
Abstract
The nature versus nurture debate has intrigued scientific circles for decades. Although extensive research has established a clear relationship between genetics and disease development, recent evidence has highlighted the insufficiency of attributing adverse health outcomes to genetic factors alone. In fact, it has been suggested that environmental influences, such as socioeconomic position (SEP), may play a much larger role in the development of disease than previously thought, with extensive research suggesting that low SEP is associated with adverse health conditions. In relation to oral health, a higher prevalence of caries (tooth decay) exists among those of low SEP. Although little is known about the biological mechanisms underlying this relationship, epigenetic modifications resulting from environmental influences have been suggested to play an important role. This review explores the intersection of health inequalities and epigenetics, the role of early-life social adversity and its long-term epigenetic impacts, and how those living within the lower hierarchies of the socioeconomic pyramid are indeed at higher risk of developing diseases, particularly in relation to oral health. A deeper understanding of these mechanisms could lead to the development of targeted interventions for individuals of low SEP to improve oral health or identify those who are at higher risk of developing oral disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
‘Nature or nurture?’ is a question that has been the subject of many scientific debates. Remarkably, geneticists can now precisely identify and locate certain genes in the DNA sequence that are associated with disease [1]. However, recent breakthroughs have established that the paradigm of thought that defines ‘nature’, or an individual’s DNA sequence, to be the sole determinant of disease outcome is insufficient. In fact, it has become apparent that other factors stemming from the environment and functioning above the genome may be at play [2]. In this regard, there is considerable interest in the field of epigenetics, which may explain the differences in health inequalities between individuals throughout the socioeconomic pyramid. The question of the relevance of early experiences of social adversity throughout life and how these impact biological processes has been at the forefront of epigenetic research over the past few decades.
In 1957, Conrad Hal Waddington, a developmental biologist, first put forward the ‘epigenetic landscape’, in which a ball could follow different paths as it rolls down a slope due to the roughness of the surface. The ball in this case represents the cell, whilst the slope represents cell differentiation. The variable roughness of the surface symbolises extracellular environmental factors which can influence the path the cell follows during differentiation. In other words, the ‘epigenetic landscape’ describes the process of cell differentiation during development. This explains how 200 different cell types can exist in the human body, with different functions, despite having the same DNA sequence [3]. Waddington later went on to define epigenetics as ‘the branch of biology which studies the causal interactions between genes and their products which bring the phenotype into being’ [4]. ‘Epi’ is a Greek prefix that means ‘above’, and so ‘epigenetics’ refers to factors functioning ‘above’ the genome. The epigenome, following signals from intra- and extra-cellular influences, results in modifications to the chromatin of the cell, which influences gene expression without causing alterations to the DNA sequence. In summary, it is a bridge between genotype and phenotype [4, 5].
Review
The genome and the epigenome
To understand how the epigenome links psychosocial stimuli to ‘get under the skin’ and become embedded into the genome to influence biological processes involved in diseases, a basic understanding of the structure of chromatin is required.
Chromatin, a complex of DNA and protein found in all eukaryotic cells, consists of building blocks known as nucleosomes, each of which consists of 146 base pairs of nucleotides and one core histone complex, known as histone octamer [6,7,8]. Histones are involved in the tight packing of nucleosomes which enables 2 m of DNA to fit into the nucleus of every cell [9]. This packing formation also enables regulation of gene expression, as transcription factors are generally unable to bind to the condensed DNA, ultimately resulting in reduced production of proteins as required by the cell [10] (Fig. 1a). Epigenetic post-translational modifications (PTMs), most notably the addition of methyl or acetyl groups, take place around the N-terminal amino acid tails of the core histones [11, 12] and are capable of condensing or relaxing the nucleosome structure in a manner which affects gene expression.

a Overview of chromatin organisation. Chromosomal DNA is tightly wrapped around histone proteins to form nucleosomes, which are further condensed into chromatin. Various modifications to the DNA and histones in this structure can stimulate or repress gene expression as required by the cell. b Histone acetylation. Acetyl groups can be added to or removed from histones to alter gene expression. This process is mediated by HATs and HDACs. c DNA methylation. Methyl groups are added to gene promoters in order to repress gene expression
The tight packing of nucleosomes that prevents gene expression can be induced by deacetylation, catalysed by a group of enzymes known as histone deacetylases (HDACs) which remove an acetyl group (−COCH3) from histone amino acid tails. In contrast, a relaxed chromatin conformation exposes transcription factor binding sites for gene expression and is mediated by the addition of an acetyl group to amino acid tails in the process of acetylation, catalysed by histone acetyltransferases (HATs) (Fig. 1b). The balance of histone acetylation and deacetylation is critical for normal cellular function, as evidenced by aberrant histone acetylation in many cancers [13]. Notably, extensive research has established histone deacetylase inhibitors (HDACi)—a class of synthetically produced biological agents—to have considerable therapeutic potential due to their ability to inhibit deacetylation [14, 15]. A number of HDACis have already been FDA-approved, such as suberoylanilide hydroxamic acid (SAHA) and panobinostat [16].
Methylation of histones is a process which involves the addition of a methyl group (–CH3) to lysine molecules on H3 and H4 core histones. This process most commonly occurs on H3 lysines K4 and K9, which results in gene expression and silencing, respectively. These chromatin modifications do not alter the DNA sequence itself, only its accessibility, resulting in potentially heritable alterations to gene expression [7].
