
Abstract
The NLRP3 inflammasome is an intracellular complex that regulates the release of proinflammatory cytokines such as interleukin-1β in response to exogenous pathogens and endogenous danger signals. Evidence from studies involving human genetics, human ex vivo mononuclear cell responses, and in vivo and in vitro murine models confirms the importance of the inflammasome and interleukin-1β in the pathogenesis of several inherited and complex diseases. The availability of several effective interleukin-1β targeted therapies has allowed for successful proof-of-concept studies in several of these disorders. However, many other diseases are likely to be mediated by the inflammasome and interleukin-1β, providing additional targets in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There are two classes of innate immune receptors: Toll-like receptors (TLRs), located on cell membranes, and nucleotide-binding oligomerization domain-like receptors (NLRs) located in the cytoplasm of cells. Both classes of receptors are programmed to recognize microbial pathogen-associated molecular patterns (PAMPs), which can stimulate secretion of proinflammatory and other cytokines. The focus of this discussion is on describing diseases that are confirmed or potentially associated with the unique NLRP3 inflammasome (Fig. 1), which, in addition to stimulation by PAMPs, can also be stimulated by danger-associated molecular patterns (DAMPs) to secrete interleukin (IL)-1β and other IL-1 cytokine family members, such as IL-18. DAMPs include extracellular adenosine triphosphate, several crystals (including uric acid/urate, calcium pyrophosphate, silica), and reactive oxygen species. Although members of the IL-1 cytokine family have significant and in some cases parallel biological activities, this discussion focuses on IL-1β secretion because of its known ability to induce neutrophilic inflammatory responses. The induction of neutrophilic inflammation by IL-1β has been documented in the cryopyrinopathies and other inherited autoinflammatory diseases. The study of hereditary syndromes has confirmed that IL-1β can initiate downstream robust neutrophilic inflammation, which can be reversed and controlled with IL-1β targeted therapy (TT). Consequently, several investigators have recognized that targeting IL-1β may have beneficial effects in the treatment of inflammatory conditions with predominant neutrophilia or neutrophilia mixed with other inflammatory cellular infiltrates. In this review, we consider inflammatory disorders in which IL-1β has been implicated as a pathogenic mediator and in which IL-1β TT has been used successfully or is being considered as a therapeutic modality in the future (Table 1).

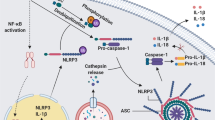
The inflammasome. The inflammasome is an intracellular protein complex consisting of NRLP3, apoptosis-associated speck protein with a caspase activation and recruitment domain (ASC), and caspase-1. Pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) activate the inflammasome, resulting in activation of caspase-1 and cleavage of pro-interleukin (IL)-1β to form active IL-1β, which leaves the cell and can then bind to the IL-1 receptor, resulting in inflammation. Several IL-1β targeted therapies have been developed. mAb—monoclonal antibody. Figure design by S. Brydges
The NLRP3 Inflammasome
The NLRP3 inflammasome is an intracellular, multiprotein complex composed of an NLR protein (NLRP3, also known as cryopyrin or NALP3), at least one adaptor protein (apoptosis-associated speck protein with a caspase activation and recruitment domain [ASC]), and caspase-1 (IL-1β-cleaving enzyme). NLRP3 serves as a protein complex scaffold, and ASC bridges NRLP3 to caspase-1, allowing for its activation. Once activated, caspase-1 can cleave pro-IL-1β to IL-1β, its mature, active, and secretable form (Fig. 1). Once IL-1β leaves the cell, it binds to the IL-1 receptor, resulting in inflammation. Therefore, the primary role of the inflammasome is to direct inflammation by controlling unregulated IL-1β release that can be damaging to the host while allowing for efficient cytokine-mediated inflammation when the host is in danger. The NLRP3 inflammasome has been implicated in the host response to bacteria such as Staphylococcus aureus and Neisseria gonorrhoeae, viruses such as Vaccinia and Influenza, fungal products such as hyphae of Candida albicans, and parasite-associated byproducts such as malarial hemozoin [1]. However, the unique features of NLRP3 are the response to DAMPs, which indicate cell death, such as adenosine triphosphate or uric acid, or cell stress, such as the formation of reactive oxygen species [2•] and the presence of disease-causing mutations in patients with cryopyrinopathies [3].
