
Opinion statement
Acute lymphoblastic leukemia (ALL) is the most frequent type of pediatric cancer with a peak incidence at 2–5 years of age. ALL frequently begins in utero with the emergence of clinically silent, preleukemic cells. Underlying leukemia-predisposing germline and acquired somatic mutations define distinct ALL subtypes that vary dramatically in treatment outcomes. In addition to genetic predisposition, a second hit, which usually occurs postnatally, is required for development of overt leukemia in most ALL subtypes. An untrained, dysregulated immune response, possibly due to an abnormal response to infection, may be an important co-factor triggering the onset of leukemia. Furthermore, the involvement of natural killer (NK) cells and T helper (Th) cells in controlling the preleukemic cells has been discussed. Identifying the cell of origin of the preleukemia-initiating event might give additional insights into potential options for prevention. Modulation of the immune system to achieve prolonged immunosurveillance of the preleukemic clone that eventually dies out in later years might present a future directive. Herein, we review the concepts of prenatal origin as well as potential preventive approaches to pediatric B cell precursor (BCP) ALL.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Leukemia is a life-threatening disease caused by uncontrolled proliferation of blood and blood precursor cells. Depending on the cell type of clonal expansion, it can be segregated into different subtypes that have quite distinct incidences, pathogenesis, treatment options, and outcomes [1]. Approximately one-third of all cancers diagnosed below the age of 18 are leukemia, with about 74% of these being acute lymphoblastic leukemia (ALL, 4.3/100,000 children <15 years) in Germany [2]. ALL peaks between the age of 2 and 5 years and has a good outcome in most cases. However, about 10% of children present with poor prognosis, based on subtype and risk factors like advanced age [3, 4]. Herein, we review and discuss recent studies and concepts of prenatal pathogenesis of leukemia, with a special focus on infections or microbiota influencing anti-leukemic immunosurveillance.
Genetic susceptibility to childhood B cell precursor ALL (BCP-ALL)
Childhood B-ALL arises through a complex interplay between inherited genetic background and acquired somatic alterations [4]. The genetic background of patients includes alterations in cancer-predisposing genes, single nucleotide polymorphisms (SNPs), and cancer predisposition syndromes that confer susceptibility to leukemia [5]. In addition to the underlying inherited genetics, prenatal chromosomal aberrations, such as aneuploidy and interchromosomal translocations [6], give rise to preleukemic cells. Further oncogenic events in these clinically silent cell clones, most likely triggered by environmental factors in early childhood, are required to ultimately lead to overt leukemia [4].
Leukemia-predisposing germline mutations
Several germline mutations which confer susceptibility to leukemia development have been described [7••]. Most of the affected genes are also targets of somatic alterations in ALL.
Cancer-predisposing gene mutations
The transcription factor ETV6 is an important regulator of hematopoiesis [8]. Families with ETV6 germline mutations often present with thrombocytopenia and susceptibility to hematologic malignancies, among which ALL is the most frequent [9, 10]. Most ALL cases with germline ETV6 mutations belong to the hyperdiploid subtype [9]. ETV6 germline mutations include missense, frameshift, nonsense mutations, deletions, and insertions, leading to a loss of function of ETV6 [5, 11]. A cluster of mutations occurs in the DNA-binding E26 transformation-specific (ETS) domain of ETV6, leading to dominant negative effects and transcriptional repression [5, 7, 12].
PAX5, located at 9p13, encodes for the B cell lineage transcription factor PAX5 which is important for B-lymphoid lineage maturation [13]. So far, only few families with germline PAX5 mutations have been described, presenting with incomplete penetrance [14, 15]. Reported missense mutations of PAX5 occur at amino acid positions G183 (c547G>A, p.Gly183Ser) or R38 (c113G>A, p.Arg38His), both resulting in decreased PAX5-mediated transcriptional repression [14,15,16]. Carriers of germline PAX5 mutations are susceptible to acquiring ALL, but the presence of the mutation does not seem to be sufficient for development of overt leukemia. A second mutational hit is required, e.g., inactivation of the wild-type PAX5 allele by deletion of 9p, formation of a 9q isochromosome, or dicentric 9q chromosome [14,15,16].
IKZF1 encodes for the hematopoietic zinc-finger (ZF) transcription factor IKAROS. Germline IKZF1 mutations have been described in families with common variable immunodeficiency (CVID) [17] and in cases of familial and sporadic ALL [18]. Mutations include missense, nonsense, and frameshift variants and are located mostly outside the ZF motifs [5]. IKZF1 mutations within its DNA-binding domain affect transcriptional activation of its target genes, whereas truncating mutations may have an impact on dimerization [18]. The majority of identified IKZF1 germline variants are not restricted to specific functional domains and were shown to impact subcellular localization, adhesion, and anti-leukemic drug efficacy [18].
Cancer predisposition syndromes
Li-Fraumeni syndrome is an autosomal dominant disorder [19], usually caused by TP53 germline mutations, that presents with high susceptibility to cancers like breast cancer, brain tumors, and ALL, predominantly low hypodiploid ALL [7, 20, 21]. Low hypodiploidy is characterized by 32–39 chromosomes and is present in approximately 1% of childhood ALL cases [7, 22]. Occurrence of germline TP53 mutations is associated with older age at diagnosis and poor outcome [23]. TP53 encodes the tumor suppressor protein p53 and is one of the most frequently mutated genes in cancer. The majority of TP53 mutations occur in its DNA-binding or nuclear export domains [7, 20].
Children with Down syndrome or Noonan syndrome are also at higher risk of developing acute leukemia, primarily acute myeloid leukemia (AML) [24, 25]. Down syndrome is characterized by trisomy of chromosome 21, which may affect leukemia development [24]. About 1% of children with Down syndrome will develop ALL or AML [24]. Noonan syndrome is an autosomal dominant disorder that belongs to the family of RASopathies and presents with symptoms including facial dysmorphologies, growth retardation, heart defects, and skin manifestations [25]. Rarely, germline mutations in PTPN11, encoding the phosphatase SHP2, and in SOS1, encoding the guanine nucleotide exchange factor SOS1, have been observed in patients with Noonan syndrome, who subsequently developed ALL [25].
Leukemia-predisposing SNPs
In addition to the rare but highly penetrant germline mutations and cancer predisposition syndromes described here, genome-wide association studies (GWASs) have identified further germline variations that are frequent but show low penetrance. These are mostly SNPs, which, cumulatively, may confer a higher risk for ALL development. Although these risk alleles individually produce a modest effect and may be of limited clinical significance, in aggregate they can give rise to as much as a ninefold increase in leukemia risk for subjects with risk alleles in multiple genes compared to subjects with no risk alleles [26]. Genes involved include IKZF1, CDKN2A, PIP4K2A, LHPP, ELK3, GATA3, ARID5B, CEBPE, MYC, ERG, and TP63 [7, 27,28,29,30], with the SNPs being located in the vicinity of these genes and influencing gene expression. Some of these SNPs are associated with distinct ALL subtypes or genetic ancestry. Examples are an intronic SNP in GATA3 (dbSNP: rs3824662) that is associated with Philadelphia chromosome (Ph)-like ALL and poor outcome [31] and a risk locus in TP63 (dbSNP: rs17505102) that is associated with ETV6-RUNX1+ ALL [28].
Prenatal somatic mutations in childhood BCP-ALL
Fusion genes generated by interchromosomal translocations are recurrent genetic alterations in pediatric BCP-ALL [32]. Several studies indicate that these translocations frequently arise in utero, giving rise to preleukemic cells. The first indications that ALL has prenatal origins were reports of concordant BCP-ALL in monozygotic twins [33,34,35,36,37]. In these cases, preleukemic cell clones arising in one twin spread to the other twin via the monochorionic placenta, as confirmed via the identification of shared genetic lesions, immunoglobulin (Ig), or T cell receptor (TCR) rearrangements in the leukemic cells of both twins [38]. Identification of genomic breakpoints in neonatal blood spots (Guthrie cards) or cord blood further corroborates the prenatal origin of preleukemic lesions [39,40,41,42,43,44,45]. Altogether, in utero development has been shown for several BCP-ALL subtypes, including high hyperdiploid ALL, ETV6-RUNX1, BCR-ABL1, TCF3-PBX1, and KMT2A rearrangements (as reviewed in [3•]).
