
Opinion statement
Inflammatory myofibroblastic tumor (IMT), characterized by intermediate malignancy and a propensity for recurrence, has presented a formidable clinical challenge in diagnosis and treatment. Its pathological characteristics may resemble other neoplasms or reactive lesions, and the treatment was limited, taking chemotherapies as the only option for those inoperable. However, discovering anaplastic lymphoma kinase (ALK) protein expression in approximately 50% of IMT cases has shed light on a new diagnostic approach and application of targeted therapies. With the previous success of combating ALK+ non-small-cell lung cancers with ALK tyrosine kinase inhibitors (TKIs), crizotinib, a first-generation ALK-TKI, was officially approved by the U.S. Food and Drug Administration in 2020, to treat unresectable ALK+ IMT. After the approval of crizotinib, other ALK-TKIs, such as ceritinib, alectinib, brigatinib, and lorlatinib, have proven their efficacy on ALK+ IMT with sporadic case reports. The sequential treatments of targeted therapies in may provide the insight into the choice of ALK-TKIs in different lines of treatment for unresectable ALK+ IMT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammatory myofibroblastic tumor (IMT) is a locally aggressive mesenchymal tumor with lymphocyte infiltration [1,2,3,4], and myofibroblast spindle cell proliferation [5], that presents as a solitary lesion [6]. Only 150–200 cases are reported in the USA annually [7]. IMT was first discovered in the lungs in 1939 [8], and was considered benign at the time [9]. However, as the understanding of IMT progressed, it was re-classified as an “intermediate” myofibroblastic tumor by the World Health Organization (WHO) due to the discovered chromosomal alteration, nature of local aggressiveness, high recurrence rate, and low metastatic potential [10]. IMT has a recurrence rate of approximately 25%, but it is highly dependent on the IMT location [11, 12]. Studies have reported higher recurrence rates in lesions located in the abdominal space when the tumor size is > 8 cm [6, 13, 14]. In a study evaluating metastatic potential, among 59 documented cases, metastasis was only restricted to six anaplastic lymphoma kinase ALK− IMT cases (10.2%), whereas none of the ALK+ IMT cases exhibited metastasis [13]. Younger age, larger tumors, and detection at abdominopelvic and pulmonary sites were indicative of higher metastatic potential [13]. Nevertheless, distant metastasis has only occurred in approximately 5% of cases [12, 15••]. This result was later confirmed by Fu et al., who reported that only five patients with IMT (5.4%) had metastasis among 92 cases [16].
From a clinical perspective, IMT is usually disclosed during routine health checkups [17, 18] because patients can be entirely asymptomatic until the tumor has grown to a size that can cause complications [19]. The presenting symptoms include weight loss and general fatigue [17]. Therefore, the type and severity of symptoms presented by patients with IMT mainly depend on the location of the primary site and size of the tumor [18, 20]. Furthermore, tumor size and patient age are considered prognostic factors; in younger patients or in patients with tumor sizes < 6.5 cm, survival rates tend to be better [16].
In most cases, surgical resection is the best approach for IMT treatment, which generally results in a better prognosis [17, 21], especially if the tumor is completely resected with a negative margin [22]. However, if the tumor is inoperable, other treatments should be considered, such as steroids or chemotherapy regimens (e.g., methotrexate, anthracycline-based, or ifosfamide-based) [23,24,25]. In the 13 of paranasal sinus and nasopharynx IMT cases presented by Zhu et al., better overall survival was correlated with the use of prednisone alone among 10 patients undergoing systemic treatment with a combination of surgery, prednisone, radiotherapy, chemotherapy, or observation alone [26]. However, the number of cases was limited. Therefore, a large cohort study should be conducted to validate this finding. In another study, chemotherapy (anthracycline-based and methotrexate plus/minus vinorelbine/vinblastine regimens) exhibited an objective response rate (ORR) of 47.6% and 53.8% for anthracycline-based or methotrexate-based chemotherapy respectively [27]. The median PFS and OS were 6.3 months and 21.2 months respectively for patients treated with anthracycline-based, and not reached and 83.4 months for methotrexate-based chemotherapy (Fig. 1). No prospective study of chemotherapy in IMT was reported.

Treatment choices for both operable and inoperable IMT
Here, we comprehensively reviewed IMT epidemiology and the current methodology for pathological and molecular diagnosis and treatment, particularly for targeted therapy against ALK+ IMT.
Epidemiology
The most common sites of IMT occurrence vary among studies; the lungs [1, 28], abdomen [13, 29], and soft tissues of the limbs or hips [16] have been reported. However, other anatomical sites [29], including the meninges of lobes, spinal cord, orbit, mandible, throat, thorax, heart, liver, duodenum, small intestine, colon, and uterus, have also been reported [16].
In terms of demographics, despite IMT can be diagnosed at any age, it seems to have a predilection for children and young adults [30,31,32]. Prevalence according to sex has been inconsistent among studies [13, 16, 28, 29]. Overall, the general prevalence of IMT ranges from 0.04 to 0.7%, irrespective of sex or race [33, 34].
Owing to the rarity of IMT, its risk factors are not fully understood. Smoking, minor trauma, and IgG4-related disease are thought to be risk factors for tumorigenesis in IMT [8, 35].
Pathogenesis
Since it was first described in 1939, the cause of IMT pathogenesis remains unclear. Rohrlich et al. proposed that IMT may be a consequence of cytokine production dysregulation following infection [36]. More investigations had supported this hypothesis, suggesting that IMT may be an unusual immunological response to viruses (such as human herpesvirus 8, and Epstein-Barr virus) [13, 37, 38], surgery, or autoimmune diseases [39]. However, an increasing number of studies have considered IMT to be a tumor rather than a reactive process [33, 40]. The recent discovery of chromosomal abnormalities may indicate that IMT is more of a tumor than an inflammatory result or a pseudotumor [41, 42]. Lovly et al. later confirmed that IMT is a largely oncogene-driven neoplasia [43], and that tumorigenesis is associated with the translocation of receptor tyrosine kinase genes, such as ALK and ROS-1 [22].