IL-1β Targeted Therapy
Since the discovery and cloning of IL-1β, this critical proinflammatory mediator implicated in many diseases has been a focus of the pharmaceutical industry. The first successful drug in this class was a recombinant form of the IL-1 receptor antagonist (RA), a natural inhibitor of IL-1β-mediated inflammation. This drug, known as anakinra, was originally developed for use in sepsis, but studies in this severe systemic and often fatal disease were challenging. Studies of anakinra in rheumatoid arthritis demonstrated modest efficacy [4], but the clinical response was sufficient for US Food and Drug Administration approval in 2001. Inconsistent results, a short half-life requiring daily dosing, and painful injection site reactions have discouraged widespread use of this drug. At least three additional IL-1-targeted drugs with longer half-lives and fewer injection site reactions have been developed, including a recombinant protein with high affinity for IL-1β, known as IL-1 TRAP, or rilonacept [5], and two humanized monoclonal antibodies, canakinumab [6] and XOMA 052 (Berkeley, CA). Rilonacept and canakinumab were studied in cryopyrinopathy patients with significant and consistent responses and were subsequently approved by the US Food and Drug Administration under its orphan drug program [7, 8]. Both drugs are also currently being developed for treatment of gout [9], and XOMA 052 is being developed for treatment of type 2 diabetes mellitus.
Disorders with IL-1β Involvement with Recognized Association with the NLRP3 Inflammasome
Conditions Improved with IL-1β TT
Cryopyrinopathies
The cryopyrinopathies are a continuum of inherited inflammatory disorders of increasing severity, including familial cold autoinflammatory syndrome, Muckle-Wells syndrome, and neonatal-onset, multisystem inflammatory disease [10]. All of these disorders are characterized by fever, an urticaria-like rash, and joint symptoms resulting in significant clinical overlap. However, several unique features distinguish the three cryopyrinopathy phenotypes. Familial cold autoinflammatory syndrome is associated with episodes that are often induced by exposure to cold. Muckle-Wells syndrome is associated with episodes induced by several triggers but also with a progressive sensorineural hearing loss and a significant risk of developing type AA amyloidosis. Neonatal-onset, multisystem inflammatory disease is associated with severe central nervous system involvement and unique overgrowth of the distal femur and patella.
In 2001, positional cloning demonstrated that heterozygous mutations in NLRP3 are the cause of most cases of cryopyrinopathy [3]. These mutations are hypothesized to be gain of function, resulting in a hyperactive or constitutively active inflammasome with subsequent systemic IL-1 release resulting in disease-associated symptoms [10]. Translational research led to successful trials and US Food and Drug Administration approval of IL-1β TT in the cryopyrinopathies, with life-altering results [7, 8, 11].
Gout
Gout is an inflammatory condition that leads to accumulation of uric acid/urate crystals in acral joints. The formation of uric acid/urate crystals in acral joints may occur because these anatomic regions are exposed and affected by low ambient temperatures, which encourage uric acid/urate crystallization. Uric acid/urates crystallize at low temperatures, as they have a solubility of 4.5 mg/dL at 30°C, compared with 7.0 mg/dL at 37°C. The concept of reversing gout crystal-induced inflammation with IL-1β TT was based on observations by several investigators who recognized that uric acid/urate crystals can stimulate secretion of IL-1β via the NLRP3 inflammasome [12]. Evidence also indicates that uric acid/urate crystals can stimulate secretion of IL-1β via TLR2 and TLR4. Results from several studies support IL-1β TT as beneficial in refractory acute and chronic gout [9].
Type 2 Diabetes Mellitus
Inflammation of pancreatic islets of patients with type 2 diabetes mellitus is associated with IL-1β secretion and reduction of IL-1RA. High glucose leads to an increase in IL-1β, impairment of insulin secretion, and apoptosis. Many studies in animals and humans have demonstrated that IL-1β TT can reverse this process with improved glycemia, β-cell function, and reduced markers of inflammation [13, 14]. Recent work has demonstrated that Nlrp3 knockout animals have improved glucose tolerance and insulin sensitivity, but no studies have shown the inflammation of pancreatic islet cells associated with type 2 diabetes mellitus is reduced in Nlrp3 inflammasome knockout animals [13, 14].