Hyperdiploidy
With up to 30% of cases, high hyperdiploidy is the most common genetic subtype in childhood BCP-ALL, characterized by the gain of chromosomes (>50 chromosomes) [22, 46]. While other tri- or tetrasomies have been reported, chromosomal gains typically include chromosomes X, 4, 6, 10, 14, 17, 18, and 21 [47]. The hyperdiploid genotype is likely generated by a single abnormal mitosis leading to simultaneous gain of chromosomes [48]. Leukemia susceptibility in high hyperdiploid ALL is driven by gene dosage effects [47, 49, 50] that impact chromatin architecture, e.g., by weakening topologically associating domain (TAD) boundaries [51•].
ETV6-RUNX1
The most common chromosomal translocation of pediatric ALL, accounting for about 20% of cases, is t(12;21)(p13;q22) [52]. This translocation leads to the fusion of two transcription factors involved in normal hematopoiesis, ETV6 and RUNX1. Although the ETV6-RUNX1 translocation has been detected in a large number of healthy neonates (1-5%), leukemia incidence among carriers is much lower (0.2–1%) [3, 43]. The fusion gene has weak oncogenic potential that manifests itself in a low concordance rate of about 10% in monozygotic twins [38]. ETV6-RUNX1 acts as an oncogenic transcription factor and leads to a specific preleukemic phenotype characterized by a partial block of B cell differentiation and aberrant co-expression of myeloid markers [53]. Recurrent postnatal, leukemia-inducing mutations include ETV6 deletions (≈70% of cases), RUNX1 gain (23%), and extra der(21)t(12;21) (10%) [54].
BCR-ABL1
BCP-ALL with t(9;22)(q34;q11), also referred to as Ph+ ALL, is present in ≈2% of pediatric ALL, but is significantly more common in adults [22, 55]. The majority of pediatric patients with BCR-ABL1 fusion genes harbor the p190 BCR-ABL1 subtype [56]. This chromosomal translocation leads to the formation of the BCR-ABL1 oncogene, encoding for a tyrosine kinase. While high hyperdiploidy and ETV6-RUNX1 are associated with a favorable treatment outcome [57], BCR-ABL1 confers a poorer outcome [58]. A common cooperating oncogenic lesion in BCR-ABL1+ ALL is the deletion of the B-lineage transcription factor IKZF1 (in >80% of cases) [59].
TCF3-PBX1
The t(1;19)(q23;p13) translocation encoding the TCF3-PBX1 fusion gene is present in ≈4% of childhood ALL cases [55, 60]. TCF3-PBX1+ ALL is associated with a good prognosis but frequent central nervous system (CNS) relapse [61]. Like ETV6-RUNX1, the TCF3-PBX1 fusion protein has low oncogenic potential and requires secondary, cooperating mutations for overt leukemia to develop [62].
KMT2A rearrangements
KMT2A (or MLL: mixed-lineage leukemia) rearrangements of 11q23 with other chromosomes are typically found in infant BCP-ALL (children <1 year) [34, 63]. KMT2A-rearranged leukemia often present with CNS involvement and are associated with poor treatment outcome [63]. Fusion genes involving KMT2A are likely sufficient for leukemia development, as suggested by a high concordance rate in monozygotic twins [38] and rare detection of secondary, cooperative mutations [64].
The preleukemic cell of origin in childhood BCP-ALL
Investigation of early BCP-ALL development is invaluable in identifying new targeted treatment options and approaches to preventing leukemic transformation. BCP-ALL originates in a single cell, with subsequent clonal expansion of premalignant cells that may acquire more malignant traits. Due to the covert early etiology of the disease and the complexity of prenatal leukemic development, identifying and characterizing the BCP-ALL cell of origin remains challenging. Several studies have tried to narrow down the cell in which the first preleukemia-initiating event preferentially occurs (Table 1). Although B cell blasts of different BCP-ALL subtypes often correspond to distinct developmental stages of normal B cell hematopoiesis, the first oncogenic event might occur at a different developmental stage. A subsequent differentiation arrest at a later cell stage or dedifferentiation of preleukemic cells place them downstream or upstream of their cell of origin. Dedifferentiation of preleukemic cells was for instance proposed for TCF3-PBX1 translocations [65, 66].
An increasing number of studies provide evidence for the in utero origin of common BCP-ALL chromosome aberrations (as reviewed in [3•]). This suggests that preleukemic cells may arise in an early progenitor cell during fetal development, e.g., in the bone marrow or fetal liver.
Ig and TCR gene rearrangements in BCP-ALL blast cells have been used as markers to investigate the clonal origin of leukemic cells. These markers have been identified in a large number of BCP-ALL patients (>90%) [79, 80]. However, given that recombination activating gene (RAG)-driven rearrangements take place continually during clonal evolution of BCP-ALL [81], Ig/TCR gene status may not reflect the preleukemia-initiating cell. Shared clonal Ig and TCR gene rearrangements in twins with concordant BCP-ALL might give better insight, as shown in studies of twins with concordant ETV6-RUNX1+ ALL that identified pro B cells or RAG1/2− stem cells as potential cells of origin [72, 82].
Lineage switching upon relapse has been described in BCP-ALL, mostly for KMT2A-rearranged or BCR-ABL1+ ALLs [83, 84]. In the latter case, a subgroup of patients carrying the fusion gene presented with chronic myeloid leukemia (CML)-like disease, pointing to a multipotent progenitor cell [75]. Likewise, ambiguous expression of lymphoid and myeloid lineage markers, as observed in many BCP-ALL patients [85], might point to an early progenitor cell with lympho-myeloid potential. Recently, lympho-myeloid precursor origin has been suggested for ETV6-RUNX1+ ALL, due to aberrant co-expression of myeloid markers observed in an ETV6-RUNX1+ human-induced pluripotent stem cell (hiPSC) model [53].
Interleukin-7 receptor α (IL-7Rα) mutations in BCP-ALL development
IL-7Rα (encoded by the IL7R gene) is an important factor for lymphoid development. Together with the interleukin-2 receptor gamma (IL-2Rγ), it forms the IL-7 receptor (IL-7R) [86]. Recently, several groups have described activating mutations in IL7R as being involved in the initiation and development of BCP-ALL [87,88,89]. Inactivating mutations of IL7R are associated with severe combined immunodeficiency (SCID). SCID patients lack T cells. In mice, SCID manifests in both B and T cell absence [90]. In contrast, activating IL7R mutations have been observed in ALL, especially in Ph-like and PAX5 P80R subtypes. Using a conditional knock-in mouse model, Almeida et al. showed that physiological levels of mutant IL-7Rα were sufficient to generate preleukemic B cell precursors and to initiate leukemia resembling the human Ph-like and PAX5 P80R ALL subtypes [87]. Thomas et al. generated a genetically engineered mouse model with B cell-intrinsic expression of mutant IL7R that presented with development of BCP-ALL [88]. In an elegant study, Geron et al. transduced human CD34+ hematopoietic cells with mutant IL-7Rα. After transplantation into NOD/LtSz-scid IL-2Rγnull mice, a preleukemic state with retained self-renewal capacity developed [89••]. In all three studies, additional mutations acquired during leukemia development were observed. These led to upregulation of IL-7R signaling (via the JAK/STAT5 or the PI3K/mTOR pathway), upregulation of oncogenes (e.g., MYC, BCL2), and downregulation of tumor suppressors (including IKZF1) [87,88,89]. Additionally, CDKN2A was silenced [89••], and recurrent somatic KRAS mutations which cooperate with mutant IL7R were observed [87, 88].
Taking all this together, a clear leukemia-initiating effect of constitutively active IL-7Rα could be observed in different mouse models as well as in human hematopoietic progenitors, with similarities to Ph-like and/or PAX5 P80R BCP-ALL subtypes. However, further studies are needed to fully understand how the interplay with other mutations leads to the development of overt leukemia.
