mofetil, and glucocorticoids; despite these therapies, 30 % of patients may still progress to end stage renal disease. Immune system abnormalities in SLE include B cell and T cell dysfunction, and abnormal expression of cytokines including TNF-α, interferon-α, interferon-γ, IL-2, IL-4, IL-5, IL-6, and IL-10 [4].
In 2011, the EBMT conducted a review of data collected on approximately 200 patients worldwide with mild to severe SLE, who had undergone autologous HSCTs, and reported a beneficial effect. Sustained clinical remissions were reported even in patients with severe refractory SLE. Disease free survival at 5 years was estimated at 50–70 %, while overall and transplant-related mortality ranged from 0 to 25 % [38]. Recently, Song et al. reported 7-year follow-up data on 17 patients with SLE refractory to available therapies (including cyclophosphamide) who underwent CD34+ selected autologous HSCT. SLE manifestations included refractory, transfusion-dependent cytopenias, severe and recurrent pericardial or myocardial involvement, pulmonary inflammatory disease, central nervous system involvement, and glomerulonephritis. Progression free survival in the transplanted group was 64.7 ± 11.6 % compared to patients on the control arm receiving conventional therapy in which progression free survival was 24.7 ± 10.3 %. Overall survival in the HSCT group was 82.4 ± 9.2 % compared to 66.7 ± 11.4 % in patients receiving conventional therapy. In addition, Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) scores improved in the transplanted group from 32.3 ± 9.2 at baseline to 0.76 ± 0.92 at 5 years [39].
The impact of HSCT on future fertility in women with ARDs has been a significant concern. Mycophenolate mofetil and cyclophosphamide are often used to treat severe SLE manifestations, and both medications are highly teratogenic. Cyclophosphamide can cause premature gonadal failure and infertility in men and women, and risk increases with cumulative exposure. [40]. Hence, these agents are used with caution in young men and women. Autologous HSCT, ideally without cyclophosphamide mobilization, may be an attractive option for young adults with SLE who have severe disease and may be at risk for infertility due to previous cumulative cyclophosphamide exposure. Data are limited concerning pregnancies after HSCT for ARD. In two small studies including 14 total female patients, 3 women had healthy pregnancies after autologous HSCT [39, 41].
Recent animal studies have investigated the use of MSCT in lupus nephritis. Umbilical (U)-MSCT in murine lupus nephritis models has been reported to delay the development of proteinuria, decrease levels of antibodies to double-stranded DNA, lessen renal injury, and prolong the lives of transplanted mice [4]. Immunologic profiling has shown decreased T-helper (Th) 1 cytokines (IFN-γ, IL-2) and pro-inflammatory cytokines (TNF-α, IL-6, IL-12) and increased Th2 cytokines (IL-4, IL-10) [4]. While MSCT shows promise in murine lupus models, controlled clinical trials in human lupus nephritis are needed.
Rheumatoid Arthritis
Rheumatoid arthritis is a chronic inflammatory arthritis affecting 1 % of the population [42]. It is a largely T cell-mediated disease characterized by synovial inflammation and articular destruction [43]. Rheumatoid arthritis is also characterized by the production of auto-antibodies including rheumatoid factor and anti-cyclic citrullinated peptide antibody. High levels of the pro-inflammatory cytokines IL-1β, IL-6,and TNF-α may also be present in the synovial fluid and the serum of patients with rheumatoid arthritis [44]. The observation that these cytokines play a role in the pathogenesis of this disease has led to the use of highly effective biologic therapies that have revolutionized the treatment of rheumatoid arthritis over the last 15 years.
The efficacy of biologic agents is likely the reason that there are limited investigations of SCT in rheumatoid arthritis compared to SSc or SLE. An observational study by Snowden et al. measured disease improvement with the American College of Rheumatology (ACR) Criteria amongst RA patients previously treated with a mean of 5 disease-modifying anti-rheumatic agents (DMARDs) with average disease duration of 8 years. Of 73 refractory rheumatoid arthritis patients treated with autologous HSCT, 67 % achieved an American College of Rheumatology 50 % improvement (ACR 50) response [45].
In murine collagen-induced arthritis models, umbilical MSCT has been reported to decrease disease severity [46]. In vitro studies have demonstrated an anti-proliferative effect of umbilical mesenchymal stem cells on fibroblast-like synoviocytes which are key players in inflammation and joint destruction in rheumatoid arthritis. The anti-proliferative effect appears to be mediated by anti-IL-10, 1-methyl-DL-tryptophan, and anti-transforming growth factor (TGF)-β1, and by down-regulation of mixed metalloproteinase 9 [46]. Umbilical mesenchymal stem cells also directly suppress T lymphocytes through TGF-β1, prostaglandin E2, and nitric oxide. In addition, they can induce regulatory T cells, which are known to play an important role in immune self-tolerance [46]. Some animal models have also shown a benefit from the combination of MSCT with either TNF-α blockade (to down-regulate pro-inflammatory cytokines and T cell proliferation) or TGF-β [44, 47].