Conditions with IL-1β Involvement Considered for IL-1β TT
Bleomycin-Induced Lung Injury
Bleomycin, a commonly used treatment of solid neoplasms, is associated with the common side effect of early pulmonary neutrophilic inflammation and later fibrosis. Gasse et al. [15] studied a murine model and established that bleomycin effects occur via uric acid/urate formation and stimulation of the NLRP3 inflammasome, leading to IL-1β secretion and neutrophilic inflammation. In a different murine model, IL-1β TT reversed neutrophilic pulmonary inflammation caused by bleomycin [16]. To date, use of IL-1β TT to prevent or reverse bleomycin-induced neutrophilic pulmonary inflammation in humans has not been reported.
Silicosis and Asbestosis
The chronic histopathology in these occupational-related disorders is made up of neutrophils, monocytes, and other inflammatory cells with resultant interstitial fibrosis. The chronicity of these disorders is thought to result from several immunopathologic pathways, including secretion of IL-1β from silica and asbestos stimulation of the NLRP3 inflammasome [17]. Based on these results, there is rationale to treat these conditions with IL-1β TT.
Contact Hypersensitivity
Exposure to skin sensitizers, such as trinitrochlorobenzene, causes type IV Gells and Coombs delayed hypersensitivity reactions. Elegant studies have demonstrated that delayed reactions of contact hypersensitivity require IL-1β /IL-18 activation by the NLRP3 inflammasome. Knockout mice for Nlrp3 or other essential components of this inflammasome displayed impaired response to trinitrochlorobenzene [18]. Future use of IL-1β TT may be appropriate to reduce and/or prevent contact hypersensitivity responses.
Disorders with IL-1β Involvement with Unconfirmed Association with the NLRP3 Inflammasome
Conditions Improved with IL-1β TT
Familial Mediterranean Fever
Familial Mediterranean fever (FMF) is the most common inherited recurrent fever disorder. Affected patients experience 2- to 3-day-long inflammatory episodes characterized by fever, abdominal pain, and joint symptoms. Rash and chest pain can also occur during episodes. However, the most significant morbidity in these patients is systemic AA amyloidosis that often results in end-stage renal disease. The standard treatment is maintenance colchicine, which has been shown to reduce the frequency and severity of episodes but also significantly reduces the risk of amyloidosis [19•]. Most FMF patients have homozygous mutations in the MEFV gene that codes for the protein pyrin. Pyrin has some structural similarities to the NLR proteins and may interact with NLR inflammasomes or form its own inflammasome, resulting in dysregulated caspase-1 activation and IL-1β release [19•]. In addition, mononuclear cells from FMF patients release higher levels of IL-1β [19•]. For this reason, some FMF patients who do not respond to or who cannot tolerate colchicine have been treated with IL-1β TT, with promising results; however, colchicine remains the treatment of choice in most patients [20].
Tumor Necrosis Factor Receptor-Associated Periodic Syndrome
This autosomal dominantly inherited recurrent fever disorder, originally known as familial Hibernian fever, is associated with 1- to 3-week-long episodes of fever, abdominal pain, and joint symptoms that are often associated with rash and periorbital swelling. As in FMF, some patients develop AA amyloidosis and kidney disease. Symptoms usually respond to high-dose corticosteroids [19•], but the discovery of heterozygous mutations in the TNFR1a gene prompted the use of tumor necrosis factor (TNF)-α TT (etanercept), with good results initially [21]. However, incomplete abrogation of episodes and persistent chronic inflammation despite proper dosing and evidence suggesting that TNF-α-mediated inflammation is dependent on IL-1β provided the rationale for IL-1β TT, with superior results to TNF-α TT [22, 23].
Hyper IgD Syndrome
Patients with hyper IgD syndrome experience recurrent episodes of fever, lymphadenopathy, splenomegaly, and abdominal pain lasting 3–7 days. This autosomal recessive disorder is caused by homozygous mutations in the gene (MVK) resulting in decreased mevalonate kinase activity, with effects on the isoprenoid pathway of cholesterol biosynthesis [19•]. Mechanisms responsible for inflammation are believed to be due to the deficiency of isoprenylated proteins, which intersect with the IL-1 pathway. In support of this mechanism, mononuclear cells from hyper IgD patients release more IL-1β [19•], and patients have responded to IL-1 TT [24].
Pyogenic Arthritis, Pyoderma Gangrenosum, and Acne
This rare autosomal dominant disease is characterized by joint inflammation, cutaneous ulcers, and cystic acne. Neutrophils are the predominant cell involved in tissue inflammation. Mutations in the proline-serine-threonine phosphatase interacting protein-1 are responsible for disease and are believed to affect interactions with the pyrin protein, resulting in IL-1-mediated inflammation [19•]. Mononuclear cells from patients release increased IL-1β, and patients have responded to IL-1 TT [25].
