
Opinion statement
Antibody drug-conjugates (ADCs) have revolutionized the treatment of many types of cancer, including breast cancer. Recently, two new ADCs have been approved, trastuzumab deruxtecan and sacituzumab govitecan; both have demonstrated impressive improvements in overall survival, trastuzumab deruxtecan in all three subtypes of metastatic breast cancer and sacituzumab govitecan in luminal and triple negative metastatic breast cancer. These drugs are the results of significant progress and innovation in the construction of the three components of an ADC, the monoclonal antibody, the payload, and the linker, and of the discovery of new target antigens. ADC engineering has profoundly changed the paradigm of cancer treatment, on one side being effective on tumors considered inherently resistant to the payload class of drugs and on the other side demonstrating activity in tumors with very low target expression. Yet, it is likely that we are just at the beginning of a new era as the identification of new targets and the introduction of new ADC constructs and combinations will expand the field of ADC rapidly over the coming years.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
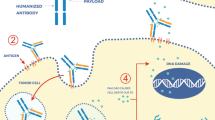
Over the past decade, the rapid evolution of hybridoma technology, along with the development of more hydrophilic and blood-stable linkers and the introduction of a wider spectrum of highly potent payloads have led to the development of a third generation of antibody–drug conjugates (ADCs) presenting several advantages over traditional therapies [1,2,3,4]. Given the complex interactions of each component with the tumor and the tumor microenvironment, the activity of the new generation of ADCs goes beyond the selective tumor delivery of high cytotoxic payloads and expands to tumors that are considered inherently resistant to the payload class of therapeutics. Indeed, the antibody backbone maintains its own functions of tumor target modulation, since the linker conjugation does not affect the antigen-binding site, and of immune effector, being able to activate antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis, and complement-dependent cytotoxicity (CDC) through its Fc region [5]. Furthermore, the growing use of cleavable linkers allows the diffusion of the lipophilic payloads across the cell membrane with the possibility of killing the neighboring cells not expressing the ADC target, the so-called bystander effect [6]. Finally, the rapid development of new conjugation technologies and the growing use of hydrophilic linkers have led to more homogenous and stable ADCs, with a higher drug to antibody ratio and a lower immunogenicity [3]. Currently, 14 ADCs have obtained approval across different countries for solid tumors and hematologic malignancies, 3 of them are approved for the treatment of early or metastatic breast cancer (BC), upon demonstration of impressive improvements in overall survival (OS) among all three BC subtypes [7,8,9,10,11,12]. However, we are just at the beginning of a new era, it is likely that the ADC landscape will be rapidly enriched with new promising compounds based on novel technologies and target antigens. Unfortunately, ADCs have also resulted in new toxicities, that can take their toll on the patients’ quality of life and requires education for their optimal management [9, 13]. Currently, a plethora of trials is evaluating new ADCs and ADC-based combinations to further improve the efficacy outcomes and to better understand unidentified resistance mechanisms, specific toxicity, and lack of predictive biomarkers. In this review, we discuss the road ahead of ADCs in BC.
Currently approved ADCs in breast cancer
Targeting HER2
Ado-trastuzumab emtansine (T-DM1), a second-generation anti-HER2 ADC, was the first ADC to be approved for the treatment of patients with HER2-positive BC [14]. T-DM1, a trastuzumab-based ADC bearing a cytotoxic microtubule inhibitor as payload, through a thioether uncleavable linker significantly improved progression-free survival (PFS) and OS over standard therapy, in patients with HER2-positive BC treated in 2nd line and beyond. In the phase III trial TH3RESA, 606 heavily pretreated (median of 4 lines) patients were randomized to T-DM1 (n = 404) or the treatment of physician choice (n = 198) [15]. The median OS was significantly longer in the experimental arm (22.7 vs 15.8 months; HR 0.68, 95%CI 0.54–0.85), despite 47% of the patients in the control arm received T-DM1 at progression. In the phase III EMILIA trial T-DM1 was compared to lapatinib plus capecitabine, the most used treatment at that time in 2nd line, in 991 patients with HER2-positive BC previously treated with trastuzumab and taxanes [8, 16]. T-DM1 outperformed standard therapy with a substantial improvement of objective response rate (ORR, 43.6% vs 30.8%), median PFS (9.6 vs 6.4 months; HR 0.65, 95%CI 0.55–0.77) and OS (29.9 vs 25.9 months; HR 0.75, 95%CI 0.64–0.88) [16]. Given the maytansinoid payload, the most frequent adverse events (AEs) with T-DM1 were thrombocytopenia (grade 3, 14%) and liver toxicity (grade 3, 5%). Relying on the positive results in metastatic setting, T-DM1 was tested as adjuvant therapy in patients with residual invasive disease after neoadjuvant chemotherapy for HER2-positive BC. The practice-changing KATHERINE trial showed that adjuvant T-DM1 halved the risk of recurrences in comparison to adjuvant trastuzumab [7].
More recently, a second anti-HER2 ADC, trastuzumab-deruxtecan (T-DXd) has revolutionized the treatment of metastatic breast cancer. T-DXd is composed of a cleavable tetrapeptide linker and a payload derivative of exatecan, a camptothecin analogues, that inhibits topoisomerase I (TOPO1), leading to DNA breaks [9]. After the striking results of the phase II single-group DESTINY-Breast01 study that led to FDA accelerated approval in December 2019, further studies confirmed the greater efficacy of T-DXd over standard therapy in patients with HER2-positive and HER2-low (immunohistochemistry 1 + or 2 + and negative results on in situ hybridization) [9]. The pivotal phase 3 DESTINY-Breast03 trial comparing T-DXd and T-DM1 in metastatic HER2-positive BC randomized 524 patients who had previously been treated with a taxane and trastuzumab. The median PFS was 4 times longer with T-DXd as compared to T-DM1 (28.8 vs 6.8 months; HR 0.33, 95% CI 0.26–0.43). At a median study follow-up of 28.4 months (range, 0.0–46.9 months), median OS was not achieved in both groups, but the difference was significant (HR 0.64, 95% CI 0.47–0.87) [10]. Furthermore, the phase III DESTINY-Breast02 study that compared T-DXd to treatment of physician choice in patients with HER2-positive metastatic BC who have received prior T-DM1, confirmed the greater efficacy of T-DXd also in later lines of treatment, with a median PFS of 17.8 months compared to 6.9 months with standard of care (HR 0.36, 95% CI 0.28–0.45).
Relying upon preclinical evidence on the T-DXd bystander effect and the preliminary data from phase I, the Phase III DESTINY-Breast04 trial evaluated the efficacy of T-DXd in pre-treated patients with HER2-low metastatic BC who had received one or two lines of chemotherapy. A total of 557 patients were randomized to receive T-DXd or physician-selected chemotherapy. In the subgroup of patients with hormone receptor (HR)-positive (n = 494), T-DXd improved the median PFS (10.1 vs 5.4 months; HR 0.51, 95%CI 0.40–0.64) and OS (23.9 vs 17.5 months; HR 0.64, 95%CI 0.48–0.86). These benefits were also seen in patients with HR-negative BC, with a median PFS of 9.9 ( vs 5.1 months; HR 0.50, 95%CI 0.40–0.63) and OS of 23.4 (vs 16.8 months; HR 0.64, 95%CI 0.49–0.84), though with the limits of an exploratory analysis of 58 patients [17]. Interestingly, T-DXd showed activity also in patients with HER2-negative BC, as demonstrated in the phase II DAISY study, in which patients with metastatic BC were divided into three cohorts based on HER2 expression to receive T-DXd. Among patients enrolled in the high HER2 expression cohort (immunohistochemistry [IHC] 3 + or IHC2 + /ISH +), the ORR was 69.1% and the median PFS was 11.1 months. Patients in the HER2-low-expression cohort (IHC1 + or IHC2 + /ISH- tumors) had an ORR of 33.3% and a median PFS of 6.7 months. In the HER2-non-expressing cohort (IHC 0), the ORR was 30.6% and the median PFS was 4.2 months [18]. Overall, T-DXd had a generally manageable safety profile, with hematologic, low-grade gastrointestinal AEs being the most common. Interstitial lung toxicity (ILD) is the most concerning AE. In a pooled analysis of nine phase I and II T-DXd monotherapy, the overall incidence of ILD was 15.4%, most of which was low grade; 77.4% was grade 1 or 2, and in rare cases was lethal (grade 5, 2.2%). Factors potentially associated with an increased risk of ILD were age < 65 years, recruitment in Japan, T-DXd dose > 6.4 mg/kg, oxygen saturation < 95%, moderate/severe renal insufficiency, presence of pulmonary comorbidities, and time since initial diagnosis > 4 years [19]. Although the ILD pathogenesis is still unclear and under investigation, it is likely that a target-independent uptake of T-DXd into alveolar macrophages could be the key mechanism [20].
