
Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare autosomal dominant genetic condition characterised by progressive extra-skeletal bone formation in connective tissues. Over time, heterotopic ossification entombs patients within a second skeleton, drastically impairing their mobility and autonomy. Mutations in the ACVR1 gene have been identified as the cause of FOP. The single nucleotide missense mutation in ACVR1, c.617G > A, causes a single amino acid substitution, p.R206H, and is found in >90% of all patients. Heterotopic bone formation in FOP mimics embryonic skeletal endochondral ossification, with cartilage forming after fibroproliferative tissue condensation as an intermediate stage prior to osteogenesis and tissue ossification. In contrast to normal embryonic endochondral ossification, heterotopic ossification in FOP involves an inflammatory phase that precedes cartilage and bone formation. New insights into the mechanisms of action of heterotopic bone formation in FOP have led to the discovery of new potential treatment targets including inhibitors of BMP signalling, activin A inhibitors, and mTOR inhibitors. This review summarises the current knowledge on mutations causing FOP, as well as the molecular basis of heterotopic ossification and the therapeutic options that result from these discoveries.
Zusammenfassung
Fibrodysplasia ossificans progressiva (FOP) ist eine äußerst seltene Erkrankung, die autosomal-dominant vererbt wird und zu einer extraskelettalen Ossifikation von Bindegewebe führt. Die über die Zeit akkumulierenden Ossifikationen zwingen Patienten in ein zweites Skelett, welches ihre Mobilität und Autonomie drastisch einschränkt. Als Ursache dieser Erkrankung wurden Mutationen im ACVR1-Gen identifiziert. Die am häufigsten auftretende Mutation ist eine Punktmutation, c.617G > A, welche eine Substitution der Aminosäure p.R206H hervorruft. Dies führt zu einer veränderten Signalweitergabe durch den ACVR1-Rezeptor, welcher Teil des osteogenen bone morphogenetic protein (BMP)-Signalwegs ist und Ossifikationen auch außerhalb des Knochens verursacht. Durch die kürzlich erworbenen Erkenntnisse über die der heterotopen Ossifikation bei FOP zugrunde liegenden Mechanismen konnten neue potenzielle Therapiestrategien entwickelt werden. Diese umfassen den Einsatz von Hemmern des BMP-Signalwegs sowie Activin A‑ und mTOR-Inhibitoren. Dieser Übersichtsartikel fasst die aktuellen Erkenntnisse zur Pathogenese der FOP zusammen und erläutert die sich daraus ergebenden neuen Therapieansätze.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Fibrodysplasia ossificans progressiva (FOP, OMIM #135100) is an autosomal dominant genetic disease that has an estimated prevalence of 1 in 1–2 million and affects individuals regardless of gender, race, ethnicity or geographical location. In France, which has well-maintained national registries for rare diseases, the prevalence was determined to be 1 in 1.36 million [1]. Most cases of FOP are due to de novo mutations in the activin receptor 1 (ACVR1) gene, which is a type I bone morphogenetic protein (BMP) receptor [2].
Affected patients suffer from excessive heterotopic ossification in skeletal muscle, tendons, ligaments, and fascia that episodically occur in a non-predictable manner and is often preceded by flare-ups, which are soft-tissue swellings that are associated with pain and inflammation. In addition, all patients with the classical clinical presentation of FOP display characteristic malformations of the great toes, which are often short, deviated, and monophalangic, but otherwise patients appear normal at birth.
The sporadically occurring flare-ups usually start within the first decade of life. Flare-ups may also occur as a result of minor trauma such as intramuscular immunisations, falls, bruises, and mandibular blocks for dental work. In particular, surgeries to remove or sample heterotopic bone must be avoided, as this tissue trauma provokes further extensive and painful ossifications [3]. Interestingly, heterotopic ossification in FOP patients appears to follow a specific pattern, starting from cranial to caudal, from proximal to distal, from dorsal to ventral, and from axial to appendicular [4]. Some other muscles, including the diaphragm, tongue, and extra-ocular muscles, as well as the cardiac muscle and smooth muscles, are spared from heterotopic ossifications. During a patient’s lifetime, heterotopic ossification continues to irreversibly accumulate, first leading to limitations of the daily activities of life, such as brushing teeth or combing hair, and later leading to progressive loss of mobility and autonomy. Patients are often bound to a wheel-chair and dependent on lifelong assistance by their second to third decade of life. Based on a cross-sectional study of 371 patients, the median age of survival was estimated to be about 56 years [5]. Death often results from complications of thoracic insufficiency syndrome, such as pneumonia or right-sided heart failure. To date, there have been no specific therapies for FOP.