Clinical and pathological features
In the early stages of IMT exploration, a thorough understanding of IMT was difficult to achieve owing to its rarity and similarities with other illnesses, and IMT was commonly confused with inflammatory pseudotumor, fibromyxoid lesions, plasma cell granuloma, or other diseases presenting as inflammatory reactions [1]. Further research on the morphology of IMT, inflammatory spindle cell lesions, revealed a high resemblance to common inflammatory conditions, such as nodular fasciitis and inflammatory fibroid polyps [13, 15••, 31, 44, 45]. Thus, the obstacles mentioned above have made the accurate diagnosis of IMT difficult [3].
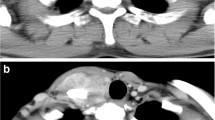
To address this problem, different examination methods have been applied to identify distinctive features of IMT that distinguish it from other similar diseases (Fig. 2). In blood testing, IMT demonstrates leukocytosis, neutrophilia, elevation of C-reactive protein and erythrocyte sedimentation rate [46, 47], microcytic anemia, thrombocytosis, and hypergammaglobulinemia [48]. However, these characteristics are not specific and can be observed in other differential diagnoses, as they are general parameters for inflammation [46]. The radiological morphologies of IMTs located in the soft tissue and bones were similar to those of benign tumors; however, peritumoral edema, parosteal soft tissue, and the invasive rim of IMT were similar to those of malignant tumors [49]. To further resolve this issue, computed tomography (CT) and magnetic resonance imaging (MRI) have been used to differentiate IMT from other neoplasms. Tine-density curves of contrast enhancement by dynamic enhanced CT scanning [50], incidence of calcification, and incidence of the burr sign [51] may help distinguish between peripheral lung cancer and IMT. However, there are various CT/MRI demonstrations of IMTs in other organs, including the mesentery and the musculoskeletal system [52,53,54,55]. As seen on CT imaging, IMT morphologies can range from infiltrating lesions to well-delineated lesions with divergent extents of inflammatory and fibrotic components in the mass [20]. MRIs can detect low signal intensity on T1 and T2 weighted images, to reveal IMT fibrosis, with a defined diffusion border [56]. Additional morphological details were discovered using microscopes to aid in the diagnosis. The most identifiable feature is the proliferation of fusiform spindle cells along one to three nucleoli within round nuclei [15] in the collagenous matrix [57], which can be associated with malignant myofibroblasts and dense polymorphic infiltration of mononuclear inflammatory cells [58].

Characteristics of IMT from different aspects
With the increasing importance of differentiating IMT from other spindle cell tumors, IMT has also been examined using immunohistochemistry (IHC), fluorescent in situ hybridization (FISH), and other technologies, such as next-generation sequencing (NGS). The discovery of ALK expression in IMT in 1999 [59] was a breakthrough in IMT diagnosis. It was later found that approximately 50% of the patients with IMT had ALK rearrangements [58]. Furthermore, IMT also presents with wild-type p53 [60], positivity for smooth muscle actin (SMA) [22], and cytokeratin AE1/3 focal positivity. Negative expression of S-100 protein, myogenin, cluster of differentiation 117, and epithelial membrane antigen was reported by IHC [31, 60]. FISH was performed to clarify the reason for ALK overexpression as a result of gene translocation [43, 61]. However, false-negative FISH results can occur for several reasons [61]. This issue can be addressed using NGS. NGS can provide evidence of kinase fusion, and identify the exact fusion partner [43]. Furthermore, NGS has also been proven to be a more reliable method for diagnosing ALK fusion-positive IMT than IHC [22].
In summary, pathological and immunohistochemical tests are considered the gold standard for IMT diagnosis [29]; however, NGS can provide genetic information for more appropriate treatments.
Genetic alterations in IMT
Following the first identification of ALK in IMT in 1999 [59], Coffin et al. discovered that approximately 50% of patients harbored ALK gene rearrangements [58]. This result was later confirmed by Casanova et al., who reviewed 60 IMT cases, and 40 patients (66.7%) were ALK+ [62]. In addition to the discovery of ALK rearrangements, ALK fusion partners have also been identified using NGS [43]. RNA binding protein 2 (RNABP2) [63, 64], insulin-like growth factor binding protein 5 (IGFBP5) [65], tropomyosin 4 (TPM4) [66], sequestosome-1 (SQSTM1) [67], and other fusion partners have been identified [22].
Although ALK rearrangements are significant in IMT studies, other gene rearrangements have also been observed. Antonescu et al. found that 85% of IMTs contain kinase fusions, two-third of which include ALK or ROS proto-oncogene 1 (ROS1)–related fusions [68]. Subsequently, Yamamoto et al. reviewed 40 IMTs diagnosed by FISH and reverse transcription polymerase chain reaction; 72.5% had ALK fusion, 5% harbored ROS1 fusions, 5% displaced neurotropic tyrosine receptor kinase 3, and the rest of the cases were quadruple negative [69]. Given the diagnostic importance of ALK expression in IMT, ALK rearrangement detection has become an approach for differentiating IMT from other conditions [3, 13, 70,71,72,73,74].
These discoveries have been used to predict the prognosis of IMT, and a few studies have suggested that ALK positivity may be an indicator of a better prognosis [11, 13, 37, 75]. In a study by Chun et al., four pediatric patients with IMT underwent incomplete surgical resection; both ALK+ patients were successfully treated with follow-up radiotherapy, while those who were ALK− died of the disease [37]. However, the link between fusion partners and the nature of the disease remains unclear [68].
Current treatment for ALK+ non-small-cell lung cancer
Non-small-cell lung cancer (NSCLC) is a general term that describes various morphologies, including adenocarcinoma and squamous cell carcinoma [76]. Moreover, 85% of lung cancers fall into this category [77]. NSCLC usually has a delayed diagnosis because patients are often unaware of the disease and symptoms resemble those of respiratory infections [78]. Thus, among diagnosed NSCLC cases, 40–65% present with distant metastases [79], with an unfavorable 5-year survival rate of merely 5–10% [80].
The treatment of NSCLC depends largely on the stage of the disease, and with delayed diagnosis, surgery is sometimes deemed impossible [76]. In 2007, an ALK rearrangement (EML4-ALK) was identified in NSCLC [81]. Later, abundant research suggested that ALK rearrangements accounted for 5% of cases [82, 83], ushering in a new era in the treatment of NSCLC.