Targeting TROP-2
The trophoblast cell-surface antigen 2 (Trop-2) is a transmembrane glycoprotein that acts as a calcium signal transducer. Present at low levels in normal tissue but overexpressed in epithelial tumors, including 80% of patients with BC [21], Trop-2 overexpression has been associated to tumor invasiveness and growth, as a result of the mesenchymal-to-epithelial transcription regulation [22]. Sacituzumab govitecan is a first-in-class ADC directed against Trop-2, in which the anti-Trop-2 antibody is coupled via a hydrolysable linker to SN-38, the active metabolite of the topoisomerase I inhibitor, irinotecan [23]. In the ASCENT trial, in patients with metastatic triple negative breast cancer (TNBC) who had received at least two prior systemic therapies, sacituzumab govitecan demonstrated, compared with physician-selected single-agent chemotherapy, an increase in median PFS (5.6 months versus 1.7 months; HR 0.41, 95% CI 0.32–0.52), OS (12.1 months vs 6.7 months (HR 0.48, 95% CI 0.38–0.59), and ORR (35% vs 5%) [11]. In the TROPiCS-02 trial, in patients with metastatic HR-positive BC who had received at least one endocrine-based therapy, including a prior CDK4/6 inhibitor, and 2 to 4 lines of chemotherapy for metastatic disease, sacituzumab govitecan was compared with investigator's choice. In the interim analysis, sacituzumab govitecan showed a statistically significant PFS improvement (5.5 vs 4.0 months: HR 0.66, 95% CI 0.53–0.83) and OS improvement (14.4 vs 11.2 months; HR 0.79, 95%CI 0.65–0.96, p = 0.02) over standard chemotherapy [24]. In the phase III ASCENT and TROPiCS-02 trials, the most clinically relevant grade ≥ 3 AEs were neutropenia, diarrhea and alopecia, primarily associated with SN-38 [11, 12]. While pharmacogenomic screening is not required when prescribing sacituzumab govitecan, severe neutropenia was doubled in patients homozygous for the UGT1A1*28 allele in the ASCENT trial [25].
Future ADCs in breast cancer
Over the past decade, we have witnessed an unprecedented fast-paced expansion of ADCs and a substantial number of novel ADC constructs have entered the clinical development, leveraging new antibody backbones, often targeting multiple antigens or different epitopes, new payload concepts that include also non-chemotherapeutic agents, and a broader range of suitable targets, not only in the tumor but also in the tumor microenvironment. Here below, we summarize the main properties of novel ADC constructs, that are currently being explored in BC, with a particular focus on further potential clinical development (Table 1).
New ADC constructs targeting known antigens
Several ongoing trials have been evaluating new ADC constructs directed towards known antigens, namely HER2 and Trop-2, with the aim of improving treatment efficacy and tolerability.
HER2
At least 8 different ADCs targeting HER2 have recently entered the clinical development, showing different profile of activity and toxicity.
Vic-trastuzumab duocarmazine (SYD985) is composed of a DNA-alkylating agent payload attached to trastuzumab via a cleavable linker [26, 27]. After a phase I trial that showed a remarkable clinical activity in heavily pretreated patients with HER2-positive and HER2-low BC and led to a FDA fast track designation, SYD985 compared favorably with standard of care in the phase III TULIP trial. The study met its primary endpoint by increasing median PFS (7.0 vs 4.9 months; HR 0.64, 95%CI 0.49–0.84) and showing a trend for OS benefit (HR 0.83, 95%CI 0.62–1.09). The most common treatment-emergent AE was ocular toxicity, with any grade conjunctivitis and keratitis reported in 76% of patients and grade 3 or higher keratitis and conjunctivitis (5.6 vs 0.0%) in 12.2 and 5.6% of patients. Notably, ILD was reported in 7.6% (5.2% grade 1–2) of patients treated with Vic-trastuzumab duocarmazine, including two grade 5 patients. events [27]. Different mitigation measures, such as prophylactic use of eye drops are being evaluated.
Disitamab Vedotin (RC48) is composed of a novel humanized monoclonal antibody targeting HER2, hertuzumab, characterized by a higher affinity for HER2 and a more potent ADCC [28], and a monomethyl auristatin E (MMAE) payload, conjugated by a cleavable linker [29]. In the dose escalating C001 CANCER phase I trial RC48 showed activity across patients with different level of HER2 expression. In the HER2-positive subgroup, RC48 1.5, 2.0, and 2.5 mg/kg doses yielded an ORR of 22.2%, 42.9%, and 40.0% and median PFS of 4.0, 5.7 and 6.3 months, respectively. In the HER2-low expressing subgroup, the ORR and median PFS were 39.6% and 5.7 months, respectively with RC48 2.0 mg/kg [30]. This compound has also obtained conditional approval by the National Medical Products Administration (NMPA) of China for the treatment of patients with HER2-positive locally advanced or metastatic gastric cancer and of patients with metastatic urothelial cancer.
ARX788 is an ADC based on a site-specific conjugation technology through a non-natural aminoacid, thus presenting high homogeneity. It is composed of a humanized HER2 antibody combined to amberstatin (AS269), a tubulin inhibitor. In a phase I trial enrolling only patients with HER2-positive BC, the ORR was 65.5% and the median PFS was 17.0 months [31]. The most common grade 3–4 AEs were ocular AEs (5.7%) and pneumonitis (4.3%). The ongoing phase 2 ACE-Breast-03 (NCT04829604) study is evaluating ARX788 activity and safety in patients with metastatic HER2-positive BC who are resistant to T-DM1, T-DXd, and/or tucatinib-containing regimens. However, the development of ARX788 is currently on hold.
Two other ADCs in clinical development containing a MMAE payload have also showed promising activity in patients with HER2 + BC. With the first, ALT-P7, the ORR was 77% and median PFS 6.2 months [32] in a phase I trial enrolling 27 heavily pretreated patients with HER2-positive BC progressing on at least two prior anti-HER2 therapies. The most frequent AEs included myalgia (33.3%), fatigue (25.9%), sensory neuropathy (22.2%), alopecia (22.2%), pruritus (22.2%) and neutropenia (22.2%). The second, MRG002, demonstrated significant activity across patients with HER2-low BC, with an ORR 34.1% and 37.5% [33] in IHC 1 + and 2 + , respectively. The reported AEs were neutropenia (53.6%), aspartate aminotransferase and alanine aminotransferase increase (46.4 and 32.1%), and alopecia (39.3%).
TROP-2
Datopotamab deruxtecan (Dato-DXd) is a third generation ADC targeting Trop-2 and composed of a IgG1 mAb coupled with a topoisomerase-I inhibitor (MAAA-1181a, an exatecan derivative) via a tetrapeptide-based cleavable linker, with a drug to antibody ratio of ~ 4:1 [34]. Multiple phase I-III studies are currently underway with this compound as single agent or in combination with immune checkpoint inhibitors. In the TROPION-PanTumor01 (NCT03401385) an ongoing phase 1 multicohort study, a 32% ORR and 80% disease control rate (DCR) was observed in 44 heavily pretreated patients with TNBC. Notably, a tumor shrinkage was reported also in 9 out of 13 patients previously treated with a topo-I-targeting ADC [35, 36]. The most frequent any grade AEs were stomatitis (73%), nausea (66%), vomiting (39%), fatigue (34%), and alopecia (36%) nausea (66%) and stomatitis (55%); grade 3 stomatitis was reported in 11% of cases. In the cohort of patients pretreated for RH + /HER2- metastatic breast cancer, the ORR was 29% and the DCR was 85%. The most common AEs were stomatitis (80%), nausea (56%), fatigue (46%), and alopecia (37%) [37]. Two phase III trials are currently evaluating the efficacy of Dato-DXd as compared to standard of care as first line therapy in patients with advanced TNBC (NCT05374512) and as 2nd or 3rd line therapy in patient with advanced HR-positive BC (NCT05104866). Also, a phase III trial is evaluating Dato-DXd with or without durvalumab in patients with stage I to III TNBC with residual invasive disease after neoadjuvant systemic therapy (NCT05629585).
Novel selected targets
HER3
HER3 is a transmembrane growth factor receptor, overexpressed in 50% of BCs. HER3 mediates resistance to EGFR, HER2, PI3K/AKT/mTOR directed therapies and endocrine therapy and its allosteric function is critical for HER2-amplified cancer growth [38,39,40]. HER3 overexpression has been associated to higher risk of cancer progression and worse prognosis across different tumor types, including BC [41]. Patritumab deruxtecan (U3-1402, HER3-DXd) is an ADC directed towards HER3 and composed of a human IgG1 monoclonal (mAb, patritumab) linked to a topoisomerase I inhibitor payload (MAAA-1181a, an exatecan derivative) via a tetrapeptide based cleavable linker, with a high drug to antibody ratio of about 8 [42]. The phase 1/2 study U3-1402-J101 study included patients with HER3 + (IHC 2 + or 3 +) heavily pretreated metastatic BC: 113 with luminal, 53 with TNBC and 14 with HER2-positive BC. The ORR was 30%, 23% and 43%, respectively. Interestingly, HER3-DXd activity was independent of HER3 expression [43]. Likewise, in the TOT-HER3 study, which tested the clinical and biological activity of a single dose of HER3-DXd in patients with treatment-naïve early luminal BC, the ORR was independent of baseline HER3 IHC and ERBB3 mRNA expression level [44]. Further phase II and III studies are underway in early and metastatic setting (NCT05569811, NCT04965766, NCT04699630).