Inflammatory Myopathies
Polymyositis and dermatomyositis are inflammatory myopathies characterized by elevated muscle enzymes, proximal muscle weakness, and other complications such as interstitial lung disease. Despite their similarities, the two diseases differ in immunologic profiles and histopathology. In polymyositis, CD8+ cytotoxic T cells invade muscle fibers expressing MHC-1 antigen and induce muscle necrosis [48]. Muscle biopsies in polymyositis show intrafasicular inflammatory infiltrates with scattered muscle fiber necrosis [49]. In contrast, in dermatomyositis, complement activation is implicated in disease pathogenesis by triggering cytokine and chemokine activation of CD4+ and CD8+ T cells, B cells, and macrophages [48]. Muscle biopsies in dermatomyositis show perivascular inflammatory infiltrates and perifascicular atrophy [49].
Investigation of SCT in inflammatory myopathies has been limited to date. In a recent single arm trial evaluating SCT in 10 patients with refractory polymyositis and dermatomyositis defined as either failure to respond to standard therapy or presence of severe systemic manifestations, allogeneic MSCT was shown to improve creatinine kinase, muscle strength, and myositis-related interstitial lung disease [16]. In another small study, one patient with polymyositis was reported to have marked improvement in muscle strength within 3 months of autologous MSCT [50].
Antiphospholipid Antibody Syndrome
Antiphospholipid antibody syndrome is a prothrombotic autoantibody-mediated disorder that can cause arterial or venous thromboses and pregnancy-associated morbidity. In severe cases, APS may lead to catastrophic consequences with widespread thromboses in multiple organ systems. APS can be associated with other immune disorders such as SLE. Conventional therapy for APS includes anti-platelet agents and anticoagulation. Successful treatment of primary or catastrophic antiphospholipid antibody syndrome with immunomodulating agents including rituximab and cyclophosphamide has been reported, but has not been studied in a controlled, clinical trial setting [51, 52].
Most published data on SCT in antiphospholipid antibody syndrome are derived from subsets of patients with SLE. Secondary analyses of these studies show it is possible to eliminate antiphospholipid autoantibodies, including both the lupus anticoagulant and anticardiolipin antibodies, with HSCT. In an open label trial of 22 patients with SLE and antiphospholipid antibody syndrome, 18 patients on chronic anticoagulation prior to autologous HSCT were able to discontinue long-term anticoagulation, with 78 % remaining thrombosis-free at a median follow-up of 15 months [53]. HSCT has also been reported to be efficacious in catastrophic antiphospholipid antibody syndrome. In one recent case report, autologous HSCT was successfully utilized to treat a 34-year-old male with catastrophic antiphospholipid antibody syndrome and an antecedent 10-year history of antiphospholipid antibody syndrome with recurrent thromboses. At 2-year follow-up, the patient remained thrombosis-free with nearly undetectable antiphospholipid antibodies [51].
Granulomatosis with Polyangiitis
There are few published reports of SCT in patients with refractory vasculitides such as granulomatosis with polyangiitis, which was formerly known as Wegener’s Granulomatosis. Granulomatosis with polyangiitis is a necrotizing vasculitis affecting small to medium blood vessels. The most commonly affected organs include the upper and lower respiratory tracts, kidneys, joints, and skin. TNFα and proteinase 3 (PR3) are believed to be critical players in the pathogenesis of the disease, and most affected patients have circulating PR3 anti-neutrophil cytoplasmic antibodies (PR3-ANCA) [54]. Standard therapies for granulomatosis with polyangiitis include glucocorticoids, cyclophosphamide, methotrexate, azathioprine, and rituximab [55].
In the past decade, at least 8 published reports have cited the use of SCT in adult patients with granulomatosis with polyangiitis. Although improvements in joint pain, fevers, pulmonary infiltrates, orbital granulomas, and decreased ANCA titers have been reported, the limited number of cases prohibits specific conclusions regarding SCT in this patient population [56–61].
Pediatric ARDs
SCT has also been investigated in pediatric patients with refractory ARDs, including juvenile idiopathic arthritis and pediatric-onset SLE. The majority of reports are of patients with juvenile idiopathic arthritis. Juvenile idiopathic arthritis represents a heterogeneous collection of inflammatory arthritides with an age of onset of less than 16 years. Conventional therapies include nonsteroidal anti-inflammatory agents, glucocorticoids, methotrexate, sulfasalazine, anti-TNF agents and abatacept.