ALK tyrosine kinase inhibitors (TKIs) were then developed as targeted therapies [84]. Crizotinib, a first-generation ALK-TKI, has gained accelerated approval from the US Food and Drug Administration (FDA) for treating either locally aggressive or metastatic NSCLC, based on two single-arm trials, which reported 50% and 61% ORRs [85]. With this success, subsequent generations of ALK-TKIs have also been developed, such as ceritinib, alectinib, brigatinib, and lorlatinib, which demonstrated superior efficacy compared to the first-generation ALK-TKI, crizotinib [86,87,88,89,90].
Due to the shared presence of ALK rearrangements in IMT and NSCLC, comparable effectiveness was anticipated based on tumor-agnostic treatment [84].
Clinical evidence of ALK-TKIs for ALK+ IMT
Crizotinib
Because IMT and NSCLC share similar ALK expression levels, the efficacy of crizotinib in IMT treatment has been an important area of research for targeted therapy. The first investigation to report satisfactory results with crizotinib in ALK+ IMT cases was conducted by Butrynski et al. in 2010 [91]. In the study, two patients with IMT were treated with crizotinib; one with ALK+ exhibited a sustained partial response, whereas the other with ALK- exhibited no observable effects [91].
Other investigations have confirmed the efficacy of crizotinib in patients with ALK+ IMT. In a time period of 4.5 years, 19 IMT cases were tracked by Schoffski et al., six ALK+ patients (50%) and one ALK− patient (14%) displayed an objective response to crizotinib [92]. Based on these results, they proposed that crizotinib may be the standard treatment for patients with locally inoperable, advanced, or metastatic ALK+ IMT [92].
In 2022, crizotinib was approved for use in adult and pediatric patients with unresectable, recurrent, or refractory ALK+ IMT based on two multicenter, single-arm, open-label trials, including 14 pediatric cases from trial NCT00939770 and seven adult patients from trial NCT01121588 [93••]. The inclusion and exclusion criteria for each study are listed in Supplementary Tables 1 (NCT00939770) and 2 (NCT01121588). In the trial NCT00939770, the ORR was assessed by an independent review committee, and among the 14 cases, 12 patients with IMT (86%) exhibited an objective response [94]. The most common adverse reactions were vomiting, nausea, diarrhea, abdominal pain, rash, cough, pyrexia, fatigue, edema, constipation, and headache [94]. For the trial NCT01121588, an objective response was observed in five (71.4%) of seven patients with IMT, and the most frequent adverse reactions were vision disorders, and edema [95•].
As crizotinib has been approved by the U.S. FDA for the treatment of ALK+ IMT, other case reports and case series have not been summarized in this review.
Ceritinib
Ceritinib, an ALK-TKI, was approved for ALK+ metastatic NSCLC with crizotinib intolerance, based on the results of a 2014 trial that included 163 patients [96], and was established as a first-line treatment for ALK+ metastatic NSCLC in 2017 based on a phase III trial [97], conducted by the U.S. FDA (recommended dosage = 750 mg orally once daily) [98].
Although it has only been approved for treating ALK+ NSCLC, ceritinib has also been reported to be effective against ALK+ IMT as an off-label treatment based on the shared characteristics of ALK expression.
Tsakiri et al. reported the case of a 33-year-old man with a TPM4-ALK fusion IMT. Two surgical resections and hyperthermic intraperitoneal chemotherapy were scheduled; however, the tumor relapsed, and crizotinib was chosen as treatment. Although there was an initial response, an activating mutation of p.G1128A in the kinase domain led to the recurrence of IMT and discontinuation of crizotinib, which initiated treatment with ceritinib. Ceritinib (750 mg/day) was prescribed, resulting in 21 months of progression-free survival (PFS) without drug-related toxicity [66]. In another case reported by Trahair et al., a 14-year-old man with an RNABP2-ALK fusion IMT was treated with crizotinib and achieved complete response (CR) as a result. Nevertheless, the patient experienced neutropenia, and the crizotinib dosage was reduced. After the fourth month of crizotinib treatment, widespread recurrence in the abdominal and thoracic spaces was observed, which was countered by increasing the dose of crizotinib to 280 mg/m2. The increased dosage stabilized the disease; however, the patient’s condition deteriorated after 1.4 years of treatment [63]. Ceritinib has been used as alternative to crizotinib with CR and PFS consistently maintained for 42 months [63].
Based on the above reports, ceritinib efficacy is similar in treatment of NSCLC and IMT and can overcome crizotinib resistance in IMT caused by previous lines of treatments.
Subsequent reports have confirmed that ceritinib can overcome resistance resulting from ALK mutations after prior ALK-TKI treatment. A 42-year-old women with proline-rich coiled-coil 2B (PRRC2B)-ALK fusion IMT was reported by Wang et al., who was initially treated with crizotinib [99]. Emergence of the ALK R1192P mutation occurred after 5 months of PFS, indicating crizotinib resistance, and the medication was changed to alectinib (600 mg, twice per day), another second-generation ALK-TKI. Alectinib was able to control the disease as partial response (PR) with 5.5 months of PFS. However, the ALK L1196M mutation was detected by NGS which had resulted in disease progression. To resolve this problem, ceritinib treatment was initiated at 450 mg/day. PR was achieved, and PFS lasted for 6 months before switching to lorlatinib [99]. Another case reported by Zhang et al. documented a 22-year-old man with ribosome binding protein 1-ALK fusion IMT who was treated with 250 mg crizotinib twice per day [100]. His condition improved, but full recovery was not achieved. Therefore, alectinib (600 mg twice daily) was prescribed and the patient’s symptoms improved. However, the tumor appeared enlarged on the CT scans. Tumor tissue was collected to identify the underlying cause of the tumor growth. A mutation in ALK L1196Q was observed, but alectinib was continued for another 4 months before substitution with ceritinib. After the initiation of ceritinib (450 mg daily) treatment, PR was observed, and PFS persisted for over 5 months at which time the study was terminated [100].
Additionally, ceritinib has also been used as a first-line treatment against ALK+ IMT. In a report by Kyi et al., a 70-year-old women with an IGFBP5-ALK fusion IMT was mis-diagnosed with uterine leiomyosarcoma and treated with pazopanib and multiple lines of chemotherapy in other institutions. After she was transferred to the organization where the authors stayed (Memorial Sloan Kettering Cancer Center, New York, NY, USA), the diagnosis was revised to IMT based on pathological features and ALK expression. After confirmation by FISH and gene fusion detection by MSK-Solid Fusion assay, ceritinib treatment was initiated with PR, and PFS observed for over 24 months. The patient remained on therapy until the study was completed [65].