External factors for the development of leukemia
For the development of overt leukemia, a multifactorial etiology is proposed where a combination of genetic susceptibility and external factors induces leukemic transformation. External factors such as radiation, smoking, and infections, amongst others, can play a role in utero or postnatally. Radiation and smoking have already been reviewed elsewhere [91, 92], associating high doses of ionizing radiation with ALL development and paternal smoking preconception and during pregnancy with an elevated risk for ALL.
Infection
Infection has been suggested to be a likely trigger for ALL development. As postulated in the two-hit or delayed infection hypothesis by Mel Greaves [4], overt BCP-ALL requires an initiating mutation in utero (first hit) as well as a second postnatal mutation (second hit) [4]. In this model, the second hit is triggered by a dysregulated immune response towards common infections. Depending on the timing, infections were suggested to either have a protective (early) or detrimental (late) effect [4]. Pre- and postnatal infections have therefore been investigated as potential risk factors for triggering ALL.
In utero cytomegalovirus (CMV) infection was found to be more prevalent in children who later developed leukemia compared to healthy controls [93]. CMV is a member of the herpesvirus family and is known to cause hearing loss and/or growth retardation in the developing child [94, 95]. CMV can cross the placenta and thus infect the child in utero. Maternal reactivation or reinfection can also play a role, probably due to influences on immune crosstalk between mother and fetus [95]. Interestingly, CMV degrades the neonatal Fc receptor (FcRn) which is responsible for the transfer of IgG through the placenta. Thereby, CMV interferes with the immunity that is conferred from mother to child [96].
Other herpesviruses, like Epstein-Barr virus (EBV) and varicella zoster virus (VZV), may also play a role in the development of childhood BCP-ALL. A significantly increased risk for ALL development could be detected for maternal EBV infection [97]; however, significant correlation of EBV infection and ALL development could not be shown in a follow-up study [98]. A higher childhood leukemia risk was also observed when the mothers were infected with varicella or rubella during pregnancy [99•].
A link between maternal influenza infection and an increased risk of leukemia development was found in several studies as early as the 1970s [100, 101]. In a current meta-analysis by He et al., maternal influenza infection was significantly associated with higher risk of developing ALL [99•].
In terms of postnatal infections, a possible connection to influenza was observed in two space-time clusters [102, 103]. In the UK, increases in ALL incidence were observed in the years 1976 and 1990, following winter influenza epidemics [102]. In Milan, Italy, seven newly diagnosed ALL cases occurred within 4 weeks. All of these children were seropositive for the AH1N1 swine flu virus, whose outbreak occurred 3 to 6 months prior to leukemia diagnosis [103]. A possible explanation could be that influenza infection led to a strong dysregulated inflammatory response in the predisposed children, resulting in leukemic transformation of preleukemic cells. However, it is unlikely that influenza plays a unique role in the development of childhood ALL. It seems to be more important that predisposed children show an abnormal immune response to common infections. Other space-time clusters with a high incidence of childhood leukemia cases, e.g., the one in Fallon, USA (1997–2003), were not linked to influenza epidemics [104].
In light of the current severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) pandemic, it will be interesting to see the influences of the virus and the infection-prevention measures (e.g., lockdown, increased hygiene) on the development of ALL cases in the future. The critical second hit for the development of overt leukemia could be infection with SARS-CoV-2, leading to an aberrant immune reaction [105]. However, it is also possible that the measures taken to prevent the spread of the disease, like closing nurseries and schools, may provide a means for reducing ALL cases. A similar scenario occurred during the SARS-CoV-1 outbreak in Hong Kong in 2003 [106]. Here, a marked decline in common infectious diseases, like chickenpox, as well as a decline in ALL incidence were observed in the same year [106]. However, the same measures could also lead to an increase of ALL cases in the next years, as children born during the current pandemic have fewer social contacts and are less exposed to common infections during the critical time period where the immune system has to be trained in order to avoid ALL development, according to the delayed infection hypothesis [4]. Thus, the next years will show the influence of the pandemic and of the lockdown measures on the development of ALL cases and may give initial insights into how to prevent the development of leukemia in the future.
Taken together, infections may promote leukemia at two different stages: (1) in utero due to the oncogenic potential of a virus or due to immune responses of a not yet fully developed fetal immune system or (2) after birth due to a dysregulated immune response.
The in utero and early-in-life development of the immune system has long-term consequences for efficient control of the preleukemic clone
The double-hit scenario of secondary events, such as infections, triggering leukemic progression is supported by epidemiological data [4]. Additionally, animal studies showed that genetically predisposed mice developed leukemia only in a pathogen-containing environment [4, 107]. The exact mechanism remains unclear, but the lack of efficient immune cell training by microbial colonization and pathogens in utero and early in life has been suggested to be crucial for the development of ALL [5, 108].
Infections shape the immune system and thereby indirectly affect the preleukemic clone. In this context, among other innate immune cells, natural killer (NK) cells have been shown to be modulated by trained immunity. Infectious stimuli induced epigenetic reprogramming towards enhanced killing capacity of NK cells [109]. Furthermore, NK cells combine an antiviral and anti-tumor killing capacity and are thus promising candidates for modulation of preleukemic cells. NK cells were shown to gain memory functions after viral infections or after stimulation with pro-inflammatory cytokines [110, 111]. Interestingly, NK cell cytotoxicity against a leukemic cell line was also significantly enhanced after CMV infection, mediated by the NKG2C(+) NK cell subpopulation [112]. In contrast, single cell RNA sequencing of ETV6-RUNX1+ ALL cases revealed significant inhibition of NK cell activity in the tumor microenvironment [113••]. This suggests that the dual role of NK cells can be explained by taking the different NK cell subtypes into account. Recent genetic studies have provided proof that a certain genetic constitution of NK cells controls BCP-ALL [114]. Killer immunoglobulin receptors (KIR) on NK cells interact with human lymphocyte antigen (HLA) class I molecules. The inhibitory NK cell receptor KIR2DL1—a high-affinity ligand for HLA-C2—is significantly increased in BCP-ALL patients. In another study, five NK cell-related factors (KIR2DL5A, NKp46, FasL, granzyme B and PI-9) were positively associated with detection of minimal residual disease at the end of induction therapy [115]. How the inhibitory NK cell receptors’ control of the preleukemic clone is determined by genetic factors or modulated by infections should be part of future studies.
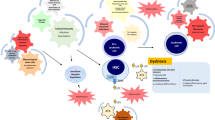
The importance of early and even prenatal immune training with microbial antigens is underlined by epidemiological data that refer to the hygiene hypothesis [116•]. Interestingly, the same epidemiological factors leading to a clean and hygienic environment, such as late introduction into day care, order and number of siblings, and early antibiotic treatment [117], have been associated with a higher incidence of autoimmune diseases and allergies as well as with a higher incidence of BCP-ALL [4, 118]. These are all diseases that are predominantly mediated by T helper (Th) cells, suggesting a certain role of Th responses in the control of the preleukemic clone. Atopic disease and childhood ALL are negatively correlated. A Th2 phenotype might be protective against ALL development [119], while pro-inflammatory Th1 cells with high interferon gamma (IFNγ) levels have been shown to migrate towards BCP-ALL cells and favor their proliferation via upregulation of CD38 and IFNγ-induced protein 10 (IP-10) production [120••] mediated by activation-induced cytidine deaminase (AID) upregulation [121]. But, what driving force skews the immune response towards one or the other direction, given the fact that early immune cell priming is lacking in both scenarios? Miedema and colleagues attributed this to a particular genetic predisposition, since they found two SNPs in the TLR6 gene associated with BCP-ALL, leading to an altered Th1/Th2 balance upon microbial exposure [122]. The immunosurveillance mechanisms are summarized in Figure 1.

Immunosurveillance of the preleukemic clone. Germline and acquired somatic mutations predispose towards leukemia and define distinct ALL subtypes. Via a dysregulated immune response, infections can trigger transformation of the preleukemic clone into overt leukemia. This process is under constant immunosurveillance. T helper (Th) 1 cells can favor leukemia development via upregulation of CD38 and interferon gamma-induced protein 10 (IP-10), mediated by activation-induced cytidine deaminase (AID). Th2 cells on the other hand can inhibit leukemia development. Natural killer (NK) cells play an important role in cancer surveillance. They can favor development of overt leukemia by up- or downregulation of different factors, such as HLA-C2, KIR2DL1, KIR2DL5A, PI-9, NKp46, FasL, and granzyme B. Apoptosis of the preleukemic clone can be mediated by NKG2C(+) NK cells. SNP, single nucleotide polymorphism; IL7R, interleukin-7 receptor alpha.