In a retrospective analysis of 34 patients with refractory systemic or polyarticular juvenile idiopathic arthritis treated with autologous HSCT with various conditioning regimens, all patients had active polyarticular arthritis and had failed to respond to high dose methotrexate. In addition, all had failed or could not tolerate one other disease-modifying anti-rheumatic drug or were corticosteroid-dependent. Ten of 34 patients had failed anti-TNF therapy, 53 % of patients achieved a complete remission, and partial responses were observed in 18 % [62]. Infections were common with 24/34 (71 %) patients experiencing at least one infection. Transplant related mortality was 5/53 (9 %), including 3 children with systemic juvenile idiopathic arthritis who developed fatal hemophagocytic syndrome after transplantation, associated with infection in two cases (histoplasmosis and Epstein-Barr Virus) [62]. In a phase II one arm prospective trial of autologous HSCT in juvenile idiopathic arthritis, 15 of 22 (68 %) children were reported to respond with 8 obtaining complete remission and 7 achieving partial response [63].
A handful of case reports of partial responses to autologous HSCT in patients with pediatric SLE have appeared in the literature [64, 65]. The largest case series of SCT for pediatric SLE was reported by the EBMT Paediatric and Working Parties Committee in 2008. This registry included 17 patients with pediatric SLE treated with autologous SCT using cyclophosphamide, anti-thymocyte globulin, and low dose total body irradiation preconditioning [64]. Details regarding SLE disease status prior to transplant were not reported. Approximately half the patients received CD34+ selected cells. Mortality at 12 months was 12 %, disease-free survival at 60 months was 55 % [64, 66–68].
Case reports suggest that SCT may also be beneficial in patients with refractory pediatric granulomatosis with polyangiitis and SSc [69]. In a retrospective review of 5 cases of pediatric SSc with lung involvement, 4 patients attained a complete remission following autologous HSCT, though 1 later relapsed 9.2 months after transplant; the fifth patient had a partial response [70].
Complications of SCT
During the cell mobilization and conditioning phases, a variety of adverse events have been reported including allergic reactions to cyclophosphamide and anti-thymocyte globulin, fever, bone pain, infection, nausea, vomiting, and elevations in liver enzymes [39]. In long-term follow-up, there have also been reports of serious infections including one case of spinal tuberculosis in an SLE patient treated with cyclophosphamide, G-CSF, and anti-thymocyte globulin [26, 39]. Macrophage activation syndrome has also been reported following SCT in the pediatric literature [62, 71].
Graft versus host disease (GVHD) is a risk that is unique to allogeneic transplants [72]. It can be limited or extensive, and results from donor allogeneic T cells attacking recipient tissues, typically the skin, liver, and GI tract [73]. It is fatal in up to 15 % of patients [74]. GVHD is often steroid-responsive. More recently, use of mesenchymal stem cells has been investigated as second line therapy for treatment of refractory GVHD [71]. Patients with refractory GVHD generally have poor outcomes with mortality rates as high as 90 % [73]. The risk of GVHD must be carefully weighed against the potential curative benefits in individual patients being considered for an allogeneic transplant. Concerns regarding this risk have led to the designation of autologous SCT being the preferred choice in patients with ARDs being considered for SCT [72].
Transplant-related mortality, defined by the Milan consensus as death within the first 3 months of transplant, remains a major concern when considering HSCT [39]. Infection and bleeding risks are highest during the aplastic phase and later during the T cell reconstitution phase infection remains a concern [33••]. Although estimates vary depending on the disease, Transplant-related mortality for most ARDs following autologous HSCT has been reported to be approximately 5 %, which is similar to that reported in the hematology/oncology literature [31•, 75].
Additional risks of particular concern in the SSc patient population undergoing HSCT is the potential development of lung toxicity from total body irradiation and of scleroderma renal crisis from high dose glucocorticoids used as prophylaxis for anti-thymocyte globulin-induced cytokine storm [33••]. Cardiac risks are also increased in patients with SSc and may be intensified with the use of cyclophosphamide for mobilization or for conditioning [76, 77]. In 2002, the EBMT Working Party for Autoimmune Diseases and the European League Against Rheumatism (EULAR) released a consensus statement recommending excluding from HSCT patients with cardiac dysfunction including pulmonary hypertension and heart failure. Electrocardiogram, transthoracic echocardiography and long-term ambulatory (Holter) cardiac monitoring are recommended to evaluate cardiac status prior to considering HSCT, and right and left heart catheterization are to be considered, particularly in SSc if initial studies show abnormalities [76].