LIV-1
LIV-1 is a transmembrane protein with metalloproteinase activity, belonging to a subfamily of ZIP (Zrt, Irt-like proteins) zinc transporters, with a heterogeneous expression across different normal tissues. Moderate/high LIV-1 expression has been found in about 90% of both HR-positive BC and TNBC [45] and it is associated with nodal and distant metastatic progression, consistently with its role in the epithelial-mesenchymal transition, through E-cadherin downregulation [46]. Ladiratuzumab vedotin (SGN-LIV1A) is an ADC composed of a humanized monoclonal antibody targeting LIV-1, bound through a protease-cleavable linker to a potent microtubule-disrupting agent, the monomethyl auristatin E (MMAE) payload, is a synthetic analogue of dolastatin 10, which determines G2/M phase cell arrest and ultimately apoptotic cell death.
A phase I open-label dose escalation and expansion study is actively recruiting heavily pretreated patients with metastatic LIV-1 positive TNBC or HR-positive endocrine-resistant BC (NCT01969643). An interim analysis of the 44 patients with TNBC included in the dose escalation and expansion cohorts showed ORR 32% and median PFS 11.3 weeks (95% CI 6.1–17.1 weeks), with the most common all-grade AEs being fatigue (59%), nausea (51%) and peripheral neuropathy (44%), while neutropenia (25%) and anemia (15%) were the most frequent grade 3 and 4 AEs. The established RP2D was 2.5 mg/kg every 3 weeks [47], however a weekly schedule is also being investigated to determine whether it widens the therapeutic window. In the adaptive platform trial I-SPY2, neoadjuvant therapy with 4 cycles of SNG-LIV1A 2.5 mg/kg every 3 weeks followed by 4 cycles of doxorucibicin + cyclophosphamide did not increase pathologic complete response rate as compared to standard arm [47]. Although normal apoptotic death is non-immunogenic, preclinical evidence demonstrated that SGN-LIV1 induced ER stress and release of immunogenic cell death (ICD) hallmarks such as ATP and HMGB1 in LIV-1 positive cells [48]. Relying upon these data, 2 ongoing studies are evaluating the combination of SGN-LIV1A with either pembrolizumab (NCT03310957) or atezolizumab (NCT03424005) in patients with TNBC as first or second line therapy, respectively, regardless of PD-L1 and LIV-1 expression.
Nectin-4
Nectin-4 is a transmembrane glycoprotein which is involved in cell proliferation, migration and adhesion. A moderate to strong staining has been reported in 60% of bladder and BCs, while its expression is low in normal tissues [49]. Enfortumab vedotin (EV) is a third generation ADC targeting nectin-4 and composed of a fully humanized IgG1 antibody coupled with a microtubule inhibitor (MMAE), via a protease-cleavable linker. In September 2021 it has obtained FDA approval for the treatment of patients with metastatic urothelial cancer [50]. Currently, a multicohort, phase 2 study (NCT04225117) is assessing EV activity and toxicity in patients with different locally advanced solid tumors, including patients with pretreated HR-positive BC and TNBC. Nectin-4 expression is not a study inclusion criterion and is evaluated as exploratory biomarker [51].
CEACAM5
Another emerging target is the carcinoembryonic antigen (CEA)-related cell adhesion molecule 5 (CEACAM5), a transmembrane glycoprotein, which presents high expression across multiple tumor types including gastrointestinal, lung, and breast, where it is involved in tumor invasion and metastasis, while its expression is low in normal tissues [52]. Tusamitamab ravtansine (SAR408701) is a new ADC targeting CEACAM5. It contains a humanized monoclonal antibody coupled to DM4, a maytansinoid payload by a cleavable Nsuccinimidyl 4-(2-pyridyldithio) butyrate (SPDB) linker. In the phase 1 study, 92 patients with different level of CEACAM 5 expression were treated with escalating doses of SAR-408701, including 1 patient with BC. Overall, 2 partial responses were observed (ORR 7.1%) in the low expression and 13 PRs (ORR 20.3%) in the high expression cohort. The most common treatment emergent AEs were asthenia, decreased appetite, keratopathy, and nausea, each of them observed in 25% of participants [53]. Both phase 2 and phase 3 clinical trials of SAR-408701 are currently ongoing in nonsmall cell lung cancer and other solid tumors (NCT04154956, NCT04659603, NCT04394624, NCT05071053, NCT04524689).
New ADC constructs
Antibody Technology
The technology of the antibody part of ADCs is constantly evolving with the objective of optimizing cell internalization rates, tumor permeability and the recruitment of cells from the tumor's microenvironment.
Bispecific ADCs, i.e. targeting two different receptors on the same cancer cell, or biparatopic ADCs, i.e. targeting two different epitopes on the same receptor, are intended to improve targeting specificity, internalization but also to minimize toxicity to healthy tissues. MEDI4276 combines a biparatopic antibody targeting two non-overlapping epitopes on HER2 site-specific conjugation to a tubulysin-based microtubule inhibitor payload. By virtue of an increased internalization and lysosomal trafficking and degradation, it has shown strong antitumor potential in in vivo models of T-DM1 resistance or low HER2 expression [54]. In a Phase I trial of patients with advanced or metastatic HER2-positive BC pretreated with anti-HER2 drugs, MEDI4276 resulted in 1/47 complete response (0.5 mg/kg) and 2/47 partial responses (0.6 and 0.75 mg/kg) but intolerable toxicity primarily on increased liver function tests [55]. Another concept is to use a poorly internalized but specific antigen with a rapidly internalized antigen. In this way, the rapidly internalized antigen accelerates the uptake of the poorly internalized antigen. In this sense, a bispecific ADC targeting HER2, and the prolactin receptor (PRLR) kills more efficiently than a HER2 targeting ADC. In contrast to HER2, PRLR is rapidly and constitutively internalized, and PRLR levels on the cell surface are sufficient to ensure internalization. Non-covalent cross-binding of HER2 and PRLR to the cell surface, using a bispecific ADC that binds to both receptors, significantly improves internalization and cytotoxic potential [56].
Probody drug conjugates are another innovative technology for expanding therapeutic windows. They are a new class of proteolysis-activated recombinant antibody prodrugs that exploit the activity of tumor proteases to deliver their therapeutic effects to the tumor microenvironment rather than to healthy tissues expressing the same target. Targeting the tumor within the tumor environment can reduce adverse effects, thereby increasing safety and/or dose [57]. Recently, CX-2009, a probody drug conjugate directed against CD166 (ALCAM) and conjugated to DM4, was studied in a phase I trial involving 92 patients, 39 of whom had advanced BC. Partial responses were observed in 5 BC patients, digestive and dose-dependent ocular toxicity being the most frequent AEs [58]. Currently, the development of the CX-2009 is interrupted.
Another development perspective is to replace antibody backbones with small molecules. Small molecule-drug conjugates (SMDCs) are still in the early stages of development, but they may have the advantage of being less immunogenic, easier to synthesize, and have a high potential for penetration of solid tumors as well as anatomical barriers [59].
New payloads
Immunostimulatory antibody conjugates (ISACs) use an immunostimulant such as Toll-like receptor (TLR) agonists, interferon stimulating gene agonists (STING) as a payload. These immunostimulants cannot be safely administered systemically, but by using a specific targeting antibody, they can be delivered to either the tumor cell or microenvironment cell. In a preclinical mouse model, BDC-1001 an ISAC comprising a dual TLR7/8 agonist conjugated to HER2-targeted antibodies elicited a localized immune response resulting in tumor reduction of myeloid and T cells and immunological memory [60]. A Phase 1/2 dose escalation/expansion study to evaluate BDC-1001 ± pembrolizumab in patients with HER2-expressing solid tumors whose disease has progressed on standard therapy is currently enrolling (NCT04278144). SBT6050 is also an ISAC composed of a TLR8 agonist conjugated to a monoclonal antibody directed against HER2. It has shown encouraging results in vitro and in vivo, particularly in combination with trastuzumab [61]. Other ADCs containing cytotoxic radioisotopes have shown clinical activity such as ibritumomab tiuxetan, an anti-CD20 antibody linked to iodine-131 against lymphoma [62]. However, to our knowledge, ADCs containing cytotoxic radioisotopes are not currently under development in BC.
Future ADCs combination strategies in breast cancer
ADCs and immune checkpoint inhibitors
A wealth of preclinical evidence support the combination of ADCs with immunotherapy. As first, most of ADCs induce ICD, which consists of cell surface exposure or dying cells release of damage associated molecular patterns (DAMPs). DAMPs are recognized by the immune system and eventually lead to the engulfment of tumor cells by antigen presenting cells and cross presentation to cytotoxic T cells [63,64,65]. Second, antibody backbones generally maintain their immune effector functions, being able of activating ADCC and CDC through their Fc portion [2, 5]. Finally, microtubule-inhibitor payloads (maytansinoids, dolastatins, auristatins) and topoisomerase-I inhibitor payloads are able to directly induce dendritic cell activation [66, 67]. Altogether, these evidence represent a strong rationale for combining ADCs and immunotherapy, a combination that could further augment the T cell infiltration within the tumor microenvironment [68,69,70,71].