Clinical presentation
Besides the defining hallmarks of classic FOP, which are the formation of postnatal heterotopic bone and malformation of the great toes, other skeletal anomalies have been reported, particularly in the thumbs and the cervical spine [6]. Stiffness of the neck due to congenital malformations, with tall narrow vertebral bodies, enlarged pedicles and large spinous processes, along with variable fusion of the posterior part, typically the facet joints of the segments C2–C7, is often an early finding in patients and precedes the development of heterotopic bone. Heterotopic ossifications occurring later may further impair mobility of the cervical spine [7]. Many patients develop severe scoliosis, which later in life can lead to thoracic insufficiency syndrome. In addition to the deformities in the vertebrae, further skeletal manifestations are short broad femoral necks, malformed thumbs and clinodactyly at the fifth digit, as well as osteochondromas, typically at the proximal medial tibia. Other reported anomalies include hearing loss, alopecia, mild mental retardation, cataracts, glaucoma, short stature, and some common facial characteristics such as reduced mandible and retrognathia, underdeveloped supra-orbital ridge, low-set ears and elongation of the lateral pterygoid plate [3, 8,9,10].
In 2009, a large study with 112 FOP patients was conducted and a classification of FOP was attempted [8]. A small proportion of FOP patients (3%) have been described with phenotypic manifestations that are unusual for classic FOP. These either occur in addition to classic FOP (termed FOP plus) or present variations in one or both of the defining features (termed FOP variants). Manifestations described in that context include persistence of primary teeth, absence of finger and toenails, sparse hair, retinal detachment, as well as mild cognitive impairment and cerebellar abnormalities [8, 11]. Building on that, a recent MRI study of 13 patients (11 with the R206H variant, 2 with non-R206H mutations) indicated various changes in the brain of FOP patients, including small lesions in the dorsal medulla and ventral pons, minor dysmorphism of the brainstem, and an enlarged origin of the vestibulocochlear nerves. However, all of these alterations were asymptomatic [12].
Diagnosis
In 2005, a survey was performed with 269 patient-members of the International FOP Association, with about half of them completing the questionnaire [13]. These patients were from 25 different countries and accounted for about half of all known FOP patients worldwide at that time. This study revealed that 87% of FOP patients were misdiagnosed at first, most frequently with cancer. It took a median of 4 years from the onset of the first symptoms until a correct diagnosis. Unnecessary invasive procedures, including biopsies, were performed in 67% of the patients and 68% received inappropriate therapies. In a more recent survey launched by the International FOP Association, with 196 enrolled patients, a mean age of 5.4 years was reported when patients noticed the first symptoms and 7.5 years when they were first diagnosed with FOP [14]. Provided that clinicians are aware of the cardinal symptoms of classic FOP, which is too often inadequately described in medical text books [13], the vast majority of cases can be correctly diagnosed if soft-tissue lesions are associated with the malformations of the great toe, even without radiography or genetic testing [15].
Genetics
All mutations associated with FOP have so far been mapped to the ACVR1 gene and can be transmitted by autosomal dominant inheritance. Most cases of FOP occur because of spontaneous new mutations, although some familial cases have been reported [16,17,18]. Even though over 90% of all patients have the recurring missense mutation c.617G > A; R206H, there is considerable phenotypic heterogeneity, likely through contributions by the varied genetic backgrounds of individual persons. In addition to genetic influences, the environment and lifestyle of the patient have significant influence, as demonstrated by the differences in disease progression among pairs of identical twins with FOP [19].
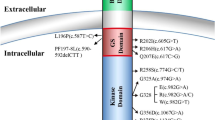
The R206H missense mutation is located in the glycine serine (GS)-rich domain of ACVR1, which is a critical site for binding and activation by ligand-activated BMP type II receptors and for FKBP12, which is an inhibitory protein that prevents the leakiness of BMP type I receptor signalling in the absence of ligand. In silico modelling supports the notion that the R206H mutation leads to a conformational change of the receptor [16]; such a structural change could weaken FKBP12 binding, bypass a requirement for ligand binding, and/or increase responsiveness to atypical ligands, thereby modulating downstream signalling events [2, 20,21,22].