A late complication of allogeneic HSCT is the development of secondary autoimmunity, attributed to repopulation of the immune system with uncontrolled autoimmune clones. Thus, new autoimmune diseases such as autoimmune hemolytic anemia, idiopathic thrombocytopenic purpura, and hemophilia may develop [33••, 78]. In the EBMT registry, 29/347 (9.8 ± 2 %) patients who underwent autologous HSCT for primary autoimmune disease developed a secondary autoimmune disease over 21 months of follow-up. Three of 16 patients undergoing allogeneic HSCT for primary autoimmune disease developed a secondary autoimmune disease. The secondary autoimmune diseases included: autoimmune hemolytic anemia, acquired hemophilia, autoimmune thrombocytopenia, antiphospholipid antibody syndrome, thyroiditis, Graves’ disease, myasthenia gravis, rheumatoid arthritis, sarcoidosis, vasculitis, psoriasis, and psoriatic arthritis. [73]. In multivariate analysis of the patients who underwent allogeneic HSCT, risk factors for developing secondary autoimmune diseases include a diagnosis of SLE prior to transplant, a <61-month interval between diagnosis of autoimmune disease and HSCT, and use of anti-thymocyte globulin in the conditioning regimen in combination with CD34+ selection [79].
Relapse of the presenting ARD has been reported in long-term follow-up. Registry data from the EBMT/EULAR database, Autologous Blood and Marrow Transplant Registry (ABMTR), and the International Bone Marrow Transplantation Registry (IBMTR) indicate a relapse rate of 32 % for adult and pediatric patients with SLE following autologous HSCT [45, 80, 81]. Relapse rates were significantly higher (73 %) in patients with rheumatoid arthritis treated with autologous HSCT. Due to the limited numbers of MSCTs performed, an accurate estimation of relapse rates in patients with ARDs following this procedure is not possible. Relapse rates likely depend in part on the type of cells transplanted (autologous vs. allogeneic, hematopoietic vs. mesenchymal stem cells) the conditioning regimen administered, and the extent and severity of the specific ARD.
Overall mortality data from published SCT trials for ARDs should be interpreted with caution as some results may be biased by disease severity. Some deaths may be attributable to advanced disease itself, as the population generally offered a transplant has had more severe disease. This is best exemplified by the SLE subset of patients undergoing SCT, in which 33 % of reported deaths have been attributed to SLE itself rather than complications from SCT [38].
Practical Application
Optimal application of SCT in the management of refractory ARDs remains an area of active investigation. Consensus statements from EULAR and the EBMT indicate a preference for autologous over allogeneic transplants due to the lower overall risk of severe toxicity with an autologous transplant that is largely due to elimination of the risk of severe refractory GVHD [82].
At the current time, HSCT should be reserved for patients with ARDs meeting the following criteria: (1) their disease is irreversible or associated with a high risk of mortality, (2) their disease is refractory to standard therapy, and (3) they have not yet developed extensive organ damage from their disease [82, 83].
Patients with SSc and associated cardiac dysfunction should be excluded as candidates for myeloablative HSCT due to an increased mortality from worsening heart failure [28]. Patients deemed appropriate candidates for SCT should be referred to medical centers with expertise and experience in SCT, which has been shown to correlate with improved outcomes [51].
Conclusions
Autologous HSCT can induce major durable responses in patients with severe ARDs refractory to conventional therapy. Despite encouraging results, transplant-related mortality remains a significant concern. Definitive conclusions regarding the role of SCT in patients with specific ARDs are not possible at this time owing to the small numbers of patients transplanted in each disease category, and to the variability in their disease involvement and severity, reliance on retrospective analyses, variability in the type of SCT performed, and conditioning regimen used, and heterogeneity in the measured outcome variables. Future larger-scale multi-center prospective randomized SCT clinical trials will need to be designed in patients with well-defined disease involvement and severity with specific ARDs disease categories to determine the safety and efficacy of mesenchymal versus hematopoietic stem cells, allogeneic versus autologous cell sources, and myeloablative versus non-myeloablative conditioning regimens. Final results from the phase III ASTIS and phase II SCOT trials are also expected to provide important insights regarding the future of SCT in ARDs.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Sullivan KM, Muraro P, Tyndall A. Hematopoietic cell transplantation for autoimmune disease: updates from Europe and the United States. Biol Blood Marrow Transplant. 2010;16(1 Suppl):S48–56.
Hönig M, Schulz A, Friedrich W. Hematopoietic stem cell transplantation for severe combined immunodeficiency. Klin Padiatr. 2011;223(6):320–5.
Kiem HP, Jerome KR, Deeks SG, et al. Hematopoietic-stem-cell-based gene therapy for HIV disease. Cell Stem Cell. 2012;10(2):137–47.
Chang JW, Hung SP, Wu HH, et al. Therapeutic effects of umbilical cord blood-derived mesenchymal stem cell transplantation in experimental lupus nephritis. Cell Transplant. 2011;20(2):245–57.