Deficiency of IL-1RA
Deficiency of IL-1RA is a rare autosomal recessive disease presenting with multifocal osteomyelitis, periostitis, and pustulosis. Mutations in the gene that codes for IL-1RA result in loss of function and unregulated IL-1 receptor-mediated inflammation. Therapy with anakinra results in complete resolution of symptoms [26].
Systemic-Onset Juvenile Idiopathic Arthritis and Adult-Onset Still’s Disease
Significant amelioration of signs and symptoms of both of these systemic inflammatory diseases occurs with use of anakinra. Results from a three-center pediatric rheumatology study and others demonstrated a reduction in large joint inflammation, corticosteroid dosage, acute-phase reactants, fever, and rashes in systemic-onset juvenile idiopathic arthritis and systemic inflammation in adult-onset Still’s disease [27, 28]. The mechanism for IL-1β secretion in this disorder is unknown, and no studies are available implicating involvement of the NLRP3 inflammasome.
Schnitzler Syndrome
Schnitzler syndrome is a rare disease characterized by fever, bone and joint pain, nonpruritic rash often mistaken for urticaria, and lymphadenopathy. Associated abnormal laboratory tests include leukocytosis, elevated erythrocyte sedimentation rate and other acute phase reactants, neutrophilic infiltration involving skin lesions, and IgM monoclonal gammopathy. Striking improvement occurs following anakinra therapy, suggesting the pathogenesis is IL-1β mediated [29]. The cause of IL-1β secretion is currently unknown, and no studies have implicated involvement of the NLRP3 inflammasome.
Conditions with IL-1β Involvement Considered for IL-1β TT
The commonality of the following diverse clinical disorders with evidence of IL-1β secretion and neutrophilic inflammation is the association of ischemia, hypoxia, and reperfusion with metabolic acidosis and catabolism of tissues. It has been hypothesized by one of the authors (Dr. Wanderer) that these pathophysiologic conditions cause an increase in byproducts (DAMPs) from breakdown of cellular nucleotides or proteins, which could stimulate innate receptors such as NLRP3 to cause secretion of IL-1β, and subsequent neutrophilic inflammation.
Ultraviolet-Induced Skin Damage
There is evidence using in vitro keratinocytes that the inflammasome can be activated by UVB radiation and cause IL-1β secretion [30]. The UVB band is similar to exposure to sunlight, and release of IL-1β may explain the fever, leukocytosis, and acute-phase responses seen with severe sunburns.
Chronic Neutrophilic Pulmonary Disorders
Examples include severe corticosteroid-resistant neutrophilic asthma and chronic obstructive pulmonary disease. It has been posited that hypoventilation with hypoperfusion and reoxygenation exhibit similarities to biphasic pathobiology of classic ischemia reperfusion injury. Low pH in exhaled breath condensates in these disorders provides evidence for the existence of acidotic milieu in affected tissues and cells, which in turn can cause DAMPs formation, IL-1β secretion, and neutrophilic inflammation. This ongoing process may explain steroid unresponsiveness [31]. Efforts are in progress to test this hypothesis by treating these diseases with IL-1β TT.
Sickle Cell Anemia
Neutrophilic inflammation is prominent in sickle cell disease and in transgenic sickle cell disease mice subjected to hypoxia and reperfusion. In addition, transgenic sickle cell disease mice compared with wild mice exhibit a heightened proinflammatory state to microbial molecules, as marked by elevated neutrophil counts, increased secretion of soluble vascular adhesion molecules, and increased secretion of proinflammatory cytokines (IL-1β and TNF-α). A similar observation of heightened inflammatory responses to microbial infections is manifested as chest injury in sickle cell disease patients. There is also evidence of DAMPs formation in sickle cell disease, as uric acid elevations are frequently observed and thought to be caused by a combination of hemolysis and nucleotide breakdown in tissues induced by ischemia and reperfusion. Ischemia reperfusion is a consequence of vasoocclusion from sequestration of sickle erythrocytes, polymerized free sickle hemoglobin, activated monocytes, neutrophils adherent to activated endothelial cells, and embolized fat from bone marrow. It is now known that PAMPs and DAMPs added to normal monocytes in vitro can cause synergistic secretion of IL-1β, which could explain the amplified inflammatory responses in these patients [32]. A trial involving sickle cell disease mice challenged by hypoxia and treated with IL-1 TT is in progress.