The most studied epigenetic process is DNA methylation, which is modulated by enzymes known as DNMTs (DNA methyltransferases). This process involves the addition of a methyl group to a cytosine base to produce 5-methylcytosine (5mC) and typically occurs at regions of the genome with a high concentration of phosphate-linked cytosine-guanine dinucleotides (CpG islands) [17]. The methylated DNA recruits methyl-binding proteins that promote chromatin compaction and induce gene silencing through transcriptional repression [18] (Fig. 1c). Approximately 28 million CpG dinucleotides are present in the human genome, 60–80% of which are normally methylated as part of processes such as chromosome inactivation, heterochromatin folding, and the maintenance of genomic stability [19,20,21]. Loss of genomic stability through aberrant methylation of certain genes may result in the activation of oncogenesis, leading to the development of numerous cancers, including oral squamous cell carcinoma (OSCC) [22,23,24]. To counteract these cases of abnormal methylation, research in DNA methyltransferase inhibitors (DNMTi), another group of exciting biological agents that are likely to play a large role in cancer therapy in the foreseeable future, has been taking place. Their ability to inhibit DNMTs has proven to be successful in reversing abnormal DNA methylation associated with cancer cells [25]. At the time of writing, two drugs have been approved by the FDA and are currently in clinical use for the treatment of myelodysplastic syndrome and acute myeloid leukaemia [26].
Epigenetic heredity
Certain mechanisms have been described to explain how epigenetic traits and modifications may be transmitted inter-generationally [27, 28]. It has been shown that immediately following fertilisation, the erasure of DNA methylation patterns of the gametes in the zygote occurs, which leads to the resetting of epigenetic marks between generations [29, 30]. However, neither DNA methylation nor histone modification patterns are fully erased between generations, which provides evidence of some potential inter-generational heritability [30]. It has also been shown that a large number of epigenetic marks also undergo reprogramming through demethylation during the meiosis of primordial germ cells, yet a substantial number of genes have been shown to retain the parental methylation patterns as a result of epigenetic transmission from primordial germ cells [31, 32].
In one study, rats exposed to endocrine disruptors demonstrated epigenetic modifications resulting in decreased male fertility through the male germline, extending from F1 to F4 generations [33]. In another study, mice subjected to specific odour fear-conditioning prior to conception later conceived F1 and F2 generations that demonstrated heightened sensitivity to that odour, despite having never been exposed to it [34].
It is not yet known how post-translational histone modifications are transmitted inter-generationally through cell mitosis [35]. However, following the disruption of nucleosomes ahead of replication, recombination of older parental histones with newer ones has been established, which may offer an insight into the method of epigenetic transmission [36], as parental histones may also modify and influence the new group of histones assembled [37].
Adversity and long-lasting epigenetic impacts
One of the earliest records demonstrating how environmental conditions during mammalian development may induce epigenetic changes that last throughout life relates to a retrospective study carried out on adults who were prenatally exposed to famine during the Dutch Hunger Winter of 1944–1945 in the Netherlands [38]. In a landmark study, individuals who experienced prenatal famine exposure were found to carry distinct epigenetic marks six decades later. Specifically, the authors discovered hypomethylation of the IGF2 (insulin-like growth factor 2) gene, which is a key factor for human growth and development, compared to same-sex siblings unexposed to prenatal malnutrition [39]. These adults who were exposed to famine during early gestation had a higher risk of developing diseases during late adulthood such as schizophrenia [40], generalised reduced cognitive ability [41], coronary artery disease [42], and obesity [43].
The role of social upbringing with regard to education and income has been shown to play a role in developing diseases throughout a person’s lifespan. The famous Whitehall study carried out first on British civil servants in 1967 demonstrated a strong inverse relationship between mortality and grade of employment across a seven-and-a-half-year follow-up. The researchers also found that men in the lower grade of employment were at higher risk of mortality specifically due to coronary heart disease (CHD). Even after controlling for other risk factors such as plasma cholesterol levels, systolic blood pressure (BP), cigarette consumption, and height, those with the lowest grade of employment still had a relative CHD risk of 2.1 compared to those with the highest grade of employment [44]. A follow-up longitudinal study conducted 20 years later—‘Whitehall II’—confirmed the inverse relationship between socioeconomic position and risk of CHD, metabolic syndrome, and diabetes [45].
The theory of biological embodiment
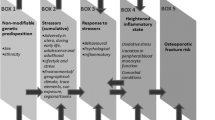
The biopsychosocial model first introduced by Engel in 1979 argued that general health is a complex outcome of biological, psychological, and social factors [46, 47]. This was succeeded by a more recent theory known as ‘biological embodiment’; a concept which posits that complex processes due to social deprivation in its many forms bring about the initiation of pathobiological sequelae that leave a long-lasting mark on the biological systems in those vulnerable [48, 49]. One of the core concepts of how biological embodiment works is through ‘allostatic load’, or the preferred term, cumulative biological risk.
Allostasis refers to the body’s ability to achieve homeostasis or ‘stability’ through internal physiological or behavioural changes [48], whilst allostatic load (AL) refers to the ‘cumulative wear and tear on body systems as a result of exposure to chronic stress’, where a person’s inability to adapt emotionally and physiologically to increasingly stressful demands results in a compounded dysregulation over time. The risk of disease occurrence is higher in such cases, resulting from a combination of excessive cortisol release and dysregulation of mechanisms that shut down cortisol release through negative feedback mechanisms once the stressor has resolved [50]. Not only does AL result in increased cardiovascular, metabolic and general physical decline, and all-cause mortality [51], but it has also been linked to alterations in brain function, which is the key organ of stress processes [52,53,54,55]. AL can be scored through biometric parameters such as BMI (body mass index), waist-to-hip ratio, resting systolic and diastolic BP, serum high-density lipoprotein, glycated haemoglobin, and C-reactive protein alongside others, all of which have been shown to be socially patterned and elevated in those of lower SEP [56,57,58].