DiPersio JF, Micallef IN, Stiff PJ, et al. Phase III Prosepective Randomized Double-Blind Placebo-Controlled Trial of Plerixafor Plus Granulocyte Colony-Stimulating Factor Compared with Placebo Plus Granulocyte Colony-Stimulating Factor for Autologous Stem-Cell Mobilization and Transplantation for Patients with Non-Hodgkin’s Lymphoma. J Clin Oncol. 2009;27(28):4767–73.
Moore J, Brooks P, Milliken S, et al. A pilot randomized trial comparing CD34-selected versus unmanipulated hematopoietic stem cell transplantation for severe refractory rheumatoid arthritis. Arthritis Rheum. 2002;46(9):2301–9.
Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276(5309):71–4.
Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–7.
Horwitz EM, Le Blanc K, Dominici M, et al. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy. 2005;7(5):393–5.
Stagg J, Galipeau J. Immune plasticity of bone marrow-derived mesenchymal stromal cells. Handb Exp Pharmacol. 2007;180:45–66.
Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol. 2001;1:147–53.
Li HW, Sykes M. Emerging concepts in haematopoietic cell transplantation. Nat Rev Immunol. 2012;12(6):403–16.
Hénon PH, Sovalat H, Bourderont D. Importance of CD34+ cell subsets in autologous PBSC transplantation: the mulhouse experience using CD34 + CD38- cells as predictive tool for hematopoietic engraftment. J Biol Regul Homeost Agents. 2001;15(1):62–7.
De Kleer I, Vasstert B, Klein M, et al. Autologous stem cell transplantation for autoimmunity induces immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4 + CD25+ immune regulatory network. Blood. 2006;107(4):1696–702.
Roord ST, de Jager W, Boon L, et al. Autologous bone marrow transplantation in autoimmune arthritis restores immune homeostasis through CD4 + CD25 + Foxp3+ regulatory T cells. Blood. 2008;111(10):5233–41.
Wang D, Zhang H, Cao M, et al. Efficacy of allogeneic mesenchymal stem cell transplantation in patients with drug-resistant polymyositis and dermatomyositis. Ann Rheum Dis. 2011;70(7):1285–8.
Djouad F, Charbonnier LM, Bouffi C, Louis-Plence P, et al. Mesenchymal stem cells inhibit the differentiation of dendritic cells through an interleukin-6-dependent mechanism. Stem Cells. 2007;25:2025–32.
Meirelles Lda S, Fontes AM, Covas DT, et al. Mechanisms involved in the therapeutic properties of mesenchymal stem cells. Cytokine Growth Factor Rev. 2009;20:419–27.
Le Blanc K, Ringden O. Immunomodulation by mesenchymal stem cells and clinical experience. J Intern Med. 2007;262:509–25.
Szodoray P, Varoczy L, Papp G, et al. Immunological reconstitution after autologous stem cell transplantation in patients with refractory systemic autoimmune diseases. Scand J Rheumatol. 2012;41(2):110–5.
Steingrimsdottir H, Gruber A, Björkholm M, et al. Immune reconstitution after autologous hematopoietic stem cell transplantation in relation to underlying disease, type of high-dose therapy and infectious complications. Haematologica. 2000;85(8):832–8.
Fuschiotti P. Role of IL-13 in systemic sclerosis. Cytokine. 2011;56(3):544–9.
Muangchan C, Pope JE. Interleukin 6 in systemic sclerosis and potential implications in targeted therapy. J Rheumatol. 2012;39(6):1120–4.
Prescott RJ, Freemont AJ, Jones CJ, et al. Sequential dermal microvascular and perivascular changes in the development of scleroderma. J Pathol. 1992;166(3):255–63.
Hu H, Stein-Streilein J. Hapten-immune pulmonary interstitial fibrosis (HIPIF) in mice requires both CD4+ and CD8+ T lymphocytes. J Leukoc Biol. 1993;54(5):414–22.
Rosa SB, Voltarelli JC, Chies JA, et al. The use of stem cells for the treatment of autoimmune diseases. Braz J Med Biol Res. 2007;40(12):1579–97.
•• Burt RK, Milanetti F. Hematopoietic stem cell transplantation for systemic sclerosis: history and current status. Curr Opin Rheumatol. 2011;23(6):519–29. Excellent overview of HSCT use in SSc.
Oyama Y, Barr WG, Statkute L, et al. Autologous non-myeloablative hematopoietic stem cell transplantation in patients with systemic sclerosis. Bone Marrow Transplant. 2007;40:549–55.
McSweeney PA, Nash RA, Sullivan KM, et al. High-dose immunosuppressive therapy for severe systemic sclerosis: initial outcomes. Blood. 2002;100:1602–10.