The detailed characteristics and treatment outcomes of other investigations and studies are summarized in Table 1.
Alectinib
In 2017, alectinib was approved by the U.S. FDA for the treatment of ALK+ metastatic NSCLC at a recommended dosage of 600 mg twice daily, based on a randomized, multicenter, open-label trial that included 303 patients, ALEX (NCT02075840) [101, 102]. Similar to ceritinib, alectinib is expected to demonstrate equivalent efficacy against ALK+ IMT.
In a study conducted by Sunga et al., a 30-year-old woman with a SQSTM1-ALK fusion IMT was successfully treated with surgical resection. However, recurrence occurred 4 months after surgery in the mesentery and omentum, along with the development of a metastatic site in the extraperitoneal space anterior to the bladder. Given the multifocal recurrence, surgical intervention was deemed impossible; thus, the patient was treated with alectinib (600 mg twice per day) owing to a unique ALK translocation. PR was achieved with no novel metastasis, and the PFS duration was > 36 months when the study was completed. The patient experienced fatigue as the only adverse event that had no effect on her livelihood post-treatment [67].
Furthermore, alectinib has also proven its ability to target crizotinib-resistant cases, as described above in the work of Wang et al. in a 42-year-old patient with PRRC2B-ALK fusion IMT [99]. However, it is noteworthy that alectinib may contribute to drug resistance and, therefore, requires supplementation with other ALK-TKIs to achieve an acceptable outcome [99, 100].
The detailed characteristics and treatment outcomes of these and other studies are summarized in Table 2.
Brigatinib
Brigatinib, another second-generation ALK-TKI, was approved for adult ALK+ metastatic NSCLC in 2020 with a recommended dosage of 90 mg daily for the first 7 days, then increased to 180 mg once daily, based on the ALTA 1L (NCT02737501) trial that targeted advanced ALK+ NSCLC in adult patients who had not previously received an ALK-TKI [103, 104].
In a report by Xu et al., a 26-year-old man was diagnosed with an RNABP2-ALK fusion IMT, which was successfully treated with crizotinib [105]. However, after 7 months of crizotinib treatment, ascites occurred and an ALK G1269A mutation was detected by Sanger sequencing. To mitigate this, brigatinib (AP26113) was administered daily at a dose of 90 mg. The tumor was 50% smaller after three months of treatment, which qualified as PR. Remission persisted throughout study duration [105].
However, owing to the rarity of IMT and the late approval of brigatinib, larger cohort studies are required to confirm its efficacy in treating ALK+ IMT. A clinical trial (Briga-PED, NCT04925609) is in progress to study the efficacy of brigatinib in pediatric and young adult (≤ 25-year-old) patients with ALK+ anaplastic large cell lymphoma, IMT, and other solid tumors, with an estimated study completion date by December 2030 [106].
The detailed characteristics and treatment outcomes are summarized in Table 3.
Lorlatinib
The third-generation ALK-TKI, lorlatinib, was approved for ALK+ metastatic NSCLC with a recommended dosage of 100 mg once daily, based on a randomized, multicenter trial, Study B7461006 (NCT03052608) [106]. Although the median PFS was not accessible, an improvement in PFS was observed, and the ORR for the central nervous system was significantly better in the lorlatinib group (82%) than in the crizotinib group (23%) [107].
A 40-year-old man with a TPM4-ALK fusion IMT was reported by Wong et al., and lorlatinib was administered as fourth-line compassionate use therapy. The patient was initially treated with prednisolone without any clinical effects and was enrolled in a clinical trial for treatment with entrectinib, a tropomyosin receptor kinase/ROS1/ALK inhibitor, which delayed disease progression by only three months. In addition, he received a combination of chemotherapy (ifosfamide- and etoposide-based) and radiotherapy for lesions in the brain and chest. Little improvement was observed, and the disease continued to progress with newly formed metastatic sites in the adrenal gland. Thus, lorlatinib was used as the fourth-line treatment, which resulted in PR after 2 months and PFS for 6 months. During lorlatinib treatment, unilateral right-sided lung consolidation was observed, which was suspected to be due to the interaction between infection, radiotherapy, and lorlatinib, requiring treatment with antibiotics and corticosteroids. After 6 months of lorlatinib treatment, the size of the existing brain lesion increased slightly, and the lesion was treated with stereotactic radiotherapy. Brigatinib was administered 3 months after disease exacerbation, ultimately resulting in death [108].
Given the late approval of lorlatinib, major cohort studies are required to verify its efficacy against ALK+ IMT. Moreover, although all reported cases of IMT used lorlatinib in later lines of treatment [65, 99, 108, 109], lorlatinib has shown superior efficacy as a first-line treatment treating ALK+ NSCLC compared to crizotinib in the CROWN trial (NCT03052608) [110]. Therefore, further studies focusing on lorlatinib as a first-line treatment for ALK+ IMT are required.
The detailed characteristics and treatment outcomes are summarized in Table 4.
Sequential treatment based on ALK mutation
As the development of ALK-TKIs has become popular in the treatment of different diseases, sequential treatment with these targeted therapies has been tested and may be crucial for maximizing patient survival [111]. Multiple studies have tested different combinations and sequences of TKIs in patients with NSCLC [111,112,113]. Development of drug resistance after the initial response has a major influence on the sequence of targeted therapies [114]. In a study by Gainor et al., among 103 patients with ALK-rearranged lung cancer, they found that a unique spectrum of ALK mutations may arise for each ALK-TKIs applied, which may result in drug resistance [115]. Moreover, they observed that lorlatinib, a third-generation ALK-TKI, was sensitive to most emerging mutation-related resistances, whereas crizotinib, a first-generation ALK-TKI, was insensitive to most mutations [115].
Given the comparability between ALK+ NSCLC and IMT, the treatment sequence for ALK+ IMT may require a pattern similar to that used for NSCLC (Fig. 3). Nonetheless, because IMT is a rare neoplasm, data supporting this idea are limited.