The role of stress in influencing the epigenome
Factors such as stress [59], diet [60], infection [61], smoking [62], maternal care during infancy [63], and even increasing age [64] may influence epigenetic modifications to alter the chromatin state and expose certain genome stretches for longer or shorter periods, subsequently resulting in variability and dysregulation of protein production. Stress, in particular, may play a detrimental role in the dysregulation of the hippocampus-pituitary-adrenal (HPA) axis [65, 66] and the sympathetic-adrenal-medullary (SAM) pathway [67]. The activation of the HPA axis due to chronic stress results in the release of cortisol into the blood via a complex signalling cascade [68], whilst the SAM pathway is activated by the stimulation of the adrenal medulla to release the catecholamines epinephrine and norepinephrine [67].
Repeated and chronic exposure to cortisol has numerous pathological sequelae, such as increased BP, reduced metabolism, and subsequent risk of obesity, alongside chronic anxiety and depression [69]. Chronic exposure to stress has numerous immunosuppressive effects, which include depression of proliferation and differentiation of immune cells (which have receptors for cortisol and epinephrine/norepinephrine) [70] and disruption of pro-inflammatory cytokine production by directly binding to immune cell surfaces at specific sites, resulting in anti-inflammatory effects throughout the body [71]. Elevated levels of cortisol have also been directly linked with increased production of pro-inflammatory cytokines, which are conducive to periodontal tissue destruction, and an increased count of active periodontal pockets in those suffering from periodontitis [72,73,74,75].
In contrast, acute episodes of stress trigger the release of norepinephrine through the SAM pathway, which binds to its receptors at cells in lymphoid organs to modulate immune cell function [67], leading to decreased blood flow to periodontal tissue, hence reducing the diffusion of cells and nutrients, resulting in poor wound healing throughout the periodontium [76, 77].
Paradoxically, individuals who are chronically stressed have been associated with increased inflammation, despite the anti-inflammatory functions of cortisol as mentioned earlier. This is due to the desensitisation of the glucocorticoid receptor pathway following long-term exposure to cortisol, rendering the immune cells insensitive to its anti-inflammatory effects [78]. Further studies have attributed this to epigenetic processes that have numerous effects including suppression of the NR3C1 gene (glucocorticoid receptor gene) [79]. Stressful experiences in early life result in disruptions to the NR3C1 gene in the hippocampus through DNA methylation, resulting in long-lasting and reduced glucocorticoid expression and HPA axis dysfunction. This low expression is linked with insufficient negative feedback mechanisms that keep cortisol in circulation for prolonged periods and result in chronic high-stress states [80,81,82].
Indeed, studies have demonstrated that under conditions of social isolation [83, 84] and low SEP [85, 86], there is an alteration to the HPA axis regulatory function. Studies have also shed light on the resultant association with reduced leukocyte sensitivity to glucocorticoid regulation [87, 88], leading to increased risk of disease susceptibility [89].
Epigenetics and oral health
It has already been established that oral diseases such as caries [90] and periodontitis [91] disproportionately affect those who are more socially disadvantaged and who occupy lower socio-economic positions. Whilst this is generally attributed to lifestyle factors associated with these groups, such as poor oral hygiene [92], increased sugar consumption [93], and high rates of smoking [94] and alcohol consumption [95], more evidence is shedding light on the long-term epigenetic effects associated with low SES during critical stages of fetal development and early childhood. Exposure to the associated social and environmental stresses leads to their embedding and imprinting into the genome at an early age, resulting in long-term cascades of side effects that place these individuals at higher risk of developing oral diseases. These experiences of early stressful events and their long-term effects on brain health and mental well-being are well documented [96,97,98,99]. In accordance with this model, a recent study found structural changes in the enamel of primary teeth in children who had experienced perinatal maternal psychosocial stress [100]. In another pertinent study, accelerated eruption of primary molars in children of lower-income families was demonstrated [101], aligning with a growing body of evidence in the literature linking experiences of early psychosocial stress to accelerated epigenetic aging [102].
Furthermore, there is compelling evidence suggesting a variety of oral microbiomes that are more consistent with varying socioeconomic groups than oral health behaviours and more consistent with certain ethnic and racial groups than diet [103]. Interestingly, elevated levels of cortisol in children with lower SEP demonstrated higher levels of the cariogenic bacteria Streptococcus mutans in the oral cavity [104], adding another link to the complex cariogenic mechanisms involved in children with lower SEP who have already been shown to be at higher risk of developing more severe forms of dental caries and tooth loss [105,106,107]. Another study found higher levels of IgG antibodies against Bacteroides forysthus bacteria, which is associated with periodontal disease, in groups with higher levels of depression and psychosocial stress [108].
In the context of periodontitis at the molecular level, a significant finding emerged regarding the epigenome of an individual suffering from the inflammatory disease, where different methylation patterns were observed between inflamed and non-inflamed sites [109]. It has also been shown that TNFA and COX2 CpG sites are hypermethylated and silenced in chronic periodontitis [110, 111], whilst in another study, hypomethylation at the promoter region of IFNY was demonstrated, which led to increased expression of IFN-γ in biopsies from tissues with periodontitis in comparison to tissues of a healthy periodontium [112]. IFN-γ cytokines have been linked to pulpal inflammation, with the IFNY promoter region demonstrated to be only partially methylated or unmethylated in 94% of inflamed pulp tissue samples, compared with healthy pulp tissue samples, of which 44% demonstrated total methylation of the gene [113]. The same authors conducted a separate study exploring the differences between methylation patterns of the TLR2 (Toll-like receptor 2) gene and CD14 (a TLR4 co-receptor) gene in inflamed and non-inflamed pulps and found no difference in methylation patterns between the two groups [114]. These findings emphasise the novelty and complexity of the research being carried out concerning epigenetic mechanisms and their dental implications and present new insights that may revolutionise our treatment of pulpitis.