In addition to the R206H mutation, which has been reported in over 200 patients, about 15 other mutations in the GS domain and the protein kinase domain of ACVR1 have been identified in FOP patients. These have been summarised in Huning et al. [23] and were also predicted by protein structure homology modelling to activate the ACVR1 protein and enhance receptor signalling [8]. With regard to the genotype–phenotype correlation, the p.G328W and p.G328E mutations show early disease onset and severe progression of heterotopic ossification without being influenced by trauma, whereas patients with the p.G356D mutation seem to have a later disease onset, but with a quick progression. Patients with p.G328R mutations seem to have a milder disease course, with the ossifications being closely associated with surgical intervention/trauma. The amino acid changes p.R258S, p.R375P, and p.L196P also seem to be associated with a milder disease course that is not related to trauma or great toe malformation [8, 24, 25]. In many cases, the number of patients with specific mutations is low; thus, these associations must be interpreted with caution. More recently, studies examining the activation of the BMP signalling pathway by FOP variant ACVR1 mutations, have supported the notion that, in general, kinase domain mutations and GS domain mutations might activate signalling through different molecular mechanisms and responsiveness to ligands [26, 27].
Current therapy
Current therapy for FOP patients includes avoiding injuries or trauma to prevent flare-ups and subsequent ossification. Once trauma-induced or spontaneous flare-ups and ossifications occur, glucocorticoids are used to alleviate pain and swelling, and may also be used preventatively before inevitable invasive procedures, especially in the jaw [6, 28, 29]. The successful use of non-steroidal anti-inflammatory drugs has also been reported in single cases, but there is no evidence showing efficacy of these drugs to modulate disease activity. Surgical interventions to remove heterotopic bone should be avoided, as it usually counterproductive and provokes further ossifications. Overall, the options for the treatment of FOP are limited at present. Guidelines for FOP treatment were established by an international clinical consortium on FOP in 2011 and updated recently to help physicians to treat their patients effectively [30, 31]. This is particularly important, as many aspects are unique when it comes to caring for patients with FOP (e.g. prophylaxis against pneumonia and influenza, dental visits, anaesthesia etc.). Specific recommendations are also provided in Pignolo et al. [6, 31].
Future therapeutic strategies based on the underlying mechanisms of FOP
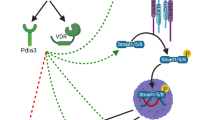
After the identification of ACVR1 as the causative gene for FOP, the door opened to investigations into the mechanisms of action of the disease. ACVR1 is a BMP type I receptor that is usually activated upon binding of BMP ligands. Together with BMP type II receptors, ACVR1 activates canonical Smad1/5/8 signalling, but also non-canonical BMP signalling, such as through MAPK, has been identified. The gain-of-function mutations in the GS domain of the ACVR1 lead to two distinct features of the protein: first, they result in the constitutive activation of the receptor, meaning that it can activate downstream Smad signalling, even in the absence of ligand [32,33,34], and second, it may alter the binding properties of ligands. As such, activin A, which normally transduces its signal via Smad2/3 through the ACVR2/ACVR1B complex, gains enhanced ability to activate the mutated ACVR1, induce Smad1/5/8 signalling, and promote heterotopic ossification similar to typical osteogenic TGF-beta family members (i.e. BMP2, BMP4, BMP6, BMP7, BMP9) [22, 27, 35, 36]. Besides Smad1/5/8 signalling, mutated ACVR1 has also been shown to induce mTOR and phosphatidylinositol 3‑kinase α (PI3Kα) signalling [37, 38]. Based on these findings, various treatment strategies have been explored, either blocking excessive BMP/Smad signalling or inhibiting activin A or activin A‑induced downstream signalling. These are discussed in more detail in the following sections.
Blocking hyperactive BMP signalling
Various approaches to blocking excessive BMP signalling have been examined. These include RNA interference approaches, blocking the activity of the mutant R206H ACVR1 receptor [39, 40]. These approaches decreased the increased Smad1/5/8 signalling in mesenchymal cells derived from FOP patients or in muscle and bone cells overexpressing mutant ACVR1. Other approaches were directed at blocking ACVR1, which belongs to the family of BMP type I receptors using dorsomorphin or other synthetic molecules (LDN-193189) [41, 42]. All of these approaches significantly reduced heterotopic ossification in mouse models of activated ACVR1 activity. However, these approaches block all three BMP type I receptors (BMPR1A/ALK3, BMPR1B/ALK6, ACVR1/ALK2), and thus do not confer specificity to the FOP mutations, possibly causing adverse effects.