Marjanovic Z, J. Snowden, M. Badoglio, et al.: Long-term outcomes of autologous haematopoietic stem cell transplantation in severe autoimmune diseases: an extended analysis of the EBMT database 1996–2011. Presented at the 38th Annual Meeting of the European-Group-for-Blood-and-Marrow-Transplantation (EBMT). Geneva, Switzerland; April 1–4, 2012.
• Burt RK, Shah SJ, Dill K, et al. Autologous non-myeloablative haematopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomized phase 2 trial. Lancet. 2011;378(9790):498–506. First randomized controlled trial regarding HSCT use in systemic sclerosis.
Henes JC, Schmalzing M, Vogel W, et al. Optimization of autologous stem cell transplantation for systemic sclerosis—a single-center long term experience in 26 patients with severe organ manifestations. J Rheumatol. 2012;39(2):269–75.
•• Tyndall A. Successes and failures of stem cell transplantation in autoimmune diseases. Hematology Am Soc Hematol Educ Program. 2011;2011:280–4. Excellent review of SCT use in autoimmune diseases.
Scleroderma clinical trials: Astis Study. Available at http://www.sclero.org/medical/treatments/clinical-trials/current/astis/a-to-z.html. Accessed June 2012.
Autologous stem cell transplantation international scleroderma(ASTIS) trial. Available at http://www.controlled-trials.com/ISRCTN54371254. Accessed June 2012.
van Laar JM, Farge D, Sont JK, et al.: The ASTIS Trial: Autologous stem cell transplantation versus IV pulse cyclophosphamide in poor prognosis systemic sclerosis, first results. [abstract LB0002]. Presented at EULAR. Berlin, Germany: June 6–9, 2012.
Musculoskeletal report: High-Dose Immune Suppression With or Without Stem Cell Transplants For Systemic Sclerosis. Available at http://www.mskreport.com/articles.cfm?articleID=1084. Accessed June 2012.
Illei GG, Cervera R, Burt RK, et al. Current state and future directions of autologous hematopoietic stem cell transplantation in systemic lupus erythematosus. Ann Rheum Dis. 2011;70(12):2071–4.
Song X, Lv HY, Sun LX, et al. Autologous stem cell transplantation for systemic lupus erythematosus: report of efficacy and safety at 7 years of follow-up in 17 patients. Transplant Proc. 2011;43(5):1924–7.
Mok CC, Ying, KY, Ng WL, et al.: Long-term Outcome of Diffuse Proliferative Lupus Glomerulonephritis Treated with Cyclophosphamide. Am J Med 2006, 119(4): 355. e25-e33.
Meng J, Wang J, Liang W, et al. Long-term remission after successful pregnancy in autologous peripheral blood stem cell transplanted system lupus erythematosus patients. Rheumatol Int. 2011;31(5):691–4.
Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358(9285):903–11.
Gonzalez-Rey E, Gonzalez MA, Varela N, et al. Human adipose-derived mesenchymal stem cells reduce inflammatory and T cell responses and induce regulatory T cells in vitro in rheumatoid arthritis. Ann Rheum Dis. 2010;69:241–8.
Wu CC, Wu TC, Liu FL, et al. TNF-α inhibitor reverse the effects of human umbilical cord-derived stem cells on experimental arthritis by increasing immunosuppression. Cell Immunol. 2012;273(1):30–40.
Snowden JA, Passweg J, Moore JJ, et al. Autologous hemopoietic stem cell transplantation in severe rheumatoid arthritis: a report from the EBMT and ABMTR. J Rheumatol. 2004;31(3):482–8.
Liu Y, Mu R, Wang S, et al. Therapeutic potential of human umbilical cord mesenchymal stem cells in the treatment of rheumatoid arthritis. Arthritis Res Ther. 2010;12(6):R210.
Park MJ, Park HS, Cho ML, et al. Transforming growth factor β-transduced mesenchymal stem cells ameliorate experimental autoimmune arthritis through reciprocal regulation of Treg/Th17 cells and osteoclastogenesis. Arthritis Rheum. 2011;63(6):1668–80.
Dalakas MC. Pathogenesis and therapies of immune-mediated myopathies. Autoimmun Rev. 2012;11(3):203–6.
Mahil S, Marks D, McComrack M, et al. Dermatomyositis. Br J Hosp Med (Lond). 2012;73(2):C18–22.
Ra JC, Kang SK, Shin IS, et al. Stem cell treatment for patients with autoimmune disease by systemic infusion of culture-expanded autologous adipose tissue derived mesenchymal stem cells. J Transl Med. 2011;9:181.
Owaidah TM, Maghrabi K, Elkarouri MA, et al. Successful treatment of a case of catastrophic antiphospholipid syndrome with autologous BMT: case report and review of literature. Bone Marrow Transplant. 2011;46(4):597–600.