In another study, hypermethylation and silencing of the TLR2 gene in tissues with periodontitis were confirmed, and the authors found a correlation between the methylation status of the TLR2 gene and probing depths [115]. Additionally, the gene encoding the IL-8 cytokine, which plays critical roles in the recruitment of neutrophils to inflamed periodontal sites, was found to be hypomethylated at the promoter region in epithelial cells of those with periodontitis [116] and aggressive periodontitis [117] compared to healthy individuals, resulting in increased expression in those suffering from periodontal disease. These findings offer a glimpse into the complexity of mechanisms underlying periodontitis and the dysregulation of genes associated with inflammation and immune response in the context of this oral condition. Epigenetic alterations influencing the inflammatory response may not be permanent. Hence, by altering the oral microbiome that was conducive to periodontal disease initially, for example, through deep cleaning of pockets, the epigenome of these inflamed sites may be reset.
Drawing parallels to tumour development, it was demonstrated that the CpG islands of tumour suppressor genes, such as P16, are often hypermethylated at the transcriptional promoter region leading to gene silencing in oral premalignant lesions, making it a potential predictive marker of the risk of malignant change in oral epithelial dysplasia [118]. Positive correlations between smoking and hypermethylation at the promoter region of P16 in patients with non-small cell lung carcinoma were demonstrated [119], whilst another study showed that consumption of nicotine-derived nitrosamine ketone in tobacco is associated with hypermethylation of the P16 gene promoter [120]. A prospective cohort study investigated the link between the methylation status of the P16 promoter region and the progression of oral epithelial dysplasia over a period of 46 months. Patients with P16-methylated dysplasia were found to be at a significantly higher risk of developing oral cancer than the control group of patients with P16-unmethylated dysplasia [121]. Interestingly, one study found that treatment with procaine, a DNMTi, reversed the hypermethylation of the PAX9 gene, which is partly involved in the differentiation of OSCC cells. The reversal of methylation and increased expression of PAX9 induced apoptosis and cell growth inhibition in OSCC tissue cells in vivo and in vitro and improved anticancer drug sensitivity [122], highlighting the potential role of DNMTi drugs in future oral cancer treatment.
There have been a limited number of studies linking histone modification to periodontal disease and pulpal inflammatory processes [123,124,125,126]; however, in relation to other inflammatory diseases, aberrant histone modification processes have been demonstrated to be involved in rheumatoid arthritis [127], diabetes mellitus [128], and asthma [129]. In addition, whilst a role for histone modifications in periodontal and pulpal inflammation has yet to be elucidated, one study found that treatment of mice with periodontitis with a HDACi increased bone volume in vivo, suggesting a potential role for HDACi in ameliorating the adverse outcomes of periodontal disease [124].
In addition to their potential therapeutic use as anti-inflammatory drugs, in vitro [130] and in vivo [131] studies have demonstrated their potential as therapeutic agents for the treatment of cancers due to their anti-tumourigenic effects in transformed cells, including growth arrest and autophagic cell death, whilst normal cells are more resistant to these changes [132,133,134]. At the time of writing, four HDACi drugs are approved by the FDA for the clinical treatment of certain tumours [135]. With regards to non-oncological normal tissue, there is great therapeutic potential for the use of HDACi and DNMTi in stimulating pulpal tissue regeneration as part of restorative dental treatment. The increased appreciation for epigenetic inhibitors in this regard is in large part due to their ability to modulate a number of complex processes which are crucial for dentinogenesis, including inflammation, mineralisation, and stem cell differentiation [136, 137].
Epigenetic mechanisms involving histone demethylases (HDMs) and histone acetyltransferases (HATs) [138] have also been shown to play crucial roles at certain stages of tooth development and may be linked to the induction of dental anomalies such as supernumeraries and hypodontia. This has been demonstrated in studies involving mice and monozygotic human twins who share a similar genetic makeup but demonstrate different dental phenotypes owing to certain environmental influences at certain stages during tooth development [139, 140].
Conclusion
Based on the current literature, evidence suggests that social adversity, particularly when experienced at an early age, plays a significant role in altering the epigenome in numerous ways, through various mechanisms. The literature describing the role of epigenetics in oral disease and dental anomalies is still in its infancy, yet there is a significant body of evidence linking psychosocial stress to many endocrine and inflammatory diseases, particularly periodontitis and oral cancer. Some of the alterations to the genome have been shown to be long-lasting with an ability to be passed down inter-generationally, at least when demonstrated through in vitro and in vivo studies, not involving humans. However, the mechanisms of epigenetic heredity are yet to be fully explained. A more comprehensive understanding of these molecular processes may enable us, through targeted interventions, to influence the genome to reduce disease risk or identify individuals who are at an increased risk of developing oral diseases.