Additional approaches include the inhibition of ligand binding to ACVR1. As such, ligand traps have been used, including ACVR2B-Fc and ACVR2A-Fc. These ligand traps are made of the extracellular domain of ACVR2B and ACVR2A and are fused to the constant region of IgG1 [43]. Both molecules blocked heterotopic ossification in mice, indicating that one or more of the ligands that are blocked by these traps are required to induce heterotopic ossification in FOP [21]. In another study, the BMP binding properties of transferrin receptor 2 were used as a ligand trap and also showed efficacy in reducing BMP-2-induced heterotopic ossification [44]. However, this latter approach requires further validation in mouse models of FOP.
Finally, one of the most promising approaches in blocking BMP signalling is palovarotene, a retinoic acid receptor agonist that has been identified to indirectly block Smad1/5/8 signalling by activating the retinoid signalling pathway through RARγ receptors and inhibiting chondrogenesis and subsequent heterotopic endochondral ossification [45, 46]. This agent showed great success in reducing the volume of new heterotopic ossifications in patients with FOP (>70%) in a phase II trial and is currently being tested in the MOVE trial (NCT03312634), a phase III clinical study. Even though palovarotene is generally well tolerated and showed the expected side effect profile of retinoids (e.g. mucocutaneous side effects, liver toxicity and abnormalities of serum lipid profiles), high levels of daily intraperitoneal treatment with palovarotene in juvenile animals carrying the R206H mutation suggested synovial joint overgrowth and long bone growth plate ablation [47]. However, lower doses of an orally administered RARγ agonist inhibited HO formation in juvenile Acvr1R206H/+ mice without these negative effects and additionally partially rescued growth plate defects induced by the FOP mutation [46]. Long-term follow-ups and monitoring of patients will be necessary to fully evaluate the spectrum of effects of palovarotene on children and adults with FOP.
Inhibiting activin A signalling
The discovery that the R206H mutation in the ACVR1 gene leads to increased Smad1/5/8 responsiveness to activin A has led to the development of neutralising antibodies for activin A as a treatment for heterotopic ossification in FOP [21]. This approach been tested in cells carrying the R206H ACVR1 mutation and in mouse models of FOP and showed great efficacy in reducing heterotopic ossification. Moreover, pSmad1/5/8 signalling by activin A has also been demonstrated in induced pluripotent stem cells (iPS) from FOP patients [22]. This antibody is now also under investigation in clinical trials (REGN2477, NCT03188666). Activin A is important in several cell and tissues, including immune cells and the reproductive system; therefore, as for palovarotene, careful monitoring and patient follow-up will provide information about the long-term effects of this treatment.
Additional treatment strategies
Another strategy to block aberrant ACVR1 signalling is focused on mTOR signalling. This pathway was identified in a large chemical screen on FOP-iPSc, which shows increased chondrogenic and osteogenic potential in vitro [48]. Blocking mTOR signalling with rapamycin prevented activin A‑induced chondrogenic and osteogenic differentiation and inhibited heterotopic bone formation in vivo [37]. In addition, BYL719, an inhibitor of PI3Kα, inhibited Smad, Akt and mTOR signalling in cells or mice carrying ACVR1 R206H or Q207D mutations [38]. Thus, these studies highlight the potential of blocking mTOR signalling for the treatment of FOP. However, mTOR signalling exerts direct functions on the skeleton that will need to be considered in the safety profile [49].
Finally, besides directly targeting aberrant pathways, the inflammatory phase of FOP is also a critical mechanism preceding heterotopic ossification and thus has been examined as a therapeutic intervention point. In particular, lesions are frequently heavily infiltrated with monocytes, macrophages and mast cells that may enhance the inflammatory immune response. Mast cells and macrophages from FOP patients have been shown to have an increased production of cytokines (IL‑3, IL‑7, IL‑8 and IL-10) and chemokines (CCL5, CCR7 and CXCL10), as well as enhanced TGF-beta, NF-κB and MAPK signalling [50, 51]. Depletion of macrophages and mast cells decreased heterotopic ossification in a mouse model of FOP, suggesting that this might be an effective approach to halting ossification in FOP patients [51].