Treatment options and outlook
Diagnosis of a severe underlying germline ALL predisposition with a high penetrance, such as TP53 mutation/Li-Fraumeni syndrome, offers the opportunity to monitor the patient closely for early cancer occurrence and clearly improves overall survival [123]. By contrast, diagnosis of a more common predisposition, like an in utero occurring somatic ETV6-RUNX1 mutation, does not provide such a benefit, as the mutation confers only a minor risk of a child developing ALL, a disease for which current chemotherapy treatment protocols achieve 80–90% overall survival without early detection being critical for its outcome. However, successful treatment comes at the price of significant acute and late toxicities, which account for a large proportion of deaths. Acute adverse effects during chemotherapy for childhood cancer can affect all organs, and two-thirds of childhood cancer survivors live with long-term effects of the toxic treatment, which can be severe (e.g., cognitive impairment, osteonecrosis, secondary cancers, infertility, depression) [124]. Therefore, there is an urgent need to employ strategies aimed at preventing children from getting cancer in the first place. In the absence of means to directly target and eliminate the preleukemic cells, general training of the immune system early in life (e.g., in child day-care, through contact with pets) is recommended and promising. More targeted approaches currently include (1) training of the innate immune response via specific vaccination or (2) modulation of the microbiome (by, e.g., probiotics) to achieve a healthier, more complex state [5]. Targeting Th1/Th2 lineage determination to prevent the clonal expansion of the preleukemic clone may be a promising alternative treatment approach to follow up on. However, differentiation programs are complex and intricately cross-linked. Side effects of pharmacologic modulation in genetically predisposed children can be severe and may outweigh potential benefits.
We believe that further studies employing larger cohorts of predisposed children are clearly necessary to understand the complex interplay of genetic predisposition and environmental factors and to finally enable us to develop targeted preventive approaches.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Inaba H, Pui C-H. Advances in the diagnosis and treatment of pediatric acute lymphoblastic leukemia. J Clin Med. 2021;10(9):1926. https://doi.org/10.3390/jcm10091926.
Erdmann F, Kaatsch P, Grabow D, Spix C. German Childhood Cancer Registry – Annual Report 2019 (1980-2018). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz, 2020.
• Hein D, Borkhardt A, Fischer U. Insights into the prenatal origin of childhood acute lymphoblastic leukemia. Cancer Metastasis Rev. 2020;39(1):161–71. https://doi.org/10.1007/s10555-019-09841-1. This reference is of importance because it gives a comprehensive overview of the most common prenatal lesions involved in childhood BCP-ALL, with a special focus on ETV6-RUNX1+ ALL
Greaves M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer. 2018;18(8):471–84. https://doi.org/10.1038/s41568-018-0015-6.
Hauer J, Fischer U, Borkhardt A. Toward prevention of childhood ALL by early-life immune training. Blood. 2021;138(16):1412–28. https://doi.org/10.1182/blood.2020009895.
Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017;35(9):975–83. https://doi.org/10.1200/jco.2016.70.7836.
•• Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer. 2021;21(2):122–37. https://doi.org/10.1038/s41568-020-00315-z. This reference is of outstanding importance because it reviews the effect of germline mutations on the development of hematologic malignancies, such as ALL, AML and myelodysplastic syndrome (MDS)
Hock H, Shimamura A. ETV6 in hematopoiesis and leukemia predisposition. Semin Hematol. 2017;54(2):98–104. https://doi.org/10.1053/j.seminhematol.2017.04.005.
Moriyama T, Metzger ML, Wu G, Nishii R, Qian M, Devidas M, Yang W, Cheng C, Cao X, Quinn E, Raimondi S, Gastier-Foster JM, Raetz E, Larsen E, Martin PL, Bowman WP, Winick N, Komada Y, Wang S, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. The Lancet Oncology. 2015;16(16):1659–66. https://doi.org/10.1016/s1470-2045(15)00369-1.
Noetzli L, Lo RW, Lee-Sherick AB, Callaghan M, Noris P, Savoia A, Rajpurkar M, Jones K, Gowan K, Balduini CL, Pecci A, Gnan C, de Rocco D, Doubek M, Li L, Lu L, Leung R, Landolt-Marticorena C, Hunger S, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nature Genetics. 2015;47(5):535–8. https://doi.org/10.1038/ng.3253.
Jarviaho T, Bang B, Zachariadis V, Taylan F, Moilanen J, Mottonen M, et al. Predisposition to childhood acute lymphoblastic leukemia caused by a constitutional translocation disrupting ETV6. Blood Adv. 2019;3(18):2722–31. https://doi.org/10.1182/bloodadvances.2018028795.
Nishii R, Baskin-Doerfler R, Yang W, Oak N, Zhao X, Yang W, Hoshitsuki K, Bloom M, Verbist K, Burns M, Li Z, Lin TN, Qian M, Moriyama T, Gastier-Foster JM, Rabin KR, Raetz E, Mullighan C, Pui CH, et al. Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood. 2021;137(3):364–73. https://doi.org/10.1182/blood.2020006164.
Medvedovic J, Ebert A, Tagoh H, Busslinger M. Pax5: a master regulator of B cell development and leukemogenesis. Adv Immunol. 2011;111:179-206. https://doi.org/10.1016/B978-0-12-385991-4.00005-2.
Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, Wechsler J, Yang J, Hayes J, Klein RJ, Zhang J, Wei L, Wu G, Rusch M, Nagahawatte P, Ma J, Chen SC, Song G, Cheng J, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45(10):1226–31. https://doi.org/10.1038/ng.2754.
Auer F, Ruschendorf F, Gombert M, Husemann P, Ginzel S, Izraeli S, et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia. 2014;28(5):1136–8. https://doi.org/10.1038/leu.2013.363.
Duployez N, Jamrog LA, Fregona V, Hamelle C, Fenwarth L, Lejeune S, Helevaut N, Geffroy S, Caillault A, Marceau-Renaut A, Poulain S, Roche-Lestienne C, Largeaud L, Prade N, Dufrechou S, Hébrard S, Berthon C, Nelken B, Fernandes J, et al. Germline PAX5 mutation predisposes to familial B-cell precursor acute lymphoblastic leukemia. Blood. 2021;137(10):1424–8. https://doi.org/10.1182/blood.2020005756.
Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, Maffucci P, Pierce KR, Abbott JK, Voelkerding KV, South ST, Augustine NH, Bush JS, Dolen WK, Wray BB, Itan Y, Cobat A, Sorte HS, Ganesan S, et al. Loss of B cells in patients with heterozygous mutations in IKAROS. N Engl J Med. 2016;374(11):1032–43. https://doi.org/10.1056/NEJMoa1512234.
Churchman ML, Qian M, Te Kronnie G, Zhang R, Yang W, Zhang H, et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell. 2018;33(5):937–48e8. https://doi.org/10.1016/j.ccell.2018.03.021.
Li FP, Fraumeni JF Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71(4):747–52. https://doi.org/10.7326/0003-4819-71-4-747.
Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma X, Zhou X, Yergeau DA, Wilkinson MR, Vadodaria B, Chen X, McGee RB, Hines-Dowell S, Nuccio R, Quinn E, Shurtleff SA, Rusch M, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336–46. https://doi.org/10.1056/NEJMoa1508054.
Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, Payne-Turner D, Churchman M, Andersson A, Chen SC, McCastlain K, Becksfort J, Ma J, Wu G, Patel SN, Heatley SL, Phillips LA, Song G, Easton J, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242–52. https://doi.org/10.1038/ng.2532.
Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020;105(11):2524–39. https://doi.org/10.3324/haematol.2020.247031.
Qian M, Cao X, Devidas M, Yang W, Cheng C, Dai Y, Carroll A, Heerema NA, Zhang H, Moriyama T, Gastier-Foster JM, Xu H, Raetz E, Larsen E, Winick N, Bowman WP, Martin PL, Mardis ER, Fulton R, et al. TP53 Germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J Clin Oncol. 2018;36(6):591–9. https://doi.org/10.1200/JCO.2017.75.5215.