References
Antonarakis SE (2021) History of the methodology of disease gene identification. Am J Med Genet A 185(11):3266–3275
Poulsen P, Esteller M, Vaag A, Fraga MF (2007) The epigenetic basis of twin discordance in age-related diseases. Pediatr Res 61(5 Pt 2):38r–42r
Goldberg AD, Allis CD, Bernstein E (2007) Epigenetics: a landscape takes shape. Cell 128(4):635–638
Peixoto P, Cartron PF, Serandour AA, Hervouet E (2020) From 1957 to nowadays: a brief history of epigenetics. Int J Mol Sci 21(20):7571. https://doi.org/10.3390/ijms21207571
Nicoglou A, Wolfe CT (2018) Introduction: sketches of a conceptual history of epigenesis. Hist Philos Life Sci 40(4):64
Szerlong HJ, Hansen JC (2011) Nucleosome distribution and linker DNA: connecting nuclear function to dynamic chromatin structure. Biochem Cell Biol 89(1):24–34
Turner BM (2002) Cellular memory and the histone code. Cell 111(3):285–291
Kornberg RD, Lorch Y (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98(3):285–294
Luger K, Mäder AW, Richmond RK et al (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389(6648):251–60
Petesch SJ, Lis JT (2012) Overcoming the nucleosome barrier during transcript elongation. Trends Genet 28(6):285–294
Bayarsaihan D (2011) Epigenetic mechanisms in inflammation. J Dent Res 90(1):9–17
Spange S, Wagner T, Heinzel T, Krämer OH (2009) Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 41(1):185–198
Audia JE, Campbell RM (2016) Histone modifications and cancer. Cold Spring Harb Perspect Biol 8(4):a019521
Bäckdahl L, Bushell A, Beck S (2009) Inflammatory signalling as mediator of epigenetic modulation in tissue-specific chronic inflammation. Int J Biochem Cell Biol 41(1):176–184
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293(5532):1074–1080
Eckschlager T, Plch J, Stiborova M, Hrabeta J (2017) Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci 18(7):1414. https://doi.org/10.3390/ijms18071414
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16(1):6–21
Jones PA, Liang G (2009) Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 10(11):805–811
Henckel A, Nakabayashi K, Sanz LA et al (2009) Histone methylation is mechanistically linked to DNA methylation at imprinting control regions in mammals. Hum Mol Genet 18(18):3375–3383
Alberti S, Nutini M, Herzenberg LA (1994) DNA methylation prevents the amplification of TROP1, a tumor-associated cell surface antigen gene. Proc Natl Acad Sci USA 91(13):5833–5837
Nasr AF, Nutini M, Palombo B et al (2003) Mutations of TP53 induce loss of DNA methylation and amplification of the TROP1 gene. Oncogene 22(11):1668–1677
Feinberg AP, Cui H, Ohlsson R (2002) DNA methylation and genomic imprinting: insights from cancer into epigenetic mechanisms. Semin Cancer Biol 12(5):389–398
Cheung HH, Lee TL, Rennert OM, Chan WY (2009) DNA methylation of cancer genome. Birth Defects Res C Embryo Today 87(4):335–350
Choi S, Myers JN (2008) Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dent Res 87(1):14–32
Hu C, Liu X, Zeng Y et al (2021) DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: mechanism and clinical application. Clin Epigenetics 13(1):166
Pechalrieu D, Etievant C, Arimondo PB (2017) DNA methyltransferase inhibitors in cancer: from pharmacology to translational studies. Biochem Pharmacol 129:1–13
Trerotola M, Relli V, Simeone P, Alberti S (2015) Epigenetic inheritance and the missing heritability. Hum Genomics 9(1):17
Pembrey M, Saffery R, Bygren LO (2014) Human transgenerational responses to early-life experience: potential impact on development, health and biomedical research. J Med Genet 51(9):563–572
Gannon JR, Emery BR, Jenkins TG, Carrell DT (2014) The sperm epigenome: implications for the embryo. Adv Exp Med Biol 791:53–66
Smith ZD, Chan MM, Humm KC et al (2014) DNA methylation dynamics of the human preimplantation embryo. Nature 511(7511):611–615
Gkountela S, Zhang KX, Shafiq TA et al (2015) DNA demethylation dynamics in the human prenatal germline. Cell 161(6):1425–1436
Tang WW, Dietmann S, Irie N et al (2015) A unique gene regulatory network resets the human germline epigenome for development. Cell 161(6):1453–1467
Anway MD, Cupp AS, Uzumcu M, Skinner MK (2005) Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308(5727):1466–1469
Dias BG, Ressler KJ (2014) Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat Neurosci 17(1):89–96
Alabert C, Barth TK, Reverón-Gómez N et al (2015) Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev 29(6):585–590
Annunziato AT (2005) Split decision: what happens to nucleosomes during DNA replication? J Biol Chem 280(13):12065–12068
Alabert C, Groth A (2012) Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol 13(3):153–167
Lumey LH, Stein AD, Kahn HS et al (2007) Cohort profile: the Dutch Hunger Winter families study. Int J Epidemiol 36(6):1196–1204
Heijmans BT, Tobi EW, Stein AD et al (2008) Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA 105(44):17046–17049
Susser E, Neugebauer R, Hoek HW et al (1996) Schizophrenia after prenatal famine. Further evidence Arch Gen Psychiatry 53(1):25–31
Scholte RS, van den Berg GJ, Lindeboom M (2015) Long-run effects of gestation during the Dutch Hunger Winter famine on labor market and hospitalization outcomes. J Health Econ 39:17–30
Painter RC, de Rooij SR, Bossuyt PM et al (2006) Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am J Clin Nutr 84(2):322–7 quiz 466–7
Ravelli GP, Stein ZA, Susser MW (1976) Obesity in young men after famine exposure in utero and early infancy. N Engl J Med 295(7):349–353
Marmot MG, Rose G, Shipley M, Hamilton PJ (1978) Employment grade and coronary heart disease in British civil servants. J Epidemiol Commun Health 32(4):244–9