The earliest trials combining immune checkpoint inhibitors to ADC in BC were conducted in patients with HER2-positive BC. The randomized phase II KATE2, enrolling 330 pretreated patients with HER2-positiveBC, has shown that the addition of atezolizumab to T-DM1 did not result in a statistically meaningful PFS and OS benefits neither in the intention to treat (HR 0.82, 95%CI 0.55–1.23 and HR 0.74, 95%CI 0.42–1.30, respectively) nor PD-L1-positive population (HR 0.60, 95%CI 0.32–1.11 and HR 0.55, 95%CI 0.22–1.38 respectively) [72]. Adverse events were more common in the combination arm, mainly involving thrombocytopenia (13 vs 4%), increased aspartate aminotransferase (8 vs 3%) and anemia (5 vs 0%) and led to study termination due to futility. Pharmacokinetic and immunogenicity results were as expected with T-DM1 and atezolizumab monotherapy; notably anti-therapeutic antibodies for T-DM1 and atezolizumab were reported in 1.7% and 12.1% [72]. A smaller phase Ib trial (NCT03032107) evaluating the combination of T-DM1 and pembrolizumab among 20 pretreated patients with HER2-positive BC has reported an ORR of 20% (95% CI 5.7–43.7%) and median PFS of 9.6 months (95% CI 2.8–16 months). All treatment related AEs were grade 2 and 3 (65% and 20%, respectively). PDL-1 and tumor infiltrating lymphocytes did not correlate with response in this small cohort [73]. More recently, a phase 1b study (NCT03523572) has evaluated the combination of T-DXd and nivolumab in patients with heavily pretreated HER2-expressng (HER2-positive or HER2-low) metastatic breast and urothelial cancers [74]. The ORR was 65.6% (95% CI, 46.8–81.4) and 50% (95% CI, 24.7–75.3) and median PFS was 11.6 months (95% CI, 6.9-not reached) and 7.0 months (95% CI, 2.3–10.8), respectively [75]. These results were nevertheless disappointing in comparison to the efficacy outcomes of T-DXd monotherapy in patients with HER2-positive (ORR 60.9% and PFS 16.4 months in DESTINY-Breast02). The safety profile was as expected with each study drug monotherapy. Treatment related AEs occurred grade ≥ 3 were reported in 48.9%; ILD occurred in seven patients (14.6%) of whom 6 were grade 2 and 1 grade 5 [74]. In contrast, the combination of T-DXd and durvalumab showed promising results in the Phase Ib study, BEGONIA, a multi-arm trial evaluating different treatment combinations in the first-line treatment of metastatic BC. In the HR-/HER2 low BC cohort, ORR was 57% (26/46) and median PFS was 12.6 months (95% CI, 8-not met), independent of PDL1 status. The most common AEs across all grades were nausea (73%), fatigue (46%), and vomiting (30%). Five patients (9%) experienced treatment-related interstitial lung disease or pneumonia, mostly grade 1 or 2, and one grade 5 case associated with COVID [76].
The combination of ADC and immune checkpoint inhibitors was also evaluated in TNBC. The combination of ladiratuzumab vedotin and pembrolizumab was evaluated in the phase Ib trial (NCT03310957) enrolling 51 treatment-naïve patients with advanced TNBC [77]. In this ongoing trial, the 26 patients with a follow up of at least 3 months had an ORR of 54% (95% CI 33.4–73.4) that exceeds the efficacy of pembrolizmab monotherapy in the patient with PDL-1-positive TNBC (ORR 21.4% [95% CI 13.9–31.4] [78]. The most common grade ≥ grade 3 were neutropenia (16%), diarrhea, fatigue, hypokalemia, and maculo-papular rash (8% each), and abdominal pain, increased alanine aminotransferase, and urinary tract infection (6% each). Similarly, in the TNBC cohort of the BEGONIA trial, 47 patients receiving the combination of Dato-DXd, and durvalumab had an ORR of 79% (95% CI, 61%-91%) independent of PD-L1 expression. 100% of patients with a complete or partial response remained in response at 6-month follow-up. The most common AEs of any grade were gastrointestinal (nausea in 55% and stomatitis in 51%). There were no reported cases of interstitial lung disease/pneumonitis or thrombocytopenia [79].
Other combinations are being evaluated in phase I-II trials, such as: T-DXd with pembrolizumab (NCT04042701) or durvalumab or durvalumab plus paclitaxel (NCT04538742), in metastatic HER2-positive BC; T-DXd with durvalumab plus paclitaxel in metastatic HER2-low BC (NCT04556773); Sacituzumab govitecan with pembrolizumab (NCT04468061) or with atezolizumab (NCT03424005).
ADCs and targeted therapies
The combination of targeted therapies and ADCs was primarily evaluated in HER2-positive BC. The combination T-DM1 and pertuzumab had shown synergistic activity in cell culture models and had an acceptable safety profile in a phase Ib/II study [80, 81]. In the MARIANNE trial enrolling pretreated patients with HER2-positive BC, the addition of pertuzumab to T-DM1 did not achieve a clinically significant OS benefit (51.8 vs 53.7 months) and the treatment related adverse event were similar between the two groups (48.6 vs 47.1%) [82]. The combination of T-DM1 and pertuzumab was also evaluated in the neoadjuvant setting in two trials [83, 84]. The KRISTINE study compared neoadjuvant T-DM1 plus pertuzumab with docetaxel, carboplatin, trastuzumab plus pertuzumab in 444 patients with stage II/III HER2 + BC. The combination of T-DM1 and pertuzumab resulted in shorter event-free survival than docetaxel-carboplatin-trastuzumab-pertuzumab (TCHP) therapy (HR 2.61, 95%CI 1.36–4.98) in relation to higher locoregional progression before surgery (6.7 vs. 0%) in T-DM1-pertuzumab-treated patients. In contrast, invasive disease-free survival after surgery was similar (HR 1.11, 95%CI 0.52–2.40) [83]. The pathological complete response rates with the T-DM1 arm was 44.4% compared to 55.7% with the standard regimen [83]. Similar results were achieved in the I-SPY phase II trial that randomized patients with clinical stage II/III HER2 + BC to T-DM1 plus pertuzumab, paclitaxel plus pertuzumab and trastuzumab, or paclitaxel plus trastuzumab followed by doxorubicin/cyclophosphamide, then surgery. The 3-year event free survival of the T-DM1 arm was 88% (95% CI 79–99%), compared to 92% (95% CI 84–100%) for paclitaxel plus pertuzumab and trastuzumab and 87% (95% CI 75–100%) for paclitaxel plus trastuzumab [84]. Notably, the degree of HER2 pathway signaling and phosphorylation at baseline are associated with response to the T-DM1 plus pertuzumab combination and can further identify highly responsive to this combination. In addition, the under-efficacy of T-DM1 combined with pertuzumab may be driven by intratumoral HER2 heterogeneity. Indeed, HER2 heterogeneity, defined as an area with ERBB2 amplification in > 5% but < 50% of tumor cells, or a HER2-negative area by FISH, showed complete response rates of 0% versus 55% in the non-heterogeneous subgroup with neoadjuvant therapy combining T-DM1 and pertuzumab [85].
Last, T-DM1 was combined to neratinib in the phase II NSABP Foundation Trial FB-10 [86]. Among the 27 patients with HER2 + BC enrolled in this study, 19 patients were evaluable for response and had an ORR of 63.2%. The translational research performed within this study suggested that resistance mechanisms to HER2 antibodies may be loss of HER2 receptor and high expression of p95HER2 [86].
The development of ADCs plus targeted therapy combinations in TNBC has also accelerated after the development of effective targeted therapies as well as ADCs in this setting. Poly ADP ribose polymerase (PARP) inhibitors, namely olaparib and talazoparib, approximately halved PFS in the OLYMPIAD and EMBRACA trials in patients with HER2-negative BC and BRCA germline mutations [87, 88]. The combination of rucaparib plus sacituzumab govitecan was reported in two cases of TNBC within the phase Ib SEASTAR Study [89]. The first patient had a deleterious homologous recombination repair gene (HRR) mutation and had a partial response and the second did not have an HRR mutation and had a stable disease at 24 weeks. The ongoing phase I/II trial NCT04039230 is evaluating the combination of sacituzumab govitecan plus talazoparib in patients with metastatic TNBC.
Several other phase I/II trials are ongoing and testing ADC-based combination with monoclonal antibodies, tyrosine kinase inhibitors, or endocrine therapies in the different subsets of BC.
ADCs and chemotherapies
The first clinical evidence pointing out to the role of the ADC combination with chemotherapy came from the ECHELON-1 trial showing that brentuximab vedotin improved the outcomes of patients with advanced Hodgkin’s lymphoma [90]. Thereafter, the combination of ADC and chemotherapy was evaluated in solid tumors and exhibited a promising efficacy [91,92,93]. Few trials have addressed the combinations of chemotherapy and ADCs in BC. The phase I trial, STELA, showed an ORR of 85.7% among 14 evaluable patients treated with T-DM1 plus nab-paclitaxel and lapatinib. It is noteworthy that the pharmacokinetics of T-DM1 were not affected by the combination [94]. Ongoing basket trials, including patients with metastatic TNBC, are evaluating the combination of ADCs to chemotherapy (NCT02996825, NCT03102320, NCT04538742, NCT04556773).
All clinical trials exploring ACD in combination are summarized in Table 2.