Izraeli S. The acute lymphoblastic leukemia of Down Syndrome - genetics and pathogenesis. Eur J Med Genet. 2016;59(3):158–61. https://doi.org/10.1016/j.ejmg.2015.11.010.
Cave H, Caye A, Strullu M, Aladjidi N, Vignal C, Ferster A, et al. Acute lymphoblastic leukemia in the context of RASopathies. Eur J Med Genet. 2016;59(3):173–8. https://doi.org/10.1016/j.ejmg.2016.01.003.
Xu H, Yang W, Perez-Andreu V, Devidas M, Fan Y, Cheng C, Pei D, Scheet P, Burchard EG, Eng C, Huntsman S, Torgerson DG, Dean M, Winick NJ, Martin PL, Camitta BM, Bowman WP, Willman CL, Carroll WL, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105(10):733–42. https://doi.org/10.1093/jnci/djt042.
Moriyama T, Relling MV, Yang JJ. Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood. 2015;125(26):3988–95. https://doi.org/10.1182/blood-2014-12-580001.
Ellinghaus E, Stanulla M, Richter G, Ellinghaus D, te Kronnie G, Cario G, Cazzaniga G, Horstmann M, Panzer Grümayer R, Cavé H, Trka J, Cinek O, Teigler-Schlegel A, ElSharawy A, Häsler R, Nebel A, Meissner B, Bartram T, Lescai F, et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia. 2012;26(5):902–9. https://doi.org/10.1038/leu.2011.302.
Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, Kinsey SE, Lightfoot T, Roman E, Irving JAE, Allan JM, Tomlinson IP, Taylor M, Greaves M, Houlston RS. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1006–10. https://doi.org/10.1038/ng.430.
Vijayakrishnan J, Studd J, Broderick P, Kinnersley B, Holroyd A, Law PJ, et al. Genome-wide association study identifies susceptibility loci for B-cell childhood acute lymphoblastic leukemia. Nat Commun. 2018;9(1):1340. https://doi.org/10.1038/s41467-018-03178-z.
Perez-Andreu V, Roberts KG, Harvey RC, Yang W, Cheng C, Pei D, Xu H, Gastier-Foster J, E S, Lim JYS, Chen IM, Fan Y, Devidas M, Borowitz MJ, Smith C, Neale G, Burchard EG, Torgerson DG, Klussmann FA, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45(12):1494–8. https://doi.org/10.1038/ng.2803.
Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012;122(10):3407–15. https://doi.org/10.1172/jci61203.
Ford AM, Bennett CA, Price CM, Bruin MC, Van Wering ER, Greaves M. Fetal origins of the TEL-AML1 fusion gene in identical twins with leukemia. Proc Natl Acad Sci U S A. 1998;95(8):4584–8. https://doi.org/10.1073/pnas.95.8.4584.
Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, Greaves M. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature. 1993;363(6427):358–60. https://doi.org/10.1038/363358a0.
Wiemels JL, Ford AM, Van Wering ER, Postma A, Greaves M. Protracted and variable latency of acute lymphoblastic leukemia after TEL-AML1 gene fusion in utero. Blood. 1999;94(3):1057–62.
Gill Super HJ, Rothberg PG, Kobayashi H, Freeman AI, Diaz MO, Rowley JD. Clonal, nonconstitutional rearrangements of the MLL gene in infant twins with acute lymphoblastic leukemia: in utero chromosome rearrangement of 11q23. Blood. 1994;83(3):641–4.
Maia AT, van der Velden VH, Harrison CJ, Szczepanski T, Williams MD, Griffiths MJ, et al. Prenatal origin of hyperdiploid acute lymphoblastic leukemia in identical twins. Leukemia. 2003;17(11):2202–6. https://doi.org/10.1038/sj.leu.2403101.
Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural history. Blood. 2003;102(7):2321–33. https://doi.org/10.1182/blood-2002-12-3817.
Gale KB, Ford AM, Repp R, Borkhardt A, Keller C, Eden OB, Greaves MF. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc Natl Acad Sci U S A. 1997;94(25):13950–4. https://doi.org/10.1073/pnas.94.25.13950.
Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, Saha V, Biondi A, Greaves MF. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999;354(9189):1499–503. https://doi.org/10.1016/s0140-6736(99)09403-9.
Maia AT, Ford AM, Jalali GR, Harrison CJ, Taylor GM, Eden OB, Greaves MF. Molecular tracking of leukemogenesis in a triplet pregnancy. Blood. 2001;98(2):478–82. https://doi.org/10.1182/blood.v98.2.478.
Olsen M, Hjalgrim H, Melbye M, Madsen HO, Schmiegelow K. RT-PCR screening for ETV6-RUNX1-positive clones in cord blood from newborns in the Danish National Birth Cohort. J Pediatr Hematol Oncol. 2012;34(4):301–3. https://doi.org/10.1097/MPH.0b013e3182332268.
Schäfer D, Olsen M, Lähnemann D, Stanulla M, Slany R, Schmiegelow K, Borkhardt A, Fischer U. Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood. 2018;131(7):821–6. https://doi.org/10.1182/blood-2017-09-808402.
Hein D, Korschgen L, Borkhardt A, Kogler G, Fischer U. Seven percent of cord blood transplants carry ETV6-RUNX1 translocations. Stem Cells Transl Med. 2020;9(Suppl 1):S4. https://doi.org/10.1002/sctm.12809.
Gruhn B, Taub JW, Ge Y, Beck JF, Zell R, Häfer R, Hermann FH, Debatin KM, Steinbach D. Prenatal origin of childhood acute lymphoblastic leukemia, association with birth weight and hyperdiploidy. Leukemia. 2008;22(9):1692–7. https://doi.org/10.1038/leu.2008.152.
Lampert F. Cellulärer DNS-Gehalt und Chromosomenzahl bei der akuten Leukämie im Kindesalter und ihre Bedeutung für Chemotherapie und Prognose. Klinische Wochenschrift. 1967;45(15):763–8. https://doi.org/10.1007/BF01747643.
Paulsson K, Lilljebjörn H, Biloglav A, Olsson L, Rissler M, Castor A, Barbany G, Fogelstrand L, Nordgren A, Sjögren H, Fioretos T, Johansson B. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet. 2015;47(6):672–6. https://doi.org/10.1038/ng.3301.
Paulsson K, Mörse H, Fioretos T, Behrendtz M, Strömbeck B, Johansson B. Evidence for a single-step mechanism in the origin of hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2005;44(2):113–22. https://doi.org/10.1002/gcc.20222.
Andersson A, Olofsson T, Lindgren D, Nilsson B, Ritz C, Edén P, et al. Molecular signatures in childhood acute leukemia and their correlations to expression patterns in normal hematopoietic subpopulations. Proc Natl Acad Sci U S A. 2005;102(52):19069–74. https://doi.org/10.1073/pnas.0506637102.
Zaliova M, Hovorkova L, Vaskova M, Hrusak O, Stary J, Zuna J. Slower early response to treatment and distinct expression profile of childhood high hyperdiploid acute lymphoblastic leukaemia with DNA index < 1.16. Genes Chromosomes Cancer. 2016;55(9):727–37. https://doi.org/10.1002/gcc.22374.
• Yang M, Vesterlund M, Siavelis I, Moura-Castro LH, Castor A, Fioretos T, et al. Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia. Nat Commun. 2019;10(1):1519. https://doi.org/10.1038/s41467-019-09469-3. This reference is of importance because it characterizes the proteogenomic landscape of hyperdiploid and ETV6-RUNX1+ childhood ALL. This work identifies changes in chromatin architecture, characterized by weaker TAD boundaries, and dysregulation of gene expression in hyperdiploid ALL
Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. The Lancet. 2013;381(9881):1943–55. https://doi.org/10.1016/S0140-6736(12)62187-4.