Considering that particularly the early, hypoxic inflammatory stages of FOP flare-ups involve—amongst others—activation of HIF1-alpha, c‑KIT, PDGFR-alpha and multiple MAP kinases, an off-label attempt has been made in a small series of seven children using the tyrosine kinase inhibitor imatinib, basically developed and approved for chronic myeloid leukaemia. Although not controlled study data, the results support the notion that treatment was well-tolerated and a decrease in the intensity of flare-ups was achieved in six of the patients [52]. Another approach that is being made concerns the development of a small molecule inhibitor with sufficient specificity for the ALK2 receptor to control undesired receptor activity [53].
Conclusions
Following the identification of mutations in ACVR1 as the cause of FOP in 2006, we are now beginning to gain important insights into the molecular mechanism underlying this debilitating disease. In light of the rarity and severity of the disease, the robust interest in understanding its mechanisms of action and the great efforts that are being undertaken to develop new therapeutic approaches are highly promising. Many are working to translate these efforts into effective therapies that will benefit FOP patients, allowing them to conduct simple actions of daily life and to maintain mobility and autonomous living throughout their lives.
References
Baujat G, Choquet R, Bouee S, Jeanbat V, Courouve L, Ruel A, Michot C, Le Quan Sang KH, Lapidus D, Messiaen C, Landais P, Cormier-Daire V (2017) Prevalence of fibrodysplasia ossificans progressiva (FOP) in France: an estimate based on a record linkage of two national databases. Orphanet J Rare Dis 12:123
Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38:525–527
Kaplan FS, Le Merrer M, Glaser DL, Pignolo RJ, Goldsby RE, Kitterman JA, Groppe J, Shore EM (2008) Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol 22:191–205
Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS (1993) The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J Bone Joint Surg Am 75:215–219
Kaplan FS, Zasloff MA, Kitterman JA, Shore EM, Hong CC, Rocke DM (2010) Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J Bone Joint Surg Am 92:686–691
Pignolo RJ, Shore EM, Kaplan FS (2013) Fibrodysplasia ossificans progressiva: diagnosis, management, and therapeutic horizons. Pediatr Endocrinol Rev 10(2):437–448
Schaffer AA, Kaplan FS, Tracy MR, O’Brien ML, Dormans JP, Shore EM, Harland RM, Kusumi K (2005) Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel-Feil syndrome: clues from the BMP signaling pathway. Spine (Phila Pa 1976) 30:1379–1385
Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, Delai P, Fastnacht-Urban E, Forman SJ, Gillessen-Kaesbach G, Hoover-Fong J, Koster B, Pauli RM, Reardon W, Zaidi SA, Zasloff M, Morhart R, Mundlos S, Groppe J, Shore EM (2009) Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat 30:379–390
Hammond P, Suttie M, Hennekam RC, Allanson J, Shore EM, Kaplan FS (2012) The face signature of fibrodysplasia ossificans progressiva. Am J Med Genet 158A:1368–1380 (Part A)
Carvalho DR, Farage L, Martins BJ, Speck-Martins CE (2011) Craniofacial findings in fibrodysplasia ossificans progressiva: computerized tomography evaluation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 111:499–502
Shore EM (2012) Fibrodysplasia ossificans progressiva: a human genetic disorder of extraskeletal bone formation, or—how does one tissue become another? Wiley Interdiscip Rev Dev Biol 1:153–165
Severino M, Bertamino M, Tortora D, Morana G, Uccella S, Bocciardi R, Ravazzolo R, Rossi A, Di Rocco M (2016) Novel asymptomatic CNS findings in patients with ACVR1/ALK2 mutations causing fibrodysplasia ossificans progressiva. J Med Genet 53:859–864
Kitterman JA, Kantanie S, Rocke DM, Kaplan FS (2005) Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 116:e654–661