Hypoxic-Ischemic Encephalopathy
It has been posited that intrapartum hypoxia-ischemia through stimulation of the NALP3 inflammasome by DAMPs has a significant role in causing inflammation associated with hypoxic-ischemic encephalopathy. The following is presented as evidence for this hypothesis:
-
1.
Metabolic acidosis (pH <7.0 and base deficit) in fetal umbilical arterial blood samples obtained at delivery is one of the International Cerebral Palsy Task Force putative criteria to diagnose an acute intrapartum hypoxic event sufficient to cause cerebral palsy. This indicator of metabolic stress could enhance DAMPs formation;
-
2.
A newborn pig model subjected to sustained hypoxic injury exhibited a rapid decrease in phosphocreatine/inorganic phosphorus ratios, pH, and adenosine triphosphate, all metabolic events that could lead to DAMPs formation. A perinatal hypoxia-ischemia rodent model (rat) with carotid arterial occlusion showed progressive decreases in phosphocreatine and adenosine triphosphate to values of less than 10% of the control ipsilateral cerebral hemisphere during the course of hypoxia-ischemia over a 3-hour duration. They also demonstrated decreased intracellular pH in areas affected by arterial occlusion.
-
3.
A study of premature infants with elevated uric acid concentration, noted on the first postnatal day, subsequently developed hypoxic-ischemic-induced injury (ie, intravascular hemorrhage and/or leukomalacia).
-
4.
The observation of elevated IL-1β levels in spinal fluid samples obtained from hypoxic-ischemic human newborns as compared with control newborns evaluated for possible sepsis/meningitis that was subsequently ruled out [33].
To date, there are no known ongoing studies involving animal studies comparing hypoxic-ischemic stress between wild type and Nlrp3 knockout mice or in mice with and without pretreatment using monoclonal IL-1β TT, and there are no human trials with IL-1β TT to determine if hypoxic-ischemic encephalopathy can be reduced with this adjunct therapy.
Organ Ischemia Reperfusion Injury Disorders (Renal, Myocardial, Central Nervous System, and Transplantation Organ Injury)
Increased secretion of IL-1β has been observed in many models of ischemia reperfusion, including models of transplantation. It also has been demonstrated in animal models that IL-1β TT can prevent and/or minimize the neutrophilic inflammation associated with these models. Although PAMPs can induce IL-1β secretion, in many of the animal and transplantation models, PAMPS are not detected, suggesting that other mechanisms may cause induction of IL-1β secretion. One such paradigm is the production of DAMPs formation from the pathobiological effects of ischemia reperfusion injury, which in turn could stimulate NLRP3 and cause IL-1β secretion with subsequent neutrophilic inflammation [32]. There are no known trials of IL-1β TT in these conditions.
Conclusions
The discovery of NLRP3 as the cause of rare inherited autoinflammatory disorders has had a major influence on our understanding of innate immune receptors and cytokine responses, mechanisms of IL-1β release, IL-1β-mediated inflammation, the role of IL-1β in many disorders, and IL-1β TT. The inflammasome and IL-1β now have been implicated in the pathogenesis of rare inherited inflammatory diseases and common and more complex disorders, and IL-1 TT is now being used successfully with a significant impact on these patients’ lives.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Martinon F, Burns K, Tschopp J: The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002, 10:417–426.
• Martinon F, Mayor A, Tschopp J: The inflammasomes: guardians of the body. Annu Rev Immunol 2009, 27:229–265. This is an excellent review of inflammasome function.
Hoffman HM, Mueller JL, Broide DH, et al.: Mutation of a new gene encoding a putative pyrin-like protein cause familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001, 29:301–305.
Mertens M, Singh JA: Anakinra for rheumatoid arthritis: a systematic review. J Rheumatol 2009, 36:1118–1125.
Hoffman HM: Rilonacept for the treatment of cryopyrin-associated periodic syndromes (CAPS). Expert Opin Biol Ther 2009, 9:519–531.
Savic S, McDermott MF: Inflammation: canakinumab for the cryopyrin-associated periodic syndromes. Nat Rev Rheumatol 2009, 5:529–530.
Hoffman HM, Throne ML, Amar NJ, et al.: Efficacy and safety of rilonacept (interleukin-1 TRAP) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 2008, 58:2443–2452.
Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al.: Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 2009, 360:2416–2425.