Marmot M, Brunner E (2005) Cohort profile: the Whitehall II study. Int J Epidemiol 34(2):251–256
Borrell-Carrió F, Suchman AL, Epstein RM (2004) The biopsychosocial model 25 years later: principles, practice, and scientific inquiry. Ann Fam Med 2(6):576–582
Engel GL (1979) The biopsychosocial model and the education of health professionals. Gen Hosp Psychiatry 1(2):156–165
Guidi J, Lucente M, Sonino N, Fava GA (2021) Allostatic load and its impact on health: a systematic review. Psychother Psychosom 90(1):11–27
Naumova OY, Rychkov SY, Kornilov SA et al (2019) Effects of early social deprivation on epigenetic statuses and adaptive behavior of young children: a study based on a cohort of institutionalized infants and toddlers. PLoS ONE 14(3):e0214285
McEwen BS (2000) Allostasis and allostatic load: implications for neuropsychopharmacology. Neuropsychopharmacology 22(2):108–124
Robertson T, Benzeval M, Whitley E, Popham F (2015) The role of material, psychosocial and behavioral factors in mediating the association between socioeconomic position and allostatic load (measured by cardiovascular, metabolic and inflammatory markers). Brain Behav Immun 45:41–49
López JF, Akil H, Watson SJ (1999) Neural circuits mediating stress. Biol Psychiatry 46(11):1461–1471
Armario A, Escorihuela RM, Nadal R (2008) Long-term neuroendocrine and behavioural effects of a single exposure to stress in adult animals. Neurosci Biobehav Rev 32(6):1121–1135
Cuadra G, Zurita A, Lacerra C, Molina V (1999) Chronic stress sensitizes frontal cortex dopamine release in response to a subsequent novel stressor: reversal by naloxone. Brain Res Bull 48(3):303–308
Sotiropoulos I, Cerqueira JJ, Catania C et al (2008) Stress and glucocorticoid footprints in the brain-the path from depression to Alzheimer’s disease. Neurosci Biobehav Rev 32(6):1161–1173
McEwen BS (1998) Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci 840:33–44
McEwen BS, Seeman T (1999) Protective and damaging effects of mediators of stress. Elaborating and testing the concepts of allostasis and allostatic load. Ann NY Acad Sci 896:30–47
Szanton SL, Gill JM, Allen JK (2005) Allostatic load: a mechanism of socioeconomic health disparities? Biol Res Nurs 7(1):7–15
Weaver IC, Champagne FA, Brown SE et al (2005) Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci 25(47):11045–11054
Kussmann M, Affolter M (2009) Proteomics at the center of nutrigenomics: comprehensive molecular understanding of dietary health effects. Nutrition 25(11–12):1085–1093
Fischer N (2020) Infection-induced epigenetic changes and their impact on the pathogenesis of diseases. Semin Immunopathol 42(2):127–130
Hillemacher T, Frieling H, Moskau S et al (2008) Global DNA methylation is influenced by smoking behaviour. Eur Neuropsychopharmacol 18(4):295–298
Szyf M, Weaver IC, Champagne FA et al (2005) Maternal programming of steroid receptor expression and phenotype through DNA methylation in the rat. Front Neuroendocrinol 26(3–4):139–162
Fraga MF, Ballestar E, Paz MF et al (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 102(30):10604–10609
McEwen BS, Gianaros PJ (2011) Stress- and allostasis-induced brain plasticity. Annu Rev Med 62:431–445
Black PH (2003) The inflammatory response is an integral part of the stress response: implications for atherosclerosis, insulin resistance, type II diabetes and metabolic syndrome X. Brain Behav Immun 17(5):350–364
Glaser R, Kiecolt-Glaser JK (2005) Stress-induced immune dysfunction: implications for health. Nat Rev Immunol 5(3):243–251
Tsigos C, Chrousos GP (2002) Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res 53(4):865–871
Mariotti A (2015) The effects of chronic stress on health: new insights into the molecular mechanisms of brain-body communication. Future Sci OA 1(3):FSO23. https://doi.org/10.4155/fso.15.21
Dhabhar FS, McEwen BS (1997) Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav Immun 11(4):286–306
Elenkov IJ, Chrousos GP (1999) Stress Hormones, Th1/Th2 patterns, Pro/anti-inflammatory cytokines and susceptibility to disease. Trends Endocrinol Metab 10(9):359–368
Hilgert JB, Hugo FN, Bandeira DR, Bozzetti MC (2006) Stress, cortisol, and periodontitis in a population aged 50 years and over. J Dent Res 85(4):324–328
Cakmak O, Tasdemir Z, Aral CA et al (2016) Gingival crevicular fluid and saliva stress hormone levels in patients with chronic and aggressive periodontitis. J Clin Periodontol 43(12):1024–1031
Giannopoulou C, Kamma JJ, Mombelli A (2003) Effect of inflammation, smoking and stress on gingival crevicular fluid cytokine level. J Clin Periodontol 30(2):145–153
Johannsen A, Rylander G, Söder B, Asberg M (2006) Dental plaque, gingival inflammation, and elevated levels of interleukin-6 and cortisol in gingival crevicular fluid from women with stress-related depression and exhaustion. J Periodontol 77(8):1403–1409
Felten DL, Felten SY, Carlson SL et al (1985) Noradrenergic and peptidergic innervation of lymphoid tissue. J Immunol 135(2 Suppl):755s-s765
D’Ambrosio F, Caggiano M, Schiavo L et al (2022) Chronic stress and depression in periodontitis and peri-implantitis: a narrative review on neurobiological, neurobehavioral and immune-microbiome interplays and clinical management implications. Dent J (Basel) 10(3):49. https://doi.org/10.3390/dj10030049
Miller GE, Chen E, Sze J et al (2008) A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry 64(4):266–272
de Assis PJ, Freitas FV, Borçoi AR et al (2021) Alcohol consumption, depression, overweight and cortisol levels as determining factors for NR3C1 gene methylation. Sci Rep 11(1):6768