Böiers C, Richardson SE, Laycock E, Zriwil A, Turati VA, Brown J, et al. A Human IPS Model Implicates Embryonic B-myeloid fate restriction as developmental susceptibility to B acute lymphoblastic leukemia-associated ETV6-RUNX1. Dev Cell. 2018;44(3):362–77.e7. https://doi.org/10.1016/j.devcel.2017.12.005.
Loncarevic IF, Roitzheim B, Ritterbach J, Viehmann S, Borkhardt A, Lampert F, Harbott J. Trisomy 21 is a recurrent secondary aberration in childhood acute lymphoblastic leukemia with TEL/AML1 gene fusion. Genes Chromosomes Cancer. 1999;24(3):272–7.
Mullighan CG. Genomic characterization of childhood acute lymphoblastic leukemia. Semin Hematol. 2013;50(4):314–24. https://doi.org/10.1053/j.seminhematol.2013.10.001.
Arana-Trejo RM, Ignacio G, Amador-Sánchez R, Cruz-Rico J, Hernández M-P, Saldivar I, et al. Frequency of p190 and p210 BCR-ABL fusions genes in acute lymphoblastic leukemia in a long group of adults and childhood. Blood. 2016;128(22):5273. https://doi.org/10.1182/blood.V128.22.5273.5273.
Bhojwani D, Pei D, Sandlund JT, Jeha S, Ribeiro RC, Rubnitz JE, Raimondi SC, Shurtleff S, Onciu M, Cheng C, Coustan-Smith E, Bowman WP, Howard SC, Metzger ML, Inaba H, Leung W, Evans WE, Campana D, Relling MV, Pui CH. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia. 2012;26(2):265–70. https://doi.org/10.1038/leu.2011.227.
Aricò M, Schrappe M, Hunger SP, Carroll WL, Conter V, Galimberti S, Manabe A, Saha V, Baruchel A, Vettenranta K, Horibe K, Benoit Y, Pieters R, Escherich G, Silverman LB, Pui CH, Valsecchi MG. Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol: official journal of the American Society of Clinical Oncology. 2010;28(31):4755–61. https://doi.org/10.1200/JCO.2010.30.1325.
Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, White D, Hughes TP, le Beau MM, Pui CH, Relling MV, Shurtleff SA, Downing JR. BCR–ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–4. https://doi.org/10.1038/nature06866.
Hein D, Dreisig K, Metzler M, Izraeli S, Schmiegelow K, Borkhardt A, Fischer U. The preleukemic TCF3-PBX1 gene fusion can be generated in utero and is present in ≈0.6% of healthy newborns. Blood. 2019;134(16):1355–8. https://doi.org/10.1182/blood.2019002215.
Jeha S, Pei D, Raimondi SC, Onciu M, Campana D, Cheng C, Sandlund JT, Ribeiro RC, Rubnitz JE, Howard SC, Downing JR, Evans WE, Relling MV, Pui CH. Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia. 2009;23(8):1406–9. https://doi.org/10.1038/leu.2009.42.
Sera Y, Yamasaki N, Oda H, Nagamachi A, Wolff L, Inukai T, Inaba T, Honda H. Identification of cooperative genes for E2A-PBX1 to develop acute lymphoblastic leukemia. Cancer Science. 2016;107(7):890–8. https://doi.org/10.1111/cas.12945.
Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. The Lancet. 2007;370(9583):240–50. https://doi.org/10.1016/S0140-6736(07)61126-X.
Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nature Genetics. 2015;47(4):330–7. https://doi.org/10.1038/ng.3230.
Wiemels JL, Leonard BC, Wang Y, Segal MR, Hunger SP, Smith MT, Crouse V, Ma X, Buffler PA, Pine SR. Site-specific translocation and evidence of postnatal origin of the t(1;19) E2A-PBX1 fusion in childhood acute lymphoblastic leukemia. Proc Natl Acad Sci. 2002;99(23):15101–6. https://doi.org/10.1073/pnas.222481199.
Fischer U, Forster M, Rinaldi A, Risch T, Sungalee S, Warnatz HJ, Bornhauser B, Gombert M, Kratsch C, Stütz AM, Sultan M, Tchinda J, Worth CL, Amstislavskiy V, Badarinarayan N, Baruchel A, Bartram T, Basso G, Canpolat C, et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet. 2015;47(9):1020–9. https://doi.org/10.1038/ng.3362.
Quijano CA, Moore D 2nd, Arthur D, Feusner J, Winter SS, Pallavicini MG. Cytogenetically aberrant cells are present in the CD34+CD33-38-19- marrow compartment in children with acute lymphoblastic leukemia. Leukemia. 1997;11(9):1508–15. https://doi.org/10.1038/sj.leu.2400754.
Kasprzyk A, Harrison CJ, Secker-Walker LM. Investigation of clonal involvement of myeloid cells in Philadelphia-positive and high hyperdiploid acute lymphoblastic leukemia. Leukemia. 1999;13(12):2000–6. https://doi.org/10.1038/sj.leu.2401580.
Hotfilder M, Röttgers S, Rosemann A, Jürgens H, Harbott J, Vormoor J. Immature CD34+CD19− progenitor/stem cells in TEL/AML1-positive acute lymphoblastic leukemia are genetically and functionally normal. Blood. 2002;100(2):640–6. https://doi.org/10.1182/blood.V100.2.640.
Cox CV, Evely RS, Oakhill A, Pamphilon DH, Goulden NJ, Blair A. Characterization of acute lymphoblastic leukemia progenitor cells. Blood. 2004;104(9):2919–25. https://doi.org/10.1182/blood-2004-03-0901.
Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, Tsuzuki S, Greaves M, Enver T. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008;319(5861):336–9. https://doi.org/10.1126/science.1150648.
Alpar D, Wren D, Ermini L, Mansur MB, van Delft FW, Bateman CM, Titley I, Kearney L, Szczepanski T, Gonzalez D, Ford AM, Potter NE, Greaves M. Clonal origins of ETV6-RUNX1+ acute lymphoblastic leukemia: studies in monozygotic twins. Leukemia. 2015;29(4):839–46. https://doi.org/10.1038/leu.2014.322.
Hotfilder M, Röttgers S, Rosemann A, Schrauder A, Schrappe M, Pieters R, Jürgens H, Harbott J, Vormoor J. Leukemic stem cells in childhood high-risk ALL/t(9;22) and t(4;11) are present in primitive lymphoid-restricted CD34+CD19- Cells. Cancer Research. 2005;65(4):1442–9. https://doi.org/10.1158/0008-5472.Can-04-1356.
Castor A, Nilsson L, Åstrand-Grundström I, Buitenhuis M, Ramirez C, Anderson K, Strömbeck B, Garwicz S, Békássy AN, Schmiegelow K, Lausen B, Hokland P, Lehmann S, Juliusson G, Johansson B, Jacobsen SEW. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nature Medicine. 2005;11(6):630–7. https://doi.org/10.1038/nm1253.
Hovorkova L, Zaliova M, Venn NC, Bleckmann K, Trkova M, Potuckova E, Vaskova M, Linhartova J, Machova Polakova K, Fronkova E, Muskovic W, Giles JE, Shaw PJ, Cario G, Sutton R, Stary J, Trka J, Zuna J. Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood. 2017;129(20):2771–81. https://doi.org/10.1182/blood-2016-11-749978.
Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135(6):1130–42. https://doi.org/10.1016/j.cell.2008.10.035.
Barrett NA, Malouf C, Kapeni C, Bacon WA, Giotopoulos G, Jacobsen SEW, Huntly BJ, Ottersbach K. Mll-AF4 confers enhanced self-renewal and lymphoid potential during a restricted window in development. Cell Rep. 2016;16(4):1039–54. https://doi.org/10.1016/j.celrep.2016.06.046.
• O’Byrne S, Elliott N, Rice S, Buck G, Fordham N, Garnett C, et al. Discovery of a CD10-negative B-progenitor in human fetal life identifies unique ontogeny-related developmental programs. Blood. 2019;134(13):1059–71. https://doi.org/10.1182/blood.2019001289. This reference is of importance because it investigates the origins of infant leukemia, identifying fetal-restricted CD10- pre-pro B progenitor cells as potential leukemia-initiating cells
Hübner S, Cazzaniga G, Flohr T, van der Velden VHJ, Konrad M, Pötschger U, Basso G, Schrappe M, van Dongen JJM, Bartram CR, Biondi A, Panzer-Grümayer ER. High incidence and unique features of antigen receptor gene rearrangements in TEL–AML1-positive leukemias. Leukemia. 2004;18(1):84–91. https://doi.org/10.1038/sj.leu.2403182.