Terkeltaub R, Sundy JS, Schumacher HR, et al.: The IL-1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, nonrandomized, single-blind pilot study. Ann Rheum Dis 2009, 68:1613–1617.
Aksentijevich I, Putnam CD, Remmers EF, et al.: The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum 2007, 56:1273–1285.
Hoffman HM, Rosengren S, Boyle DL, et al.: Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet 2004, 364:1779–1785.
So A: Developments in the scientific and clinical understanding of gout. Arthritis Res Ther 2008, 10:221.
Zhou R, Tardivel A, Thorens B, et al.: Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 2010, 11:136–140.
Maedler K, Dharmadhikari G, Schumann DM, Størling J: Interleukin-1 beta targeted therapy for type 2 diabetes. Expert Opin Biol Ther 2009, 9:1177–1188.
Gasse P, Riteau N, Charron S, et al.: Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med 2009, 179:903–913.
Piguet PF, Vesin C, Grau GE, Thompson RC: Interleukin 1 receptor antagonist (IL-1ra) prevents or cures pulmonary fibrosis elicited in mice by bleomycin or silica. Cytokine 1993, 5:57–61.
Dostert C, Petrilli V, Van Bruggen R, et al.: Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320:674–677.
Watanabe H, Gaide O, Petrilli V, et al.: Activation of the IL-1[beta]-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol 2007, 127:1956–1963.
• Masters SL, Simon A, Aksentijevich I, Kastner DL: Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol 2009, 27:621–668. This is a comprehensive review of autoinflammatory syndromes.
Belkhir R, Moulonguet-Doleris L, Hachulla E, et al.: Treatment of familial Mediterranean fever with anakinra. Ann Intern Med 2007, 146:825–826.
Hull KM, Aksentijevich I, Singh HK: Efficacy of etanercept for the treatment of patients with TNF receptor-associated periodic syndrome (TRAPS) [abstract]. Arthritis Rheum 2003, 46:S378.
Simon A, Bodar EJ, van der Hilst JC, et al.: Beneficial response to interleukin 1 receptor antagonist in traps. Am J Med 2004, 117:208–210.
Drewe E, Powell RJ, McDermott EM: Comment on: Failure of anti-TNF therapy in TNF receptor 1-associated periodic syndrome (TRAPS). Rheumatology 2007, 46:1865–1866.
Cailliez M, Garaix F, Rousset-Rouvière C, et al.: Anakinra is safe and effective in controlling hyperimmunoglobulinaemia D syndrome-associated febrile crisis. J Inherit Metab Dis 2006, 29:763.
Brenner M, Ruzicka T, Plewig G, et al.: Targeted treatment of pyoderma gangrenosum in PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome with the recombinant human interleukin-1 receptor antagonist anakinra. Br J Dermatol 2009, 161:1199–1201.
Aksentijevich I, Masters SL, Ferguson PJ, et al.: An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 2009, 360:2426–2437.
Lequerre T, Quartier P, Rosellini D, et al.: Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann Rheum Dis 2008, 67:302–308.
Zeft AMD, Hollister RMD, LaFleur BP, et al.: Anakinra for systemic juvenile arthritis: the Rocky Mountain experience. J Clin Rheumatol 2009, 15:161–164.
de Koning HD, Bodar EJ, Simon A, et al.: Beneficial response to anakinra and thalidomide in Schnitzler’s syndrome. Ann Rheum Dis 2006, 65:54–544.
Faustin B, Reed JC: Sunburned skin activates inflammasomes. Trends Cell Biol 2008, 18:4–8.
Wanderer AA: Corticosteroid resistance in pulmonary neutrophilic inflammatory disorders and rationale for adjunct IL-1{beta} targeted therapy. Am J Respir Cell Mol Biol 2009, 41:246–247.
Wanderer AA: Rationale for IL-1[beta] targeted therapy for ischemia-reperfusion induced pulmonary and other complications in sickle cell disease. J Pediatr Hematol Oncol 2009, 31:537–538.
Wanderer AA: Rationale for IL-1[beta]-targeted therapy to minimize hypoxic-ischemic encephalopathy. J Perinatol 2009, 29:785–787.
Disclosure
Dr. Hoffman has served as a consultant for Regeneron and Novartis pharmaceuticals. No other potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hoffman, H.M., Wanderer, A.A. Inflammasome and IL-1β-Mediated Disorders. Curr Allergy Asthma Rep 10, 229–235 (2010). https://doi.org/10.1007/s11882-010-0109-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11882-010-0109-z