Adamson R, Sangle S, Kaul A, et al. Clinical improvement in antiphospholipid syndrome after rituximab therapy. J Clin Rheumatol. 2008;14(6):359–60.
Statkute L, Traynor A, Oyama Y, et al. Antiphospholipid syndrome in patients with systemic lupus erythematosus treated by autologous hematopoietic stem cell transplantation. Blood. 2005;106(8):2700–9.
Schilder AM. Wegener’s Granulomatosis vasculitis and granuloma. Autoimmun Rev. 2010;9(7):483–7.
Langford CA. Update on the treatment of granulomatosis with polyangiitis (Wegener’s). Curr Treat Options Cardiovasc Med. 2012;14(2):164–76.
Daikeler T, Erley C, Mohren M, et al. Fever and increasing cANCA titre after kidney and autologous stem cell transplantation for Wegener’s Granulomatosis. Ann Rheum Dis. 2005;64(4):646–7.
Kunitomi A, Ishikawa T, Tajima K, et al. Bone marrow transplantation with a reduced-intensity conditioning regimen in a patient with Wegener granulomatosis and therapy-related leukemia. Int J Hematol. 2006;83(3):262–5.
Tsukamoto H, Nagafuji K, Horiuchi T, et al. A phase I-II trial of autologous peripheral blood stem cell transplantation in the treatment of refractory autoimmune disease. Ann Rheum Dis. 2006;65(4):508–14.
Daikeler T, Kötter I, Bocelli Tyndall C, et al. Haematopoietic stem cell transplantation for vasculitis including Behcet’s disease and polychondritis: a retrospective analysis of patients recorded in the European Bone Marrow Transplantation and European League Against Rheumatism databases and a review of the literature. Ann Rheum Dis. 2007;66(2):202–7.
Statkute L, Oyama Y, Barr WG, et al. Autologous non-myeloablative haematopoietic stem cell transplantation for refractory systemic vasculitis. Ann Rheum Dis. 2008;67(9):991–7.
Bornhäuser M, Aringer M, Theide C. Mixed lymphohematopoietic chimerism and response in Wegener’s granulomatosis. N Engl J Med. 2010;362(25):2431–2.
De Kleer IM, Brinkman DM, Ferster A, et al. Autologous stem cell transplantation for refractory juvenile idiopathic arthritis: analysis of clinical effects, mortality, and transplant related morbidity. Ann Rheum Dis. 2004;63(10):1318–26.
Brinkman DM, de Kleer IM, ten Cate R, et al. Autologous stem cell transplantation in children with severe progressive systemic or polyarticular idiopathic arthritis: long-term follow-up of a prospective clinical trial. Arthritis Rheum. 2007;56(7):2410–21.
Rabusin M, Andolina M, Maximova N, et al. Haematopoietic SCT in autoimmune diseases in children: rational and new perspectives. Bone Marrow Transplant. 2008;41 suppl 2:S96–9.
Chen J, Wang Y, Kunkel G, et al. Use of CD34+ autologous stem cell transplantation in the treatment of children with refractory systemic lupus erythematosus. Clin Rheumtol. 2005;24(5):464–8. 2 patients.
Jayne D, Passweg J, Marmont A, et al. Autologous stem cell transplantation for systemic lupus erythematosus. Lupus. 2004;13:168–76.
Krauss AC, Kamani NR. Hematopoietic stem cell transplantation for pediatric autoimmune disease: where we stand and where we need to go. Bone Marrow Transplant. 2009;44:137–43.
Wulffraat NM, van Rooijen EM, Tewarie R, et al. Current perspectives of autologous stem cell transplantation for severe juvenile idiopathic arthritis. Autoimmunity. 2008;41(8):632–8.
Lawitschka A, Peters C, Seidel MG, et al. Long-term remission in pediatric Wegener granulomatosis following allo-SCT after reduced intensity conditioning. Bone Marrow Transplant. 2011;46(3):462–3.
Farge D, Passweg J, van Laar JM, et al. Autologous stem cell transplantation in the treatment of systemic sclerosis: report from the EBMT/EULAR Registry. Ann Rheum Dis. 2004;63(8):974–81.
Swart JF, Lindemans CA, van Royen A, et al. Changing winds in refractory autoimmune disease in children: clearing the road for tolerance with cellular therapies. Curr Opin Rheumatol. 2012;24(3):267–73.
Hügle T, van Laar JM. Stem cell transplantation for rheumatic autoimmune diseases. Arthritis Res Ther. 2008;10(5):217.
Blazer BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012;12(6):443–58.