Different strategies of IMT sequential treatments
Tables 1, 2, 3, and 4 describe the patterns of sequential treatment for different generations of ALK-TKIs (crizotinib, ceritinib, brigatinib, and lorlatinib). Crizotinib is usually used as a first-line treatment, as it has been approved by the FDA [93]; however, the next drug to be used in the sequence remains undetermined. Although ceritinib and alectinib are effective against crizotinib-related resistance, further cohort studies are required to confirm these results. The implicit importance of repeated biopsy and genetic sequencing may direct the next choice of ALK inhibitors as what have been studied in NSCLC.
However, sequential treatment with ALK inhibitors may not be the ultimate solution for ALK-rearranged tumors. Wang et al. reported the case of a 40-year-old man with ALK-rearranged adenocarcinoma. After receiving three different ALK-TKIs (crizotinib, belizatinib, and ceritinib), the patient developed a tumor point mutation under the selective pressure of sequential targeted therapies, resulting in death [116].
Future challenges
Although some successful treatments using different combinations of ALK-TKIs are reported above, they are mostly presented as “case reports.” Cases with statistically insignificant or negative results were likely to be excluded [117]. Furthermore, confirmation bias may also be a problem. Owing to the success of ALK-TKIs in ALK+ NSCLC, IMTs that share similar traits are believed to exhibit compatible results. Hence, there is a tendency to acquire new data in accordance with previous beliefs [118]. To resolve these issues, larger studies are needed.
The diagnosis and treatment of ALK− IMT remain uncertain. Currently, pathological features and IHC tests are viewed as standard procedures to confirm the presence of IMT [29]; however, those with ALK- expression are still difficult to identify, and can be easily confused with similar diseases, such as pseudotumors [22]. In 2022, Zhu et al. reported their experience in treating eight patients with pulmonary IMT, and proposed that vimentin and SMA may be important markers for diagnosing IMT [119]. The accuracy of this result needs to be tested in larger studies, but it still brings hope for IMT diagnosis, even with negative ALK expression. Given the absence of ALK expression, ALK-TKIs are not as useful as in ALK+ IMT treatment. Although surgeries with negative margins are still considered the best approach, treatment for inoperable ALK- IMT may remain with traditional measures for neoplasms, such as chemotherapy [62].
Summary
IMT has nature of local aggressiveness, high recurrence rate, and low metastatic potential. Surgical resection is still the main therapeutic method for localized IMT. Once IMT develops to locally advanced (unresectable) or metastatic, systemic treatment should be applied. Anthracyclin- or methotrexate-based regimens are the potential options even lacking of prospective studies.
After the approval of crizotinib targeting ALK+ IMT, ceritinib and other generations of ALK-TKIs have proven their efficacy in some cases, and follow a similar pattern as in ALK+ NSCLC, which should be further confirmed with cohort studies. Moreover, the efficacy of lorlatinib as a first-line treatment should also be tested, given its success in ALK+ NSCLC. A basket trial is suggested to verify whether the efficacy of ALK-TKIs is optimal, and whether other factors have any impact on the drugs. Sequential treatment for ALK+ IMT based on mutation-related drug resistance remains to be developed.
Many questions regarding IMT remain to be answered. Although it is a rare neoplasm with a low recurrence rate, any groundbreaking advancement could be advantageous when faced with other diseases in similar contexts.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kovach SJ, et al. Inflammatory myofibroblastic tumors. J Surg Oncol. 2006;94(5):385–91.
Häusler M, et al. Inflammatory pseudotumors of the central nervous system: report of 3 cases and a literature review. Hum Pathol. 2003;34(3):253–62.
Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol. 2008;61(4):428–37.
Patnana M, et al. Inflammatory pseudotumor: the great mimicker. AJR Am J Roentgenol. 2012;198(3):W217–27.
Fletcher, C.D.M., K.K. Unni, and F. Mertens, Pathology and Genetics of tumours of soft tissue and bone. 3rd ed. Vol. 5. 2002: World Health Organization.
Coffin CM, et al. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol. 1995;19(8):859–72.
Webb TR, et al. Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev Anticancer Ther. 2009;9(3):331–56.
Palaskar S, et al. Inflammatory myofibroblastic tumor. Contemp Clin Dent. 2011;2(4):274–7.
Karnak I, et al. Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg. 2001;36(6):908–12.
Sbaraglia M, Bellan E, Dei Tos AP. The 2020 WHO classification of soft tissue tumours: news and perspectives. Pathologica. 2021;113(2):70–84.
Lawrence B, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol. 2000;157(2):377–84.
Fletcher CDM. Diagnostic histopathology of tumors. New York, Churchill, Livingstone; 1500-1501.
Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31(4):509–20.
Bennett JA, et al. Inflammatory myofibroblastic tumor of the uterus: a clinicopathological, immunohistochemical, and molecular analysis of 13 cases highlighting their broad morphologic spectrum. Mod Pathol. 2017;30(10):1489–503.
Antonescu, C.R., et al., WHO classification of tumours: soft tissue and bone tumours. 5th ed. Vol. 3. 2020, Lyon: International Agency for Research on cancer. The newest update of WHO classification, presenting the guideline of approaching IMT.
Fu GX, et al. Inflammatory myofibroblastic tumor: a demographic, clinical and therapeutic study of 92 cases. Math Biosci Eng. 2019;16(6):6794–804.
Panagiotopoulos N, et al. Inflammatory myofibroblastic tumour of the lung: a reactive lesion or a true neoplasm? J Thorac Dis. 2015;7(5):908–11.
Al-Obaidi A, et al. Inflammatory myofibroblastic tumor of the lung: an extremely rare condition in adults. Cureus. 2019;11(12):e6432.
Choi AH, et al. Inflammatory myofibroblastic tumor of the small bowel mesentery: an unusual cause of abdominal pain and uveitis. J Gastrointest Surg. 2011;15(4):584–8.
Gros L, et al. Inflammatory myofibroblastic tumour: state of the art. Cancers (Basel). 2022;14(15)
Alaggio R, et al. Inflammatory myofibroblastic tumors in childhood: a report from the Italian cooperative group studies. Cancer. 2010;116(1):216–26.
Siemion K, et al. What do we know about inflammatory myofibroblastic tumors? – a systematic review. Advan Med Sci. 2022;67(1):129–38. A systemic review on the update of kowledge regarding IMT diagnosis.