Szczepański T, Beishuizen A, Pongers-Willemse MJ, Hählen K, Van Wering ER, Wijkhuijs AJM, et al. Cross-lineage T cell receptor gene rearrangements occur in more than ninety percent of childhood precursor-B acute lymphoblastic leukemias: alternative PCR targets for detection of minimal residual disease. Leukemia. 1999;13(2):196–205. https://doi.org/10.1038/sj.leu.2401277.
Gawad C, Pepin F, Carlton VEH, Klinger M, Logan AC, Miklos DB, Faham M, Dahl G, Lacayo N. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120(22):4407–17. https://doi.org/10.1182/blood-2012-05-429811.
Bungaro S, Irving J, Tussiwand R, Mura R, Minto L, Molteni C, Citterio M, Hall A, Biondi A, Cazzaniga G. Genomic analysis of different clonal evolution in a twin pair with t(12;21) positive acute lymphoblastic leukemia sharing the same prenatal clone. Leukemia. 2008;22(1):208–11. https://doi.org/10.1038/sj.leu.2404973.
Lao ZT, Ding LW, An O, Hattori N, Sun QY, Tan KT, Mayakonda A, Chuan WG, Madan V, Lin DC, Yang H, Koeffler HP. Mutational and transcriptomic profiling of acute leukemia of ambiguous lineage reveals obscure but clinically important lineage bias. Haematologica. 2019;104(5):e200–e3. https://doi.org/10.3324/haematol.2018.202911.
Park M, Koh KN, Kim BE, Im HJ, Jang S, Park CJ, Chi HS, Seo JJ. Lineage switch at relapse of childhood acute leukemia: a report of four cases. J Korean Med Sci. 2011;26(6):829–31. https://doi.org/10.3346/jkms.2011.26.6.829.
Abdelhaleem M. Frequent but nonrandom expression of myeloid markers on de novo childhood acute lymphoblastic leukemia. Exp Mol Pathol. 2007;83(1):138–41. https://doi.org/10.1016/j.yexmp.2007.02.004.
Barata JT, Durum SK, Seddon B. Flip the coin: IL-7 and IL-7R in health and disease. Nat Immunol. 2019;20(12):1584–93. https://doi.org/10.1038/s41590-019-0479-x.
Almeida ARM, Neto JL, Cachucho A, Euzebio M, Meng X, Kim R, et al. Interleukin-7 receptor alpha mutational activation can initiate precursor B-cell acute lymphoblastic leukemia. Nat Commun. 2021;12(1):7268. https://doi.org/10.1038/s41467-021-27197-5.
Thomas KR, Allenspach EJ, Camp ND, Wray-Dutra MN, Khim S, Zielinska-Kwiatkowska A, Timms AE, Loftus JP, Liggitt HD, Georgopoulos K, Tasian SK, James RG, Rawlings DJ. Activated interleukin-7 receptor signaling drives B-cell acute lymphoblastic leukemia in mice. Leukemia. 2022;36(1):42–57. https://doi.org/10.1038/s41375-021-01326-x.
•• Geron I, Savino AM, Fishman H, Tal N, Brown J, Turati VA, et al. An instructive role for Interleukin-7 receptor α in the development of human B-cell precursor leukemia. Nat Commun. 2022;13(1):659. https://doi.org/10.1038/s41467-022-28218-7. This reference is of outstanding importance because it investigates the role of activating IL-7Rα mutations for the development of a preleukemic state in BCP-ALL. This study elegantly shows that activation of IL-7Rα in human hematopoietic stem and progenitor cells is sufficient for the development of preleukemic cells and leads to overt leukemia in combination with CDKN2A deletions
Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. Nat Genet. 1998;20(4):394–7. https://doi.org/10.1038/3877.
Schmidt JA, Hornhardt S, Erdmann F, Sanchez-Garcia I, Fischer U, Schuz J, et al. Risk factors for childhood leukemia: radiation and beyond. Front Public Health. 2021;9:805757. https://doi.org/10.3389/fpubh.2021.805757.
Cao Y, Lu J, Lu J. Paternal smoking before conception and during pregnancy is associated with an increased risk of childhood acute lymphoblastic leukemia: a systematic review and meta-analysis of 17 case-control studies. J Pediatr Hematol Oncol. 2020;42(1):32–40. https://doi.org/10.1097/MPH.0000000000001657.
Francis SS, Wallace AD, Wendt GA, Li L, Liu F, Riley LW, Kogan S, Walsh KM, de Smith AJ, Dahl GV, Ma X, Delwart E, Metayer C, Wiemels JL. In utero cytomegalovirus infection and development of childhood acute lymphoblastic leukemia. Blood. 2017;129(12):1680–4. https://doi.org/10.1182/blood-2016-07-723148.
Cheeran MC, Lokensgard JR, Schleiss MR. Neuropathogenesis of congenital cytomegalovirus infection: disease mechanisms and prospects for intervention. Clin Microbiol Rev. 2009;22(1):99–126.Table of Contents. https://doi.org/10.1128/CMR.00023-08.
Semmes EC, Hurst JH, Walsh KM, Permar SR. Cytomegalovirus as an immunomodulator across the lifespan. Curr Opin Virol. 2020;44:112–20. https://doi.org/10.1016/j.coviro.2020.07.013.
Liu X, Palaniyandi S, Zhu I, Tang J, Li W, Wu X, Ochsner SP, Pauza CD, Cohen JI, Zhu X. Human cytomegalovirus evades antibody-mediated immunity through endoplasmic reticulum-associated degradation of the FcRn receptor. Nat Commun. 2019;10(1):3020. https://doi.org/10.1038/s41467-019-10865-y.
Lehtinen M, Koskela P, Ogmundsdottir HM, Bloigu A, Dillner J, Gudnadottir M, Hakulinen T, Kjartansdottir A, Kvarnung M, Pukkala E, Tulinius H, Lehtinen T. Maternal herpesvirus infections and risk of acute lymphoblastic leukemia in the offspring. Am J Epidemiol. 2003;158(3):207–13. https://doi.org/10.1093/aje/kwg137.
Tedeschi R, Bloigu A, Ogmundsdottir HM, Marus A, Dillner J. dePaoli P et al. Activation of maternal Epstein-Barr virus infection and risk of acute leukemia in the offspring. Am J Epidemiol. 2007;165(2):134–7. https://doi.org/10.1093/aje/kwj332.
• He JR, Ramakrishnan R, Hirst JE, Bonaventure A, Francis SS, Paltiel O, et al. Maternal infection in pregnancy and childhood leukemia: a systematic review and meta-analysis. J Pediatr. 2020;217:98–109 e8. https://doi.org/10.1016/j.jpeds.2019.10.046. This reference is of importance because it thoroughly analyzes 20 studies regarding the association between maternal infection during pregnancy and childhood leukemia. The authors conclude that specific infections during pregnancy are associated with a higher risk to develop leukemia
Fedrick J, Alberman ED. Reported influenza in pregnancy and subsequent cancer in the child. Br Med J. 1972;2:485–8.
Bithell JF, Draper GJ, Gorbach PD. Association between malignant disease in children and maternal virus infections. Br Med J. 1973;1:706–8.
Kroll ME, Draper GJ, Stiller CA, Murphy MF. Childhood leukemia incidence in Britain, 1974-2000: time trends and possible relation to influenza epidemics. J Natl Cancer Inst. 2006;98(6):417–20. https://doi.org/10.1093/jnci/djj095.
Cazzaniga G, Bisanti L, Randi G, Deandrea S, Bungaro S, Pregliasco F, Perotti D, Spreafico F, Masera G, Valsecchi MG, Biondi A, Greaves M. Possible role of pandemic AH1N1 swine flu virus in a childhood leukemia cluster. Leukemia. 2017;31(8):1819–21. https://doi.org/10.1038/leu.2017.127.