Pasquini MC, Wang Z, Horowitz MM, et al. report from the Center for International Blood and Marrow Transplant Research (CIBMTR): current uses and outcomes of hematopoietic cell transplants for blood and bone marrow disorders. Clin Transplant. 2010;2010:87–105.
Stamatović D, Balint B, Tukić L, et al. Impact of stem cell source on allogeneic stem cell transplantation outcome in hematological malignancies. Vojnosanit Pregl. 2011;68(12):1026–32.
Saccardi R, Tyndall A, Coghlan G, et al. Consensus statement concerning cardiotoxicity occurring during haematopoietic stem cell transplantation in the treatment of autoimmune disease, with special reference to systemic sclerosis and multiple sclerosis. Bone Marrow Transplant. 2004;34(10):877–81.
Burt RK, Shah SJ, Gheorghiade M, et al. Hematopoietic stem cell transplantation for systemic sclerosis: if you are confused, remember: “it is a matter of the heart”. J Rheumatol. 2012;39(2):206–9.
Bohgaki T, Atsumi T, Koike T. Autoimmune disease after autologous hematopoietic stem cell transplantation. Autoimmun Rev. 2008;7:198–203.
Daikeler T, Labopin M, Di Gioia M, et al. Secondary autoimmune diseases occurring after HSCT for anautoimmune disease: a retrospective study of the EBMT Autoimmune Disease Working Party. Blood. 2011;118:1693–8.
Tyndall A, Saccardi R. Haematopoietic stem cell transplantation in the treatment of severe autoimmune disease: results from phase I/II studies, prospective randomized trials and future directions. Clin Exp Immunol. 2005;141(1):1–9.
Jayne D, Tyndall A. Autologous stem cell transplantation for systemic lupus erythematosus. Lupus. 2004;13(5):359–65.
Tyndall A, Gratwohl A. Blood and marrow stem cell transplants in auto-immune disease: a consensus report written on behalf of the European League against Rheumatism (EULAR) and the European Group for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant. 1997;19(7):643–5.
Scleroderma Cyclophosphamide or Transplantation Study (SCOT). Available at. https://www.fbo.gov/index?s=opportunity&mode=form&id=e3a055606954c423b4a4606326f96081&tab=core&_cview=0. Accessed June 2012.
Liang J, Zhang H, Hua B, et al. Allogenic mesenchymal stem cells transplantation in refractory systemic lupus erythematosus: a pilot study. Ann Rheum Dis. 2010;69(8):1423–9.
Sun L, Akiyama K, Zhang H, et al. Mesenchymal Stem Cell Transplantation Reverses Multi-Organ Dysfunction in Systemic Lupus Erythematosus Mice and Humans. Stem Cells. 2009;27(6):1421–32.
Abinun M, Flood TJ, Cant AJ, et al. Autologous T cell depleted haematopoietic stem cell transplantation in children with severe juvenile idiopathic arthritis in the UK (2000–2007). Mol Immunol. 2009;47(1):46–51.
Daikeler T, Hügle T, Farge D, et al. Allogenic hematopoietic SCT for patients with autoimmune diseases. Bone Marrow Transplant. 2009;44(1):27–33.
Alexander T, Thiel A, Rosen O, et al. Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long-term remission through de novo generation of a juvenile and tolerant immune system. Blood. 2009;113(1):214–23.
Nevskaya T, Ananieva L, Bykovskaia S, et al. Autologous progenitor cell implantation as a novel therapeutic intervention for ischaemic digits in systemic sclerosis. Rheumatology (Oxford). 2009;48(1):61–4.
Sun L, Wang D, Liang J, et al. Umbilical cord mesenchymal stem cell transplantation in severe and refractory systemic lupus erythematosus. Arthritis Rheum. 2010;62(8):2467–75.
Carrion F, Nova E, Ruiz C, et al. Autologous mesenchymal stem cell treatment increased T regulatory cells with no effect on disease activity in two systemic lupus erythematosus patients. Lupus. 2010;19(3):317–22.
van Laar JM, Farge D, Tyndall A. Autologous stem cell transplantation International Scleroderma (ASTIS) trial: hope on the horizon for patients with severe systemic sclerosis. Ann Rheum Dis. 2005;64(10):1515.
Craciunescu OI, Steffey BA, Kelsey CR, et al. Renal shielding and dosimetry for patients with severe systemic sclerosis receiving immunoablation with total body irradiation in the scleroderma: cyclophosphamide or transplant trial. Int J Radiat Oncol Biol Phys. 2011;79(4):1248–55.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mascarenhas, S., Avalos, B. & Ardoin, S.P. An Update on Stem Cell Transplantation in Autoimmune Rheumatologic Disorders. Curr Allergy Asthma Rep 12, 530–540 (2012). https://doi.org/10.1007/s11882-012-0298-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11882-012-0298-8