Conclusion and perspectives
Over the past years, we have witnessed a tremendous development of new ADCs in solid tumors, particularly in breast cancer, thanks to the availability of novel technologies for ADC production and the identification of new suitable targets. However, if on one hand the treatment landscape is being rapidly filled with new ADCs and novel ADC combinations, on the other hand we urgently need to understand ADC mechanism of action and resistance to better define future ADC sequential strategies in clinical practice. Mechanisms of resistance may involve the target antigens, ADC internalization, trafficking pathways, cell cycle modifications and signaling pathways, drug efflux pumps, lysosomal function, alteration of the target of the cytotoxic compound, and apoptotic dysregulation [95, 96]. Many of these resistance mechanisms are actively being evaluated in patients treated with third-generation ADCs. The translational research performed within the DAISY trial has shed lights on additional resistance mechanisms. Primary resistance may be associated to a high prevalence and spatial distribution of HER2 0 cells or to ERBB2 hemizygous deletion and acquired resistance involved reduction in HER2 expression and occurrence of SLX4 mutations [97, 98]. Similarly, resistance to sacituzumab govitecan was reported in 2 patients, of whom one lacked TROP2 expression and progressed rapidly, while a second one, who was a long-responder had initial high TROP-2 expression and focal amplification of TACSTD2/TROP2 gene. At progression, analysis of distinct metastatic subclones of the latest patient showed acquired TOP1 E418K resistance mutation and subsequent frameshift TOP1 mutation that conferred resistance to SN-38 and TACSTD2/TROP2 T256R missense mutation that determined a defective plasma membrane localization TROP-2 [99].
Undoubtedly, the more comprehensive knowledge of ADC mechanisms of resistance will allow in the coming future the selection of the most appropriate ADC sequential strategies for each patient according to the specific tumor characteristics upfront and the acquired resistance mutations. Being ADC modular, it is also likely that the development of ADC-platforms will furtherly evolve towards more personalized, patient-specific compounds in which the selection of the antibody backbone, the linker and the payload will be based on the specific tumor and tumor microenvironment activated pathways and expressing proteins.
Change history
18 April 2023
A Correction to this paper has been published: https://doi.org/10.1007/s11864-023-01092-1
References and Recommended Reading
Lu J, Jiang F, Lu A, Zhang G. Linkers Having a Crucial Role in Antibody-Drug Conjugates. Int J Mol Sci. 2016;17:561. https://doi.org/10.3390/ijms17040561.
Peters C, Brown S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep. 2015;35:e00225. https://doi.org/10.1042/BSR20150089.
Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther. 2022;7:1–25. https://doi.org/10.1038/s41392-022-00947-7.
Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–27. https://doi.org/10.1038/nri2744.
Vidarsson G, Dekkers G, Rispens T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front Immunol. 2014;5.
Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, Soma M, Okamoto H, Oitate M, Arakawa S, et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin Cancer Res. 2016;22:5097–108. https://doi.org/10.1158/1078-0432.CCR-15-2822.
von Minckwitz G, Huang C-S, Mano MS, Loibl S, Mamounas EP, Untch M, Wolmark N, Rastogi P, Schneeweiss A, Redondo A, et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N Engl J Med. 2019;380:617–28. https://doi.org/10.1056/NEJMoa1814017.
Diéras V, Miles D, Verma S, Pegram M, Welslau M, Baselga J, Krop IE, Blackwell K, Hoersch S, Xu J, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18:732–42. https://doi.org/10.1016/S1470-2045(17)30312-1.
Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, Andre F, Iwata H, Ito Y, Tsurutani J, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N Engl J Med. 2019. https://doi.org/10.1056/NEJMoa1914510.
Hurvitz SA, Hegg R, Chung W-P, Im S-A, Jacot W, Ganju V, Chiu JWY, Xu B, Hamilton E, Madhusudan S, et al. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: updated results from DESTINY-Breast03, a randomised, open-label, phase 3 trial. The Lancet. 2022. https://doi.org/10.1016/S0140-6736(22)02420-5.
Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, Brufsky A, Sardesai SD, Kalinsky K, Zelnak AB, et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N Engl J Med. 2021. https://doi.org/10.1056/NEJMoa2028485.
Rugo HS, Bardia A, Marmé F, Cortes J, Schmid P, Loirat D, Tredan O, Ciruelos E, Dalenc F, Gómez Pardo P, et al. Primary results from TROPiCS-02: A randomized phase 3 study of sacituzumab govitecan (SG) versus treatment of physician’s choice (TPC) in patients (Pts) with hormone receptor–positive/HER2-negative (HR+/HER2-) advanced breast cancer. J Clin Oncol. 2022;40, LBA1001–LBA1001. https://doi.org/10.1200/JCO.2022.40.17_suppl.LBA1001.
Wahab A, Rafae A, Mushtaq K, Masood A, Ehsan H, Khakwani M, Khan A. Ocular Toxicity of Belantamab Mafodotin, an Oncological Perspective of Management in Relapsed and Refractory Multiple Myeloma. Front Oncol. 2021; 11.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh D-Y, Diéras V, Guardino E, et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N Engl J Med. 2012;367:1783–91. https://doi.org/10.1056/NEJMoa1209124.
Krop IE, Kim S-B, González-Martín A, LoRusso PM, Ferrero J-M, Smitt M, Yu R, Leung ACF, Wildiers H. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:689–99. https://doi.org/10.1016/S1470-2045(14)70178-0.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh D-Y, Diéras V, Guardino E, et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N Engl J Med. 2012;367:1783–91. https://doi.org/10.1056/NEJMoa1209124.
Modi S, Jacot W, Yamashita T, Sohn J, Vidal M, Tokunaga E, Tsurutani J, Ueno NT, Prat A, Chae S, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N Engl J Med. 2022; 0, null. https://doi.org/10.1056/NEJMoa2203690.
Diéras V, Deluche E, Lusque A, Pistilli B, Bachelot T, Pierga J.-Y, Viret F, Levy C, Salabert L, Du FL, et al. Abstract PD8–02: Trastuzumab deruxtecan (T-DXd) for advanced breast cancer patients (ABC), regardless HER2 status: A phase II study with biomarkers analysis (DAISY). Cancer Res. 2022;82:PD8–02. https://doi.org/10.1158/1538-7445.SABCS21-PD8-02.
Powell CA, Modi S, Iwata H, Takahashi S, Smit EF, Siena S, Chang D.-Y, Macpherson E, Qin A, Singh J, et al. Pooled analysis of drug-related interstitial lung disease and/or pneumonitis in nine trastuzumab deruxtecan monotherapy studies☆. ESMO Open. 2022;7. https://doi.org/10.1016/j.esmoop.2022.100554.
Kumagai K, Aida T, Tsuchiya Y, Kishino Y, Kai K, Mori K. Interstitial pneumonitis related to trastuzumab deruxtecan, a human epidermal growth factor receptor 2-targeting Ab–drug conjugate, in monkeys. Cancer Sci. 2020;111:4636–45. https://doi.org/10.1111/cas.14686.
Vidula N, Yau C, Rugo HS. Trop2 gene expression (Trop2e) in primary breast cancer (BC): Correlations with clinical and tumor characteristics. J Clin Oncol. 2017;35:1075–1075. https://doi.org/10.1200/JCO.2017.35.15_suppl.1075.
Remšík J, Binó L, Kahounová Z, Kharaishvili G, Šimečková Š, Fedr R, Kučírková T, Lenárt S, Muresan XM, Slabáková E, et al. Trop-2 plasticity is controlled by epithelial-to-mesenchymal transition. Carcinogenesis. 2018;39:1411–8. https://doi.org/10.1093/carcin/bgy095.
Bardia A, Mayer IA, Vahdat LT, Tolaney SM, Isakoff SJ, Diamond JR, O’Shaughnessy J, Moroose RL, Santin AD, Abramson VG, et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N Engl J Med. 2019. https://doi.org/10.1056/NEJMoa1814213.
Rugo HS, Bardia A, Marmé F, Cortés J, Schmid P, Loirat D, Tredan O, Ciruelos EM, Dalenc F, Pardo PG, et al. LBA76 Overall survival (OS) results from the phase III TROPiCS-02 study of sacituzumab govitecan (SG) vs treatment of physician’s choice (TPC) in patients (pts) with HR+/HER2- metastatic breast cancer (mBC). Ann Oncol. 2022;33:S1386. https://doi.org/10.1016/j.annonc.2022.08.012.
Rugo HS, Tolaney SM, Loirat D, Punie K, Bardia A, Hurvitz SA, O’Shaughnessy J, Cortés J, Diéras V, Carey L, et al. Abstract PS11–09: Impact of UGT1A1 status on the safety profile of sacituzumab govitecan in the phase 3 ASCENT study in patients with metastatic triple-negative breast cancer. Cancer Res. 2021; 81:PS11–09. https://doi.org/10.1158/1538-7445.SABCS20-PS11-09.
Banerji U, van Herpen CML, Saura C, Thistlethwaite F, Lord S, Moreno V, Macpherson IR, Boni V, Rolfo C, de Vries EGE, et al. Trastuzumab duocarmazine in locally advanced and metastatic solid tumours and HER2-expressing breast cancer: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2019;20:1124–35. https://doi.org/10.1016/S1470-2045(19)30328-6.