Kaffman A, Meaney MJ (2007) Neurodevelopmental sequelae of postnatal maternal care in rodents: clinical and research implications of molecular insights. J Child Psychol Psychiatry 48(3–4):224–244
Morrison KE, Epperson CN, Sammel MD et al (2017) Preadolescent adversity programs a disrupted maternal stress reactivity in humans and mice. Biol Psychiatry 81(8):693–701
Smart C, Strathdee G, Watson S et al (2015) Early life trauma, depression and the glucocorticoid receptor gene–an epigenetic perspective. Psychol Med 45(16):3393–3410
Algamal M, Pearson AJ, Hahn-Townsend C et al (2021) Repeated unpredictable stress and social isolation induce chronic HPA axis dysfunction and persistent abnormal fear memory. Prog Neuropsychopharmacol Biol Psychiatry 104:110035
Cole SW, Hawkley LC, Arevalo JM et al (2007) Social regulation of gene expression in human leukocytes. Genome Biol 8(9):R189
Bosquet Enlow M, Sideridis G, Chiu YM et al (2019) Associations among maternal socioeconomic status in childhood and pregnancy and hair cortisol in pregnancy. Psychoneuroendocrinology 99:216–224
Clearfield MW, Carter-Rodriguez A, Merali AR, Shober R (2014) The effects of SES on infant and maternal diurnal salivary cortisol output. Infant Behav Dev 37(3):298–304
Miller GE, Cohen S, Ritchey AK (2002) Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol 21(6):531–541
Cole SW (2008) Social regulation of leukocyte homeostasis: the role of glucocorticoid sensitivity. Brain Behav Immun 22(7):1049–1055
Cohen S, Janicki-Deverts D, Doyle WJ et al (2012) Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci USA 109(16):5995–5999
Schwendicke F, Dörfer CE, Schlattmann P et al (2015) Socioeconomic inequality and caries: a systematic review and meta-analysis. J Dent Res 94(1):10–18
Boillot A, El Halabi B, Batty GD et al (2011) Education as a predictor of chronic periodontitis: a systematic review with meta-analysis population-based studies. PLoS ONE 6(7):e21508
Park JB, Han K, Park YG, Ko Y (2016) Association between socioeconomic status and oral health behaviors: the 2008–2010 Korea national health and nutrition examination survey. Exp Ther Med 12(4):2657–2664
Purohit BM, Dawar A, Bansal K et al (2023) Sugar-sweetened beverage consumption and socioeconomic status: a systematic review and meta-analysis. Nutr Health 29(3):465–77
Hiscock R, Bauld L, Amos A et al (2012) Socioeconomic status and smoking: a review. Ann N Y Acad Sci 1248:107–123
Collins SE (2016) Associations between socioeconomic factors and alcohol outcomes. Alcohol Res 38(1):83–94
Gilman SE, Hornig M, Ghassabian A et al (2017) Socioeconomic disadvantage, gestational immune activity, and neurodevelopment in early childhood. Proc Natl Acad Sci USA 114(26):6728–6733
Kingsbury M, Weeks M, MacKinnon N et al (2016) Stressful life events during pregnancy and offspring depression: evidence from a prospective cohort study. J Am Acad Child Adolesc Psychiatry 55(8):709–16.e2
Buss C, Davis EP, Shahbaba B et al (2012) Maternal cortisol over the course of pregnancy and subsequent child amygdala and hippocampus volumes and affective problems. Proc Natl Acad Sci USA 109(20):E1312–E1319
Van den Bergh BRH, van den Heuvel MI, Lahti M et al (2020) Prenatal developmental origins of behavior and mental health: the influence of maternal stress in pregnancy. Neurosci Biobehav Rev 117:26–64
Mountain RV, Zhu Y, Pickett OR et al (2021) Association of maternal stress and social support during pregnancy with growth marks in children’s primary tooth enamel. JAMA Netw Open 4(11):e2129129
McDermott CL, Hilton K, Park AT et al (2021) Early life stress is associated with earlier emergence of permanent molars. Proc Natl Acad Sci U S A 118(24):e2105304118. https://doi.org/10.1073/pnas.2105304118
Colich NL, Rosen ML, Williams ES, McLaughlin KA (2020) Biological aging in childhood and adolescence following experiences of threat and deprivation: a systematic review and meta-analysis. Psychol Bull 146(9):721–764
Renson A, Jones HE, Beghini F et al (2019) Sociodemographic variation in the oral microbiome. Ann Epidemiol 35:73-80.e2
Boyce WT, Den Besten PK, Stamperdahl J et al (2010) Social inequalities in childhood dental caries: the convergent roles of stress, bacteria and disadvantage. Soc Sci Med 71(9):1644–1652
Foley M, Akers HF (2019) Does poverty cause dental caries? Aust Dent J 64(1):96–102
Vasireddy D, Sathiyakumar T, Mondal S, Sur S (2021) Socioeconomic factors associated with the risk and prevalence of dental caries and dental treatment trends in children: a cross-sectional analysis of National Survey of Children’s Health (NSCH) Data, 2016–2019. Cureus 13(11):e19184
Zhang T, Hong J, Yu X et al (2021) Association between socioeconomic status and dental caries among Chinese preschool children: a cross-sectional national study. BMJ Open 11(5):e042908
Moss ME, Beck JD, Kaplan BH et al (1996) Exploratory case-control analysis of psychosocial factors and adult periodontitis. J Periodontol 67(10 Suppl):1060–1069
Barros SP, Offenbacher S (2014) Modifiable risk factors in periodontal disease: epigenetic regulation of gene expression in the inflammatory response. Periodontol 64(1):95–110
Zhang S, Barros SP, Niculescu MD et al (2010) Alteration of PTGS2 promoter methylation in chronic periodontitis. J Dent Res 89(2):133–137
Zhang S, Barros SP, Moretti AJ et al (2013) Epigenetic regulation of TNFA expression in periodontal disease. J Periodontol 84(11):1606–1616
Zhang S, Crivello A, Offenbacher S et al (2010) Interferon-gamma promoter hypomethylation and increased expression in chronic periodontitis. J Clin Periodontol 37(11):953–961