Favini F, et al. Inflammatory myofibroblastic tumor of the conjunctiva: response to chemotherapy with low-dose methotrexate and vinorelbine. Pediatr Blood Cancer. 2010;54(3):483–5.
Kube S, et al. Inflammatory myofibroblastic tumors-a retrospective analysis of the cooperative Weichteilsarkom Studiengruppe. Pediatr Blood Cancer. 2018;65(6):e27012.
Baldi GG, et al. Activity of chemotherapy in inflammatory myofibroblastic tumor (IMT): a retrospective analysis within the Italian rare tumours network (RTR). J Clin Oncol. 2019;37(15):e22545–5.
Zhu Z, et al. Inflammatory myofibroblastic tumors in paranasal sinus and nasopharynx: a clinical retrospective study of 13 cases. Biomed Res Int. 2018;2018:7928241.
Baldi GG, et al. The activity of chemotherapy in inflammatory myofibroblastic tumors: a multicenter. Eur Retrospective Case Series Anal Oncol. 2020;25(11):e1777–84.
Soyer T, et al. Surgical treatment of childhood inflammatory myofibroblastic tumors. Eur J Pediatr Surg. 2017;27(4):319–23.
Da M, et al. Inflammatory myofibroblastic tumors in children: a clinical retrospective study on 19 cases. Front Pediatr. 2021;9:543078.
Sanders BM, et al. Inflammatory pseudotumor of the alimentary tract: clinical and surgical experience. J Pediatr Surg. 2001;36(1):169–73.
Lindberg MR. Diagnostic pathology: soft tissue Tumors. 3rd ed; 2019.
Mocellin S. Soft tissue tumors: a practical and comprehensive guide to sarcomas and benign neoplasms. Springer Cham; 2020.
Elmadi A, et al. Pseudotumeur inflammatoire pulmonaire chez un enfant. Journal de Pédiatrie et de Puériculture. 2011;24(2):69–71.
Pinilla I, et al. Tumor inflamatorio miofibroblástico pulmonar. Radiología. 2007;49(1):53–5.
Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–51.
Rohrlich P, et al. Interleukin-6 and interleukin-1 beta production in a pediatric plasma cell granuloma of the lung. Am J Surg Pathol. 1995;19(5):590–5.
Chun YS, et al. Pediatric inflammatory myofibroblastic tumor: anaplastic lymphoma kinase (ALK) expression and prognosis. Pediatr Blood Cancer. 2005;45(6):796–801.
Gómez-Román JJ, et al. Human herpesvirus-8 genes are expressed in pulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). Am J Surg Pathol. 2001;25(5):624–9.
Hammas N, et al. A rare tumor of the lung: inflammatory myofibroblastic tumor. Diagn Pathol. 2012;7(1):83.
Zhang Y, et al. Inflammatory myofibroblastic tumor in lung with osteopulmonary arthropathy. Chin Med J. 2009;122(24):3094–6.
Su LD, et al. Inflammatory myofibroblastic tumor: cytogenetic evidence supporting clonal origin. Mod Pathol. 1998;11(4):364–8.
Treissman SP, et al. Omental-mesenteric inflammatory pseudotumor. Cytogenetic demonstration of genetic changes and monoclonality in one tumor. Cancer. 1994;73(5):1433–7.
Lovly CM, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4(8):889–95.
Lee JC, et al. ALK oncoproteins in atypical inflammatory myofibroblastic tumours: novel RRBP1-ALK fusions in epithelioid inflammatory myofibroblastic sarcoma. J Pathol. 2017;241(3):316–23.
Yamamoto H, et al. p53 mutation and MDM2 amplification in inflammatory myofibroblastic tumours. Histopathology. 2003;42(5):431–9.
Elpek G. Inflammatory myofibroblastic tumor of the liver: a diagnostic challenge. J Clin Transl Hepatol. 2014;2(1):53–7.
Mamone G, et al. Imaging of primary malignant tumors in non-cirrhotic liver. Diagn Interv Imaging. 2020;101(9):519–35.
Horger M, et al. Synchronous gastrointestinal and musculoskeletal manifestations of different subtypes of inflammatory myofibroblastic tumor: CT. MRI and pathological features. Eur Radiol. 2005;15(8):1713–6.
Zeng X, et al. The clinical and radiological characteristics of inflammatory myofibroblastic tumor occurring at unusual sites. Biomed Res Int. 2018;2018:5679634.
Liu C, et al. Using CT texture analysis to differentiate between peripheral lung cancer and pulmonary inflammatory pseudotumor. BMC Med Imaging. 2020;20(1):75.
Zheng S, et al. CT signs and differential diagnosis of peripheral lung cancer and inflammatory pseudotumor: a meta-analysis. J Healthc Eng. 2022;2022:3547070.
Chen B, et al. Inflammatory myofibroblastic tumor of the urinary system on computed tomography at a high-volume institution in China. Urol Int. 2020;104(11-12):960–7.
Liang P, et al. Inflammatory myofibroblastic tumor of the bladder: computed tomographic features. Mol Clin Oncol. 2023;18(5):40.
Oguz B, et al. Imaging of childhood inflammatory myofibroblastic tumor. Pediatr Radiol. 2015;45(11):1672–81.
Porrino J, et al. Update of pediatric soft tissue tumors with review of conventional MRI appearance-part 1: tumor-like lesions, adipocytic tumors, fibroblastic and myofibroblastic tumors, and perivascular tumors. Skelet Radiol. 2022;51(3):477–504.
Surabhi VR, et al. Inflammatory myofibroblastic tumors: current update. Radiol Clin N Am. 2016;54(3):553–63.
Weinberger, S.E. Inflammatory myofibroblastic tumo (plasma cell granuloma) of the lung. 2021 Aug, 20, 2021.
Coffin CM, et al. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol. 2001;14(6):569–76.
Griffin CA, et al. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999;59(12):2776–80.
Bennett JA, et al. Inflammatory myofibroblastic tumor of the uterus: an immunohistochemical study of 23 cases. Am J Surg Pathol. 2020;44(11):1441–9.
Rao N, et al. Inflammatory myofibroblastic tumor driven by novel NUMA1-ALK fusion responds to ALK inhibition. J Natl Compr Cancer Netw. 2018;16(2):115–21.