Manich CS, O’Shaughnessy J, Aftimos PG, Tweel, E. van den, Oesterholt M, Escrivá-de-Romaní SI, Tueux NQ, Tan TJ, Lim JS, Ladoire S, et al. LBA15 Primary outcome of the phase III SYD985.002/TULIP trial comparing [vic-]trastuzumab duocarmazine to physician’s choice treatment in patients with pre-treated HER2-positive locally advanced or metastatic breast cancer. Ann Oncol. 2021;32:S1288. https://doi.org/10.1016/j.annonc.2021.08.2088.
Li H, Yu C, Jiang J, Huang C, Yao X, Xu Q, Yu F, Lou L, Fang J. An anti-HER2 antibody conjugated with monomethyl auristatin E is highly effective in HER2-positive human gastric cancer. Cancer Biol Ther. 2016;17:346–54. https://doi.org/10.1080/15384047.2016.1139248.
Li L, Xu M.-Z, Wang L, Jiang J, Dong L.-H, Chen F, Dong K, Song H.-F. Conjugating MMAE to a novel anti-HER2 antibody for selective targeted delivery. Eur Rev Med Pharmacol Sci. 2020;24:12929–12937. https://doi.org/10.26355/eurrev_202012_24196.
Wang J, Liu Y, Zhang Q, Feng J, Fang J, Chen X, Han Y, Li Q, Zhang P, Yuan P, et al. RC48-ADC, a HER2-targeting antibody-drug conjugate, in patients with HER2-positive and HER2-low expressing advanced or metastatic breast cancer: A pooled analysis of two studies. J Clin Oncol. 2021;39:1022–1022. https://doi.org/10.1200/JCO.2021.39.15_suppl.1022.
Zhang J, Ji D, Shen W, Xiao Q, Gu Y, O’Shaughnessy J, Hu X. Phase I Trial of a Novel Anti-HER2 Antibody-drug Conjugate, ARX788, for the Treatment of HER2-positive Metastatic Breast Cancer. Clin Cancer Res, clincanres.0456.2022–2–11. 2022;09:49:17.170. https://doi.org/10.1158/1078-0432.CCR-22-0456.
Park YH, Ahn HK, Kim J-Y, Ahn JS, Im Y-H, Kim S-H, Lee S, Chung H-S, Park SJ. First-in-human phase I study of ALT-P7, a HER2-targeting antibody-drug conjugate in patients with HER2-positive advanced breast cancer. J Clin Oncol. 2020;38:3551–3551. https://doi.org/10.1200/JCO.2020.38.15_suppl.3551.
Jiang Z, Sun T, Wang X, Liu Q, Yan M, Tong Z, Geng C, Tang J, Yin Y, Yu G, et al. A multiple center, open-label, single-arm, phase II clinical trial of MRG002, an HER2-targeted antibody-drug conjugate, in patients with HER2-low expressing advanced or metastatic breast cancer. J Clin Oncol. 2022;40:1102–1102. https://doi.org/10.1200/JCO.2022.40.16_suppl.1102.
Okajima D, Yasuda S, Maejima T, Karibe T, Sakurai K, Aida T, Toki T, Yamaguchi J, Kitamura M, Kamei R, et al. Datopotamab Deruxtecan, a Novel TROP2-directed Antibody–drug Conjugate, Demonstrates Potent Antitumor Activity by Efficient Drug Delivery to Tumor Cells. Mol Cancer Ther. 2021;20:2329–40. https://doi.org/10.1158/1535-7163.MCT-21-0206.
Krop I, Juric D, Shimizu T, Tolcher A, Spira A, Mukohara T, Lisberg AE, Kogawa T, Papadopoulos KP, Hamilton E, et al. Abstract GS1–05: Datopotamab deruxtecan in advanced/metastatic HER2- breast cancer: Results from the phase 1 TROPION-PanTumor01 study. Cancer Res. 2022;82:GS1–05. https://doi.org/10.1158/1538-7445.SABCS21-GS1-05.
Aditya Bardia, Ian Krop, Funda Meric-Bernstam, and Takahiro Kogawa P6–10–03 Datopotamab Deruxtecan (Dato-DXd) in Advanced Triple-Negative Breast Cancer (TNBC): Updated Results From the Phase 1 TROPION-PanTumor01 Study.
Funda Meric-Bernstam, Ian Krop, Dejan Juric, and Aditya Bardia PD13–08 Phase 1 TROPION-PanTumor01 Study Evaluating Datopotamab Deruxtecan (Dato-DXd) in Unresectable or Metastatic Hormone Receptor–Positive/HER2–Negative Breast Cancer (BC).
Campbell MR, Amin D, Moasser MM. HER3 Comes of Age: New Insights into Its Functions and Role in Signaling, Tumor Biology, and Cancer Therapy. Clin Cancer Res. 2010;16:1373–83. https://doi.org/10.1158/1078-0432.CCR-09-1218.
Campbell MR, Ruiz-Saenz A, Peterson E, Agnew C, Ayaz P, Garfinkle S, Littlefield P, Steri V, Oeffinger J, Sampang M, et al. Targetable HER3 functions driving tumorigenic signaling in HER2-amplified cancers. Cell Rep. 2022;38. https://doi.org/10.1016/j.celrep.2021.110291.
Gala K, Chandarlapaty S. Molecular Pathways: HER3 Targeted Therapy. Clin Cancer Res. 2014;20:1410–6. https://doi.org/10.1158/1078-0432.CCR-13-1549.
Ocana A, Vera-Badillo F, Seruga B, Templeton A, Pandiella A, Amir E. HER3 Overexpression and Survival in Solid Tumors: A Meta-analysis. JNCI J Natl Cancer Inst. 2013;105:266–73. https://doi.org/10.1093/jnci/djs501.
Hashimoto Y, Koyama K, Kamai Y, Hirotani K, Ogitani Y, Zembutsu A, Abe M, Kaneda Y, Maeda N, Shiose Y, et al. A Novel HER3-Targeting Antibody-Drug Conjugate, U3–1402, Exhibits Potent Therapeutic Efficacy through the Delivery of Cytotoxic Payload by Efficient Internalization. Clin Cancer Res. 2019;25:7151–61. https://doi.org/10.1158/1078-0432.CCR-19-1745.
Krop IE, Masuda N, Mukohara T, Takahashi S, Nakayama T, Inoue K, Iwata H, Toyama T, Yamamoto Y, Hansra DM, et al. Results from the phase 1/2 study of patritumab deruxtecan, a HER3-directed antibody-drug conjugate (ADC), in patients with HER3-expressing metastatic breast cancer (MBC). J Clin Oncol. 2022;40:1002–1002. https://doi.org/10.1200/JCO.2022.40.16_suppl.1002.
Oliveira M, Andujar JMC, Vila MM, Ortega PT, Saez OM, Bofill FJS, Jurado JC, Barrera AML, de Dios MAA, Losada MJV, et al. 202TiP SOLTI-1805 TOT-HER3 trial: A window-of-opportunity trial of patritumab deruxtecan (HER3-DXd) in patients with treatment-naïve, early breast cancer. Ann Oncol. 2022;33:S628. https://doi.org/10.1016/j.annonc.2022.07.237.
Taylor KM, Morgan HE, Smart K, Zahari NM, Pumford S, Ellis IO, Robertson JFR, Nicholson RI. The Emerging Role of the LIV-1 Subfamily of Zinc Transporters in Breast Cancer. Mol Med. 2007;13:396–406. https://doi.org/10.2119/2007-00040.Taylor.
Taylor KM, Morgan HE, Johnson A, Hadley LJ, Nicholson RI. Structure–function analysis of LIV-1, the breast cancer-associated protein that belongs to a new subfamily of zinc transporters. Biochem J. 2003;375:51–9. https://doi.org/10.1042/bj20030478.
Beckwith HC, Medgyesy DC, Abraham J, Nanda R, Tkaczuk KHR, Krop IE, Pusztai L, Modi S, Mita MM, Specht JM, et al. SGNLVA-001: A phase I open-label dose escalation and expansion study of SGN-LIV1A administered weekly in breast cancer. J Clin Oncol. 2020;38, TPS1104–TPS1104. https://doi.org/10.1200/JCO.2020.38.15_suppl.TPS1104.
Cao AT, Higgins S, Stevens N, Gardai SJ, Sussman D. Abstract 2742: Additional mechanisms of action of ladiratuzumab vedotin contribute to increased immune cell activation within the tumor. Cancer Res. 2018;78:2742–2742. https://doi.org/10.1158/1538-7445.AM2018-2742.
Liu Y, Han X, Li L, Zhang Y, Huang X, Li G, Xu C, Yin M, Zhou P, Shi F, et al. Role of Nectin-4 protein in cancer (Review). Int J Oncol. 2021;59:1–14. https://doi.org/10.3892/ijo.2021.5273.
Research, C. for D.E. and (2021). FDA grants regular approval to enfortumab vedotin-ejfv for locally advanced or metastatic urothelial cancer. FDA.
Bruce JY, Pusztai L, Braiteh FS, Gorla SR, Wu C, Baranda J. EV-202: A phase II study of enfortumab vedotin in patients with select previously treated locally advanced or metastatic solid tumors. J Clin Oncol. 2020;38, TPS3647–TPS3647. https://doi.org/10.1200/JCO.2020.38.15_suppl.TPS3647.