Francis SS, Selvin S, Yang W, Buffler PA, Wiemels JL. Unusual space-time patterning of the Fallon, Nevada leukemia cluster: evidence of an infectious etiology. Chem Biol Interact. 2012;196(3):102–9. https://doi.org/10.1016/j.cbi.2011.02.019.
Lillie K. Leukaemia and lockdown: The delayed infection model of childhood acute lymphoblastic leukaemia and the COVID-19 pandemic. Pediatr Blood Cancer. 2021;68(10):e29194. https://doi.org/10.1002/pbc.29194.
Li CK, Zee B, Lee J, Chik KW, Ha SY, Lee V. Impact of SARS on development of childhood acute lymphoblastic leukaemia. Leukemia. 2007;21(7):1353–6. https://doi.org/10.1038/sj.leu.2404729.
Rodriguez-Hernandez G, Hauer J, Martin-Lorenzo A, Schafer D, Bartenhagen C, Garcia-Ramirez I, et al. Infection exposure promotes ETV6-RUNX1 precursor B-cell leukemia via impaired H3K4 demethylases. Cancer Res. 2017;77(16):4365–77. https://doi.org/10.1158/0008-5472.CAN-17-0701.
Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, Gustafsson A, Bernhardsson AK, Zhang C, Bohlin K, Brodin P. Stereotypic immune system development in newborn children. Cell. 2018;174(5):1277–92e14. https://doi.org/10.1016/j.cell.2018.06.045.
Zhang C, Yin J, Zheng J, Xiao J, Hu J, Su Y, et al. EZH2 identifies the precursors of human natural killer cells with trained immunity. Cancer Biol Med. 2021;18:1021–39. https://doi.org/10.20892/j.issn.2095-3941.2020.0791.
Hammer Q, Romagnani C. About training and memory: NK-cell adaptation to viral infections. Adv Immunol. 2017;133:171–207. https://doi.org/10.1016/bs.ai.2016.10.001.
Sun JC, Madera S, Bezman NA, Beilke JN, Kaplan MH, Lanier LL. Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J Exp Med. 2012;209(5):947–54. https://doi.org/10.1084/jem.20111760.
Bigley AB, Rezvani K, Shah N, Sekine T, Balneger N, Pistillo M, Agha N, Kunz H, O'Connor DP, Bollard CM, Simpson RJ. Latent cytomegalovirus infection enhances anti-tumour cytotoxicity through accumulation of NKG2C+ NK cells in healthy humans. Clin Exp Immunol. 2016;185(2):239–51. https://doi.org/10.1111/cei.12785.
•• Mehtonen J, Teppo S, Lahnalampi M, Kokko A, Kaukonen R, Oksa L, et al. Single cell characterization of B-lymphoid differentiation and leukemic cell states during chemotherapy in ETV6-RUNX1-positive pediatric leukemia identifies drug-targetable transcription factor activities. Genome Med. 2020;12(1):99. https://doi.org/10.1186/s13073-020-00799-2. This reference is of outstanding importance because it unravels the regulatory cell network in the bone marrow during BCP-ALL using single cell RNA sequencing. It describes a mechanism of immune cell depletion in ETV6-RUNX1+ ALL with low numbers and activity of NK cells
Babor F, Manser AR, Fischer JC, Scherenschlich N, Enczmann J, Chazara O, Moffett A, Borkhardt A, Meisel R, Uhrberg M. KIR ligand C2 is associated with increased susceptibility to childhood ALL and confers an elevated risk for late relapse. Blood. 2014;124(14):2248–51. https://doi.org/10.1182/blood-2014-05-572065.
Sullivan EM, Jeha S, Kang G, Cheng C, Rooney B, Holladay M, Bari R, Schell S, Tuggle MC, Pui CH, Leung W. NK cell genotype and phenotype at diagnosis of acute lymphoblastic leukemia correlate with postinduction residual disease. Clin Cancer Res. 2014;20(23):5986–94. https://doi.org/10.1158/1078-0432.CCR-14-0479.
• Apostol AC, Jensen KDC, Beaudin AE. Training the fetal immune system through maternal inflammation-a layered hygiene hypothesis. Front Immunol. 2020;11:123. https://doi.org/10.3389/fimmu.2020.00123. This reference is of importance because it discusses the effect of microbial exposure during pregnancy on fetal immune system development. This review gives an overview of research on the hygiene hypothesis that may play a role in the development of autoimmune diseases, allergies and BCP-ALL
Krämer U, Heinrich J, Wjst M, Wichmann HE. Age of entry to day nursery and allergy in later childhood. The Lancet. 1999;353(9151):450–4. https://doi.org/10.1016/s0140-6736(98)06329-6.
Jourdan-Da Silva N, Perel Y, Mechinaud F, Plouvier E, Gandemer V, Lutz P, et al. Infectious diseases in the first year of life, perinatal characteristics and childhood acute leukaemia. Br J Cancer. 2004;90(1):139–45. https://doi.org/10.1038/sj.bjc.6601384.
Schuz J, Morgan G, Bohler E, Kaatsch P, Michaelis J. Atopic disease and childhood acute lymphoblastic leukemia. Int J Cancer. 2003;105(2):255–60. https://doi.org/10.1002/ijc.11054.
•• Traxel S, Schadt L, Eyer T, Mordasini V, Gysin C, Munthe LA, et al. Bone marrow T helper cells with a Th1 phenotype induce activation and proliferation of leukemic cells in precursor B acute lymphoblastic leukemia patients. Oncogene. 2019;38(13):2420–31. https://doi.org/10.1038/s41388-018-0594-4. This reference is of outstanding importance because it unravels a possible mechanistic link between infections and BCP-ALL development. This study explains how pro-inflammatory Th cell responses directly favor the activation and proliferation of leukemic cells in BCP-ALL
Traxel S, Lehmann J, Richard S, Sidorov S, Niggli F, Berger C, Nadal D, Bürgler S. Support of BCP-ALL-cells by autologous bone marrow Th-cells involves induction of AID expression but not widespread AID off-target mutagenesis. Cancer Immunol Immunother. 2021;70(8):2275–89. https://doi.org/10.1007/s00262-020-02835-x.
Miedema KG, Tissing WJ, Te Poele EM, Kamps WA, Alizadeh BZ, Kerkhof M, et al. Polymorphisms in the TLR6 gene associated with the inverse association between childhood acute lymphoblastic leukemia and atopic disease. Leukemia. 2012;26(6):1203–10. https://doi.org/10.1038/leu.2011.341.
Villani A, Shore A, Wasserman JD, Stephens D, Kim RH, Druker H, Gallinger B, Naumer A, Kohlmann W, Novokmet A, Tabori U, Tijerin M, Greer MLC, Finlay JL, Schiffman JD, Malkin D. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. The Lancet Oncology. 2016;17(9):1295–305. https://doi.org/10.1016/S1470-2045(16)30249-2.
Fulbright JM, Raman S, McClellan WS, August KJ. Late effects of childhood leukemia therapy. Curr Hematol Malig Rep. 2011;6(3):195–205. https://doi.org/10.1007/s11899-011-0094-x.
Acknowledgements
The authors thank Stewart Boden for critical reading of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. A.B. was funded by the Löwenstern e.V., the Katharina-Hardt-Stiftung, and the German José Carreras Leukemia Foundation (DJCLS 07/19). A.B., U.F., and D.H were funded by the German Federal Office for Radiation Protection (BfS, 3618S32274 and 3618S32275) and A.B. and U.F. by the Foundation Unoentrecienmil. U.F. and D.H. were supported by the German José Carreras Leukemia Foundation (DJCLS 18R/2021). U.F. was supported by the German José Carreras Leukemia Foundation (DJCLS 21R/2019). D.H. was supported by the German Research Foundation (DFG, 495318549). K.L.G. was supported by the German Research Foundation (DFG, 428917761).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Leukemia
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rüchel, N., Jepsen, V.H., Hein, D. et al. In Utero Development and Immunosurveillance of B Cell Acute Lymphoblastic Leukemia. Curr. Treat. Options in Oncol. 23, 543–561 (2022). https://doi.org/10.1007/s11864-022-00963-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11864-022-00963-3