Casanova M, et al. Inflammatory myofibroblastic tumor: the experience of the European pediatric soft tissue sarcoma study group (EpSSG). Eur J Cancer. 2020;127:123–9. A multicenter study that confirmed approximately 50% of patients with IMT harboured ALK gene rearrangement, and proposed the utility of chemotherapies in IMT treatment
Trahair T, et al. Crizotinib and surgery for long-term disease control in children and adolescents with ALK-positive inflammatory myofibroblastic tumors. JCO Precis. Oncol. 2019:3.
Ono A, et al. Drastic initial response and subsequent response to two ALK inhibitors in a patient with a highly aggressive ALK-rearranged inflammatory myofibroblastic tumor arising in the pleural cavity. Lung Cancer. 2016;99:151–4.
Kyi C, et al. Uterine mesenchymal tumors harboring ALK fusions and response to ALK-targeted therapy. Gynecol Oncol Rep. 2021;37:100852.
Tsakiri K, et al. Crizotinib failure in a TPM4-ALK-rearranged inflammatory myofibroblastic tumor with an emerging ALK kinase domain mutation. JCO Precis Oncol. 2017;1:1–7.
Sunga CGG, et al. Inflammatory myofibroblastic tumor of the mesentery with a SQSTM1::ALK fusion responding to alectinib. Cancer Rep (Hoboken). 2023;6(3):e1792.
Antonescu CR, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol. 2015;39(7):957–67.
Yamamoto H, et al. Diagnostic utility of pan-Trk immunohistochemistry for inflammatory myofibroblastic tumours. Histopathology. 2020;76(5):774–8.
Cessna MH, et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol. 2002;15(9):931–8.
Cook JR, et al. Anaplastic lymphoma kinase (ALK) expression in the inflammatory myofibroblastic tumor: a comparative immunohistochemical study. Am J Surg Pathol. 2001;25(11):1364–71.
Montgomery EA, et al. Inflammatory myofibroblastic tumors of the urinary tract: a clinicopathologic study of 46 cases, including a malignant example inflammatory fibrosarcoma and a subset associated with high-grade urothelial carcinoma. Am J Surg Pathol. 2006;30(12):1502–12.
Swain RS, et al. Inflammatory myofibroblastic tumor of the central nervous system and its relationship to inflammatory pseudotumor. Hum Pathol. 2008;39(3):410–9.
Qiu X, Montgomery E, Sun B. Inflammatory myofibroblastic tumor and low-grade myofibroblastic sarcoma: a comparative study of clinicopathologic features and further observations on the immunohistochemical profile of myofibroblasts. Hum Pathol. 2008;39(6):846–56.
Chan JK, Cheuk W, Shimizu M. Anaplastic lymphoma kinase expression in inflammatory pseudotumors. Am J Surg Pathol. 2001;25(6):761–8.
Clark SB, Alsubait S. Non small cell lung cancer. StatPearls: Tresure Island (FL); 2022.
Khajuria O, Sharma N. Epigenetic targeting for lung cancer treatment via CRISPR/Cas9 technology. Advances Cancer Biol-Metastasis. 2021;3:100012.
Ellis PM, Vandermeer R. Delays in the diagnosis of lung cancer. J Thorac Dis. 2011;3(3):183–8.
Chen H, et al. The epidemiology of lung metastases. Front Med (Lausanne). 2021;8:723396.
Siegel RL, et al. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33.
Soda M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6.
Schneider JL, Lin JJ, Shaw AT. ALK-positive lung cancer: a moving target. Nat Cancer. 2023;4(3):330–43.
Du X, et al. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac Cancer. 2018;9(4):423–30.
Childress MA. Understanding oncogenic tyrosine kinase fusion driven cancer: an investigation into inflammatory myofibroblastic tumor and the non-kinase fusion partner. 2018, Vanderbilt University: Nashville. Tennessee.
Kazandjian D, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist. 2014;19(10):e5–11.
Li J, et al. Comparative efficacy of first-line ceritinib and crizotinib in advanced or metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer: an adjusted indirect comparison with external controls. Curr Med Res Opin. 2019;35(1):105–11.
Hida T, et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): an open-label, randomised phase 3 trial. Lancet. 2017;390(10089):29–39.
Peters S, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med. 2017;377(9):829–38.
Camidge DR, et al. Brigatinib versus crizotinib in ALK-positive non-small-cell lung cancer. N Engl J Med. 2018;379(21):2027–39.
Hayashi H, et al. First-line lorlatinib versus crizotinib in ALK-positive NSCLC: Japanese subgroup analysis of CROWN. JTO Clin Res Rep. 2023;4(4):100471.
Butrynski JE, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363(18):1727–33.
Schöffski P, et al. Crizotinib in patients with advanced, inoperable inflammatory myofibroblastic tumours with and without anaplastic lymphoma kinase gene alterations (European Organisation for Research and Treatment of Cancer 90101 CREATE): a multicentre, single-drug, prospective, non-randomised phase 2 trial. Lancet Respir Med. 2018;6(6):431–41.
FDA approves crizotinib for ALK-positive inflammatory myofibroblastic tumor. 2022; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-crizotinib-alk-positive-inflammatory-myofibroblastic-tumor. The approval of crizotinib to be used in ALK+ IMT, granted by the U.S. FDA.
Children's Oncology, G., I. National Cancer, and Pfizer, Crizotinib in treating younger patients with relapsed or refractory solid tumors or anaplastic large cell lymphoma. 2018.
Pfizer, An investigational drug, Crizotinib (PF-02341066), is being studied in tumors, except non-small cell lung cancer, that are positive for anaplastic lymphoma kinase (ALK). 2023. The clinical trial that contributed to the approval of crizotinib in treating ALK+ IMT.
Khozin S, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res. 2015;21(11):2436–9.
Novartis, P. and Novartis, LDK378 versus chemotherapy in previously untreated patients with ALK rearranged non-small cell lung cancer. 2016.
FDA broadens ceritinib indication to previously untreated ALK-positive metastatic NSCLC. 2017; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-broadens-ceritinib-indication-previously-untreated-alk-positive-metastatic-nsclc.
Wang Z, et al. Durable clinical response to ALK tyrosine kinase inhibitors in epithelioid inflammatory myofibroblastic sarcoma harboring PRRC2B-ALK rearrangement: a case report. Front Oncol. 2022;12:761558.