Zhang X, Han X, Zuo P, Zhang X, Xu H. CEACAM5 stimulates the progression of non-small-cell lung cancer by promoting cell proliferation and migration. J Int Med Res. 2020;48:0300060520959478. https://doi.org/10.1177/0300060520959478.
Gazzah A, Bedard PL, Hierro C, Kang Y-K, Razak AA, Ryu M-H, Demers B, Fagniez N, Henry C, Hospitel M, et al. Safety, pharmacokinetics, and antitumor activity of the anti-CEACAM5-DM4 antibody–drug conjugate tusamitamab ravtansine (SAR408701) in patients with advanced solid tumors: first-in-human dose-escalation study. Ann Oncol. 2022;33:416–25. https://doi.org/10.1016/j.annonc.2021.12.012.
Li JY, Perry SR, Muniz-Medina V, Wang X, Wetzel LK, Rebelatto MC, Hinrichs MJM, Bezabeh BZ, Fleming RL, Dimasi N, et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell. 2016;29:117–29. https://doi.org/10.1016/j.ccell.2015.12.008.
Pegram MD, Hamilton EP, Tan AR, Storniolo AM, Balic K, Rosenbaum AI, Liang M, He P, Marshall S, Scheuber A, et al. First-in-Human, Phase 1 Dose-Escalation Study of Biparatopic Anti-HER2 Antibody-Drug Conjugate MEDI4276 in Patients with HER2+ Advanced Breast or Gastric Cancer. Mol Cancer Ther. 2021. https://doi.org/10.1158/1535-7163.MCT-20-0014.
Andreev J, Thambi N, Perez Bay AE, Delfino F, Martin J, Kelly MP, Kirshner JR, Rafique A, Kunz A, Nittoli T, et al. Bispecific Antibodies and Antibody-Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol Cancer Ther. 2017;16:681–93. https://doi.org/10.1158/1535-7163.MCT-16-0658.
Autio KA, Boni V, Humphrey RW, Naing A. Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance On-Target Effects with Reduced Off-Tumor Toxicity for Use in Immuno-Oncology. Clin Cancer Res. 2020;26:984–9. https://doi.org/10.1158/1078-0432.CCR-19-1457.
Boni V, Burris HA III, Liu JF, Spira AI, Arkenau H-T, Fidler MJ, Rosen LS, Sweis RF, Uboha NV, Sanborn RE, et al. CX-2009, a CD166-directed probody drug conjugate (PDC): Results from the first-in-human study in patients (Pts) with advanced cancer including breast cancer (BC). J Clin Oncol. 2020;38:526–526. https://doi.org/10.1200/JCO.2020.38.15_suppl.526.
Zhuang C, Guan X, Ma H, Cong H, Zhang W, Miao Z. Small molecule-drug conjugates: A novel strategy for cancer-targeted treatment. Eur J Med Chem. 2019;163:883–95. https://doi.org/10.1016/j.ejmech.2018.12.035.
Ackerman SE, Pearson CI, Gregorio JD, Gonzalez JC, Kenkel JA, Hartmann FJ, Luo A, Ho PY, LeBlanc H, Blum LK, et al. Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nat Cancer. 2021;2:18–33. https://doi.org/10.1038/s43018-020-00136-x.
Metz H, Childs M, Brevik J, Winship D, Brender T, Comeau M, Moyes K, Chang J, Adamo J, Setter B, et al. SBT6050, a HER2-directed TLR8 therapeutic, as a systemically administered, tumor-targeted human myeloid cell agonist. J Clin Oncol. 2020;38:3110–3110. https://doi.org/10.1200/JCO.2020.38.15_suppl.3110.
Leahy MF, Seymour JF, Hicks RJ, Turner JH. Multicenter Phase II Clinical Study of Iodine-131–Rituximab Radioimmunotherapy in Relapsed or Refractory Indolent Non-Hodgkin’s Lymphoma. J Clin Oncol. 2006;24:4418–25. https://doi.org/10.1200/JCO.2005.05.3470.
Muller P, Rios-Doria J, Harper J, Cao A. Combining ADCs with immunooncology agents. In: Damelin M, editor. Innovations for next-generation antibody-drug conjugates. Totowa (NJ): Humana Press; 2018. p. 11–44.
Müller P, Martin K, Theurich S, Schreiner J, Savic S, Terszowski G, Lardinois D, Heinzelmann-Schwarz VA, Schlaak M, Kvasnicka H-M, et al. Microtubule-depolymerizing agents used in antibody-drug conjugates induce antitumor immunity by stimulation of dendritic cells. Cancer Immunol Res. 2014;2:741–55. https://doi.org/10.1158/2326-6066.CIR-13-0198.
Cao A, Heiser R, Law C-L, Gardai SJ. Abstract 4914: Auristatin-based antibody drug conjugates activate multiple ER stress response pathways resulting in immunogenic cell death and amplified T-cell responses. Cancer Res. 2016;76:4914. https://doi.org/10.1158/1538-7445.AM2016-4914.
Müller P, Martin K, Theurich S, Schreiner J, Savic S, Terszowski G, Lardinois D, Heinzelmann-Schwarz VA, Schlaak M, Kvasnicka H-M, et al. Microtubule-Depolymerizing Agents Used in Antibody-Drug Conjugates Induce Antitumor Immunity by Stimulation of Dendritic Cells. Cancer Immunol Res. 2014;2:741–55. https://doi.org/10.1158/2326-6066.CIR-13-0198.
Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-Targeting Antibody-Drug Conjugate, Trastuzumab Deruxtecan (DS-8201a), Enhances Antitumor Immunity in a Mouse Model. Mol Cancer Ther. 2018;17:1494–503. https://doi.org/10.1158/1535-7163.MCT-17-0749.
Rios-Doria J, Harper J, Rothstein R, Wetzel L, Chesebrough J, Marrero A, Chen C, Strout P, Mulgrew K, McGlinchey K, et al. Antibody-Drug Conjugates Bearing Pyrrolobenzodiazepine or Tubulysin Payloads Are Immunomodulatory and Synergize with Multiple Immunotherapies. Cancer Res. 2017;77:2686–98. https://doi.org/10.1158/0008-5472.CAN-16-2854.
Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-Targeting Antibody-Drug Conjugate, Trastuzumab Deruxtecan (DS-8201a), Enhances Antitumor Immunity in a Mouse Model. Mol Cancer Ther. 2018;17:1494–503. https://doi.org/10.1158/1535-7163.MCT-17-0749.
Hoimes CJ, Flaig TW, Milowsky MI, Friedlander TW, Bilen MA, Gupta S, Srinivas S, Merchan JR, McKay RR, Petrylak DP, et al. Enfortumab Vedotin Plus Pembrolizumab in Previously Untreated Advanced Urothelial Cancer. J Clin Oncol. 2022;JCO.22.01643. https://doi.org/10.1200/JCO.22.01643.
Rosenberg JE, Milowsky M, Ramamurthy C, Mar N, McKay RR, Friedlander T, Ferrario C, Bracarda S, George S, Moon H, et al. LBA73 Study EV-103 Cohort K: Antitumor activity of enfortumab vedotin (EV) monotherapy or in combination with pembrolizumab (P) in previously untreated cisplatin-ineligible patients (pts) with locally advanced or metastatic urothelial cancer (la/mUC). Ann Oncol. 2022;33:S1441. https://doi.org/10.1016/j.annonc.2022.08.079.
Emens LA, Esteva FJ, Beresford M, Saura C, De Laurentiis M, Kim S-B, Im S-A, Wang Y, Salgado R, Mani A, et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): a phase 2, multicentre, randomised, double-blind trial. Lancet Oncol. 2020;21:1283–95. https://doi.org/10.1016/S1470-2045(20)30465-4.
Waks AG, Keenan TE, Li T, Tayob N, Wulf GM, Richardson ET, Attaya V, Anderson L, Mittendorf EA, Overmoyer B, et al.. Phase Ib study of pembrolizumab in combination with trastuzumab emtansine for metastatic HER2-positive breast cancer. J Immunother Cancer. 2022;10:e005119. https://doi.org/10.1136/jitc-2022-005119.
Hamilton EP, Shapiro CL, Boni V, Jimenez MM, Conte GD, Cortés J, Agrawal L, Arkenau H-T, Tan AR, Debruyne PR, et al. 162O Primary analysis from DS8201-A-U105: A 2-part, open label, phase Ib trial assessing trastuzumab deruxtecan (T-DXd) with nivolumab (nivo) in patients (pts) with HER2-expressing advanced breast cancer. Ann Oncol. 2022;33:S196. https://doi.org/10.1016/j.annonc.2022.03.181.
Hamilton EP, Kaklamani V, Falkson C, Vidal GA, Ward PJ, Patre M, Chui SY, Rotmensch J, Gupta K, Molinero L, et al. Abstract PD1–05: Atezolizumab in combination with trastuzumab emtansine or with trastuzumab and pertuzumab in patients with HER2-positive breast cancer and atezolizumab with doxorubicin and cyclophosphamide in HER2-negative breast cancer: Safety and biomarker outcomes from a multi-cohort Phase Ib study. Cancer Res. 2020;80, PD1-PD1–05. https://doi.org/10.1158/1538-7445.SABCS19-PD1-05.