Cardoso FP, Viana MB, Sobrinho AP et al (2010) Methylation pattern of the IFN-gamma gene in human dental pulp. J Endod 36(4):642–646
Cardoso FP, de Faria Amormino SA, Dutra WO et al (2014) Methylation pattern of the CD14 and TLR2 genes in human dental pulp. J Endod 40(3):384–386
de Faria Amormino SA, Arão TC, Saraiva AM et al (2013) Hypermethylation and low transcription of TLR2 gene in chronic periodontitis. Hum Immunol 74(9):1231–1236
Oliveira NF, Damm GR, Andia DC et al (2009) DNA methylation status of the IL8 gene promoter in oral cells of smokers and non-smokers with chronic periodontitis. J Clin Periodontol 36(9):719–725
Andia DC, de Oliveira NF, Casarin RC et al (2010) DNA methylation status of the IL8 gene promoter in aggressive periodontitis. J Periodontol 81(9):1336–1341
Hall GL, Shaw RJ, Field EA et al (2008) p16 Promoter methylation is a potential predictor of malignant transformation in oral epithelial dysplasia. Cancer Epidemiol Biomarkers Prev 17(8):2174–2179
Zhang B, Zhu W, Yang P et al (2011) Cigarette smoking and p16INK4α gene promoter hypermethylation in non-small cell lung carcinoma patients: a meta-analysis. PLoS ONE 6(12):e28882
Saatci C, Caglayan AO, Ozkul Y et al (2009) Detection of p16 promotor hypermethylation in “Maras powder” and tobacco users. Cancer Epidemiol 33(1):47–50
Cao J, Zhou J, Gao Y et al (2009) Methylation of p16 CpG island associated with malignant progression of oral epithelial dysplasia: a prospective cohort study. Clin Cancer Res 15(16):5178–5183
Bhol CS, Mishra SR, Patil S et al (2022) PAX9 reactivation by inhibiting DNA methyltransferase triggers antitumor effect in oral squamous cell carcinoma. Biochim Biophys Acta Mol Basis Dis 1868(9):166428
Ari G, Cherukuri S, Namasivayam A (2016) Epigenetics and periodontitis: a contemporary review. J Clin Diagn Res 10(11):Ze07-ze9
Cantley MD, Bartold PM, Marino V et al (2011) Histone deacetylase inhibitors and periodontal bone loss. J Periodontal Res 46(6):697–703
Larsson L, Castilho RM, Giannobile WV (2015) Epigenetics and its role in periodontal diseases: a state-of-the-art review. J Periodontol 86(4):556–568
Hui T, Peng A, Zhao Y et al (2014) EZH2, a potential regulator of dental pulp inflammation and regeneration. J Endod 40(8):1132–8
Angiolilli C, Kabala PA, Grabiec AM et al (2017) Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann Rheum Dis 76(1):277–285
Christensen DP, Dahllöf M, Lundh M et al (2011) Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol Med 17(5–6):378–390
Ren Y, Su X, Kong L et al (2016) Therapeutic effects of histone deacetylase inhibitors in a murine asthma model. Inflamm Res 65(12):995–1008
Li X, Zhang Y, Jiang Y et al (2017) Selective HDAC inhibitors with potent oral activity against leukemia and colorectal cancer: design, structure-activity relationship and anti-tumor activity study. Eur J Med Chem 134:185–206
Nagumo T, Takaoka S, Yoshiba S et al (2009) Antitumor activity of suberoylanilide hydroxamic acid against human oral squamous cell carcinoma cell lines in vitro and in vivo. Oral Oncol 45(9):766–770
Ungerstedt JS, Sowa Y, Xu WS et al (2005) Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA 102(3):673–678
Konsoula Z, Velena A, Lee R et al (2011) Histone deacetylase inhibitor: antineoplastic agent and radiation modulator. Adv Exp Med Biol 720:171–179
Herold C, Ganslmayer M, Ocker M et al (2002) The histone-deacetylase inhibitor trichostatin A blocks proliferation and triggers apoptotic programs in hepatoma cells. J Hepatol 36(2):233–240
Daśko M, de Pascual-Teresa B, Ortín I, Ramos A (2022) HDAC inhibitors: innovative strategies for their design and applications. Molecules 27(3):715. https://doi.org/10.3390/molecules27030715
Duncan HF, Smith AJ, Fleming GJ, Cooper PR (2011) HDACi: cellular effects, opportunities for restorative dentistry. J Dent Res 90(12):1377–1388
Kearney M, Cooper PR, Smith AJ, Duncan HF (2018) Epigenetic approaches to the treatment of dental pulp inflammation and repair: opportunities and obstacles. Front Genet 9:311
Wang T, Liu H, Ning Y, Xu Q (2014) The histone acetyltransferase p300 regulates the expression of pluripotency factors and odontogenic differentiation of human dental pulp cells. PLoS ONE 9(7):e102117
Townsend GC, Richards L, Hughes T et al (2005) Epigenetic influences may explain dental differences in monozygotic twin pairs. Aust Dent J 50(2):95–100
Brook AH (2009) Multilevel complex interactions between genetic, epigenetic and environmental factors in the aetiology of anomalies of dental development. Arch Oral Biol 54(1):S3-17
Funding
Open Access funding provided by the IReL Consortium
Author information
Authors and Affiliations
Contributions
Dr. Sakr Khalid: writing—original draft preparation and writing—review and editing; Dr. Michaela Kearney: writing—review and editing; Dr. David McReynolds: writing—review and editing, conceptualisation, project administration, and supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This review article is expanded from a final year dissertation presented at Dublin Dental University Hospital in fulfilment of the award of an undergraduate Dental Science degree.
Clinical relevance
It is thought that the biological consequences of social adversity can get as deep as leaving imprints on the epigenome that can trigger oral disease, which may be passed down inter-generationally.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khalid, S., Kearney, M. & McReynolds, D.E. Can social adversity alter the epigenome, trigger oral disease, and affect future generations?. Ir J Med Sci (2024). https://doi.org/10.1007/s11845-024-03697-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11845-024-03697-3