Zhang C, et al. Efficacy and resistance of ALK inhibitors in two inflammatory myofibroblastic tumor patients with ALK fusions assessed by whole exome and RNA sequencing. Onco Targets Ther. 2020;13:10335–42.
Alectinib approved for (ALK) positive metastatic non-small cell lung cancer (NSCLC). 2017; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/alectinib-approved-alk-positive-metastatic-non-small-cell-lung-cancer-nsclc.
Hoffmann-La R. A study comparing alectinib with crizotinib in treatment-naive anaplastic lymphoma kinase-positive advanced non-small cell lung cancer participants; 2017.
FDA approves brigatinib for ALK-positive metastatic NSCLC. 2020; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-brigatinib-alk-positive-metastatic-nsclc.
Ariad, P. and Takeda, ALTA-1L study: a study of brigatinib versus crizotinib in anaplastic lymphoma kinase positive (ALK+) advanced non-small cell lung cancer (NSCLC) participants. 2020.
Xu X, et al. ALK-G1269A mutation in epithelioid inflammatory myofibroblastic sarcoma after progression on crizotinib: a case report. Oncol Lett. 2019;17(2):2370–6.
Princess Maxima Center for Pediatric, O. and Takeda, Brigatinib in pediatric and young adult patients with ALK+ ALCL, IMT or other solid tumors. 2021.
Pfizer, A study of lorlatinib versus crizotinib in first line treatment of patients with ALK-positive NSCLC. 2020.
Wong HH, et al. Lorlatinib for the treatment of inflammatory myofibroblastic tumour with TPM4-ALK fusion following failure of entrectinib. Anti-Cancer Drugs. 2020;31(10):1106–10.
Yuan C, et al. Metastatic anaplastic lymphoma kinase-1 (ALK-1)-rearranged inflammatory myofibroblastic sarcoma to the brain with leptomeningeal involvement: favorable response to serial ALK inhibitors: a case report. Am J Case Rep. 2017;18:799–804.
Shaw AT, et al. First-line lorlatinib or crizotinib in advanced ALK-positive lung cancer. N Engl J Med. 2020;383(21):2018–29.
Elsayed M, et al. Feasibility and challenges for sequential treatments in ALK-rearranged non-small-cell lung cancer. Front Oncol. 2021;11:670483.
Ito K, et al. Sequential therapy of crizotinib followed by alectinib for non-small cell lung cancer harbouring anaplastic lymphoma kinase rearrangement (WJOG9516L): a multicenter retrospective cohort study. Eur J Cancer. 2021;145:183–93.
Miriam AG, et al. ALK non-small cell lung cancer sequence of treatment: a case report. Precision Cancer Med. 2021:5.
Mologni L, et al. NPM/ALK mutants resistant to ASP3026 display variable sensitivity to alternative ALK inhibitors but succumb to the novel compound PF-06463922. Oncotarget. 2015;6(8):5720–34.
Gainor JF, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6(10):1118–33.
Wang HY, Ho CC, Shih JY. Multiple acquired resistance mutations of the ALK tyrosine kinase domain after sequential use of ALK inhibitors. J Thorac Oncol. 2017;12(5):e49–51.
Nair AS. Publication bias - importance of studies with negative results! Indian J Anaesth. 2019;63(6):505–7.
Allahverdyan AE, Galstyan A. Opinion dynamics with confirmation bias. PLoS One. 2014;9(7):e99557.
Zhu X, et al. Treatment, pathological characteristics, and prognosis of pulmonary inflammatory myofibroblastic tumor-a retrospective study of 8 cases. Front Oncol. 2022;12:840886.
Mittal A, et al. Near-complete response to low-dose ceritinib in recurrent infantile inflammatory myofibroblastic tumour. Ecancermedicalscience. 2021;15:1215.
Brivio E, Zwaan CM. ALK inhibition in two emblematic cases of pediatric inflammatory myofibroblastic tumor: efficacy and side effects. Pediatr Blood Cancer. 2019;66(5):e27645.
Michels SYF, et al. ALK(G1269A) mutation as a potential mechanism of acquired resistance to crizotinib in an ALK-rearranged inflammatory myofibroblastic tumor. NPJ Precis Oncol. 2017;1(1):4.
Mansfield AS, et al. Chromoplectic TPM3-ALK rearrangement in a patient with inflammatory myofibroblastic tumor who responded to ceritinib after progression on crizotinib. Ann Oncol. 2016;27(11):2111–7.
Takeyasu Y, et al. Impact of ALK inhibitors in patients with ALK-rearranged nonlung solid tumors. JCO Precis Oncol. 2021:5.
Saiki M, et al. Dramatic response to alectinib in inflammatory myofibroblastic tumor with anaplastic lymphoma kinase fusion gene. Jpn J Clin Oncol. 2017;47(12):1189–92.
Honda K, et al. Durable response to the ALK inhibitor alectinib in inflammatory myofibroblastic tumor of the head and neck with a novel SQSTM1-ALK fusion: a case report. Investig New Drugs. 2019;37(4):791–5.
Han Q, et al. Case report: early distant metastatic inflammatory myofibroblastic tumor harboring EML4-ALK fusion gene: study of two typical cases and review of literature. Front Med (Lausanne). 2022;9:826705.
Spafford M, Lunn D, Graham P. Malignant inflammatory myofibroblastic tumor: a rare case presentation. J Surg Case Reports. 2022;2022(9)
Fujiki T, et al. Pediatric inflammatory myofibroblastic tumor of the bladder with ALK-FN1 fusion successfully treated by alectinib. Pediatr Blood Cancer. 2023;70(4):e30172.
Funding
This work was supported by grants from Linkou Chang-Gung Memorial Hospital (CMRPG3M0971-2 and CMRPG3L0911-3 to CEW), National Science and Technology Council (Grant No. 109-2314-B-182-080-MY3, 111-2811-B-182-017 to CEW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 27 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, QA., Chen, HW., Wu, RC. et al. Update of Diagnosis and Targeted Therapy for ALK+ Inflammation Myofibroblastic Tumor. Curr. Treat. Options in Oncol. 24, 1683–1702 (2023). https://doi.org/10.1007/s11864-023-01144-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11864-023-01144-6