Peter Schmid, Piotr Wysocki, Yeon H. Park, and Zbigniew Nowecki PD11–08 Trastuzumab deruxtecan (T-DXd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic hormone receptor-negative (HR−), HER2low breast cancer: updated results from BEGONIA, a phase 1b/2 study.
Han H. (Heather), Diab S, Alemany C, Basho R, Brown-Glaberman U, Meisel J, Pluard T, Cortes J, Dillon P, Ettl J, et al. Abstract PD1–06: Open label phase 1b/2 study of ladiratuzumab vedotin in combination with pembrolizumab for first-line treatment of patients with unresectable locally-advanced or metastatic triple-negative breast cancer. Cancer Res. 2020;80:PD1–06. https://doi.org/10.1158/1538-7445.SABCS19-PD1-06.
Adams S, Loi S, Toppmeyer D, Cescon DW, Laurentiis MD, Nanda R, Winer EP, Mukai H, Tamura K, Armstrong A, et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase II KEYNOTE-086 study. Ann Oncol. 2019;30:405–11. https://doi.org/10.1093/annonc/mdy518.
Peter Schmid, Piotr Wysocki, Cynthia Ma, and Zbigniew Nowecki PD11–09 Datopotamab deruxtecan (Dato-DXd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic triple-negative breast cancer (a/mTNBC): updated results from BEGONIA, a phase 1b/2 study.
Krop IE, Modi S, LoRusso PM, Pegram M, Guardino E, Althaus B, Lu D, Strasak A, Elias A. Phase 1b/2a study of trastuzumab emtansine (T-DM1), paclitaxel, and pertuzumab in HER2-positive metastatic breast cancer. Breast Cancer Res. 2016;18:34. https://doi.org/10.1186/s13058-016-0691-7.
Phillips GDL, Fields CT, Li G, Dowbenko D, Schaefer G, Miller K, Andre F, Burris HA, Albain KS, Harbeck N, et al. Dual targeting of HER2-positive cancer with trastuzumab emtansine and pertuzumab: critical role for neuregulin blockade in antitumor response to combination therapy. Clin. Cancer Res. Off J Am Assoc Cancer Res. 2014;20:456–68. https://doi.org/10.1158/1078-0432.CCR-13-0358.
Perez EA, Barrios C, Eiermann W, Toi M, Im Y-H, Conte P, Martin M, Pienkowski T, Pivot XB, Burris HA, et al. Trastuzumab emtansine with or without pertuzumab versus trastuzumab with taxane for human epidermal growth factor receptor 2-positive advanced breast cancer: Final results from MARIANNE. Cancer. 2019;125:3974–84. https://doi.org/10.1002/cncr.32392.
Hurvitz SA, Martin M, Jung KH, Huang C-S, Harbeck N, Valero V, Stroyakovskiy D, Wildiers H, Campone M, Boileau J-F, et al. Neoadjuvant Trastuzumab Emtansine and Pertuzumab in Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer: Three-Year Outcomes From the Phase III KRISTINE Study. J Clin Oncol Off J Am Soc Clin Oncol. 2019;37:2206–16. https://doi.org/10.1200/JCO.19.00882.
Clark AS, Yau C, Wolf DM, Petricoin EF, van ‘t Veer, L.J., Yee, D., Moulder, S.L., Wallace, A.M., Chien, A.J., Isaacs, C., et al. Neoadjuvant T-DM1/pertuzumab and paclitaxel/trastuzumab/pertuzumab for HER2+ breast cancer in the adaptively randomized I-SPY2 trial. Nat Commun. 2021;12:6428. https://doi.org/10.1038/s41467-021-26019-y.
Filho OM, Viale G, Stein S, Trippa L, Yardley DA, Mayer IA, Abramson VG, Arteaga CL, Spring LM, Waks AG, et al. Impact of HER2 Heterogeneity on Treatment Response of Early-Stage HER2-Positive Breast Cancer: Phase II Neoadjuvant Clinical Trial of T-DM1 Combined with Pertuzumab. Cancer Discov. 2021;11:2474–87. https://doi.org/10.1158/2159-8290.CD-20-1557.
Abraham J, Montero AJ, Jankowitz RC, Salkeni MA, Beumer JH, Kiesel BF, Piette F, Adamson LM, Nagy RJ, Lanman RB, et al. Safety and Efficacy of T-DM1 Plus Neratinib in Patients With Metastatic HER2-Positive Breast Cancer: NSABP Foundation Trial FB-10. J Clin Oncol Off J Am Soc Clin Oncol. 2019;37:2601–9. https://doi.org/10.1200/JCO.19.00858.
Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017;377:523–33. https://doi.org/10.1056/NEJMoa1706450.
Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee K-H, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N Engl J Med. 2018;379:753–63. https://doi.org/10.1056/NEJMoa1802905.
Yap TA, Hamilton E, Bauer T, Dumbrava EE, Jeselsohn R, Enke A, Hurley S, Lin KK, Habeck J, Giordano H, et al. Phase Ib SEASTAR Study: Combining Rucaparib and Sacituzumab Govitecan in Patients With Cancer With or Without Mutations in Homologous Recombination Repair Genes. JCO Precis Oncol. 2022;6:e2100456. https://doi.org/10.1200/PO.21.00456.
Connors JM, Jurczak W, Straus DJ, Ansell SM, Kim WS, Gallamini A, Younes A, Alekseev S, Illés Á, Picardi M, et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N Engl J Med. 2018;378:331–44. https://doi.org/10.1056/NEJMoa1708984.
Lassman AB, Pugh SL, Wang TJC, Aldape K, Gan HK, Preusser M, Vogelbaum MA, Sulman EP, Won M, Zhang P, et al. Depatuxizumab-mafodotin in EGFR-amplified newly diagnosed glioblastoma: a phase III randomized clinical trial. Neuro-Oncol. 2022; noac173. https://doi.org/10.1093/neuonc/noac173.
Van Den Bent M, Eoli M, Sepulveda JM, Smits M, Walenkamp A, Frenel J-S, Franceschi E, Clement PM, Chinot O, De Vos F, et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncol. 2020;22:684–93. https://doi.org/10.1093/neuonc/noz222.
Padovan M, Eoli M, Pellerino A, Rizzato S, Caserta C, Simonelli M, Michiara M, Caccese M, Anghileri E, Cerretti G, et al. Depatuxizumab Mafodotin (Depatux-M) Plus Temozolomide in Recurrent Glioblastoma Patients: Real-World Experience from a Multicenter Study of Italian Association of Neuro-Oncology (AINO). Cancers. 2021;13:2773. https://doi.org/10.3390/cancers13112773.
Patel TA, Ensor J, Rodriguez AA, Belcheva A, Darcourt JG, Niravath PA, Kuhn JG, Kaklamani VG, Li X, Boone T, et al. Phase ib study of trastuzumab emtansine (TDM1) in combination with lapatinib and nab-paclitaxel in metastatic HER2-neu overexpressed breast cancer patients: Stela results. J Clin Oncol. 2018;36:1035–1035. https://doi.org/10.1200/JCO.2018.36.15_suppl.1035.
Rassy E, Rached L, Pistilli B. Antibody drug conjugates targeting HER2: Clinical development in metastatic breast cancer. The Breast. 2022;66:217–26. https://doi.org/10.1016/j.breast.2022.10.016.
García-Alonso S, Ocaña A, Pandiella A. Resistance to Antibody-Drug Conjugates. Cancer Res. 2018;78:2159–65. https://doi.org/10.1158/0008-5472.CAN-17-3671.
Mosele MF, Lusque A, Dieras V, Deluche E, Ducoulombier A, Pistilli B, Bachelot T, Viret F, Levy C, Signolle N, et al. LBA1 Unraveling the mechanism of action and resistance to trastuzumab deruxtecan (T-DXd): Biomarker analyses from patients from DAISY trial. Ann Oncol. 2022;33:S123. https://doi.org/10.1016/j.annonc.2022.03.277.
Mosele MF, Lusque A, Dieras VC, Deluche E, Ducoulombier A, Pistilli B, Bachelot T, Viret F, Levy C, Pradat YC, et al. LBA72 Unraveling the mechanism of action and resistance to trastuzumab deruxtecan (T-DXd): Biomarker analyses from patients from DAISY trial. Ann Oncol. 2022;33:S1440. https://doi.org/10.1016/j.annonc.2022.08.078.
Coates JT, Sun S, Leshchiner I, Thimmiah N, Martin EE, McLoughlin D, Danysh BP, Slowik K, Jacobs RA, Rhrissorrakrai K, et al. Parallel Genomic Alterations of Antigen and Payload Targets Mediate Polyclonal Acquired Clinical Resistance to Sacituzumab Govitecan in Triple-Negative Breast Cancer. Cancer Discov. 2021;11:2436–45. https://doi.org/10.1158/2159-8290.CD-21-0702.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Pistilli, El-Rassy and Grinda has nothing to disclose.
Human and Animal Rights
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Grinda, T., Rassy, E. & Pistilli, B. Antibody–Drug Conjugate Revolution in Breast Cancer: The Road Ahead. Curr. Treat. Options in Oncol. 24, 442–465 (2023). https://doi.org/10.1007/s11864-023-01072-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11864-023-01072-5