Mantick N, Bachman E, Baujat G, Brown M, Collins O, De Cunto C, Delai P, Eekhoff M, Zum Felde R, Grogan DR, Haga N, Hsiao E, Kantanie S, Kaplan F, Keen R, Milosevic J, Morhart R, Pignolo R, Qian X, di Rocco M, Scott C, Sherman A, Wallace M, Williams N, Zhang K, Bogard B (2018) The FOP connection registry: design of an international patient-sponsored registry for fibrodysplasia ossificans progressiva. Bone 109:285–290
Kaplan FS, Xu M, Glaser DL, Collins F, Connor M, Kitterman J, Sillence D, Zackai E, Ravitsky V, Zasloff M, Ganguly A, Shore EM (2008) Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics 121:e1295–1300
Janoff HB, Muenke M, Johnson LO, Rosenberg A, Shore EM, Okereke E, Zasloff M, Kaplan FS (1996) Fibrodysplasia ossificans progressiva in two half-sisters: evidence for maternal mosaicism. Am J Med Genet 61:320–324
Kaplan FS, McCluskey W, Hahn G, Tabas JA, Muenke M, Zasloff MA (1993) Genetic transmission of fibrodysplasia ossificans progressiva. Report of a family. J Bone Joint Surg Am 75:1214–1220
Rogers JG, Chase GA (1979) Paternal age effect in fibrodysplasia ossificans progressiva. J Med Genet 16:147–148
Hebela NM, Shore EM, Kaplan FS (2005) Three pairs of monozygotic twins with fibrodysplasia ossificans progressiva. Clinical reviews in bone and mineral. Metabolism 3–4:205–208
Groppe JC, Shore EM, Kaplan FS (2007) Functional modeling of the ACVR1 (R206H) mutation in FOP. Clin Orthop Relat Res 462:87–92
Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, Wen X, Nannuru KC, Jimenez J, Xie L, Das N, Makhoul G, Chernomorsky R, D’Ambrosio D, Corpina RA, Schoenherr CJ, Feeley K, Yu PB, Yancopoulos GD, Murphy AJ, Economides AN (2015) ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med 7:303ra137
Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, Sekiguchi K, Shibata M, Nagata S, Matsuda S, Toguchida J (2015) Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci U S A 112:15438–15443
Huning I, Gillessen-Kaesbach G (2014) Fibrodysplasia ossificans progressiva: clinical course, genetic mutations and genotype-phenotype correlation. Mol Syndromol 5:201–211
Bocciardi R, Bordo D, Di Duca M, Di Rocco M, Ravazzolo R (2009) Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements. Eur J Hum Genet 17:311–318
Nakahara Y, Katagiri T, Ogata N, Haga N (2014) ACVR1 (587T>C) mutation in a variant form of fibrodysplasia ossificans progressiva: second report. Am J Med Genet 164A:220–224 (Part A)
Chaikuad A, Alfano I, Kerr G, Sanvitale CE, Boergermann JH, Triffitt JT, von Delft F, Knapp S, Knaus P, Bullock AN (2012) Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem 287:36990–36998
Haupt J, Xu M, Shore EM (2018) Variable signaling activity by FOP ACVR1 mutations. Bone 109:232–240
Pignolo RJ, Bedford-Gay C, Liljesthrom M, Durbin-Johnson BP, Shore EM, Rocke DM, Kaplan FS (2016) The natural history of flare-ups in fibrodysplasia ossificans progressiva (FOP): a comprehensive global assessment. J Bone Miner Res 31:650–656
Rhen T, Cidlowski JA (2005) Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 353:1711–1723
Kaplan FS et al (2019) The medical management of fibrodysplasia ossificans progressiva: current treatment considerations. Proc Intl Clin Council FOP 1:1–111. https://www.fopsverige.se/wp-content/uploads/2019/03/Medicinska-riktlinjer-f%C3%B6r-FOP-2019-GUIDELINES-3.8.19-FINAL-1.pdf
Kaplan FS, Mukaddam AM, Baujat G, Brown M, Cali A, Cho T‑J, Crowe C, De Cunto C, Delai P, Diecidue R, Di Rocco M, Eekhoff EMW, Friedman C, Grunwald Z, Haga N, Hsiao E, Keen R, Kitterman J, Levy C, Morhart R, Netelenbos C, Scott C, Shore EM, Zasloff M, Zhang K, Pignolo RJ (2019) The medical management of fibrodysplasia ossificans progressiva: current treatment considerations. Proc Intl Clin Council FOP 1:1–111
Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, Mundlos S, Seemann P, Kaplan FS, Mullins MC, Shore EM (2009) The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest 119:3462–3472
Fukuda T, Kanomata K, Nojima J, Kokabu S, Akita M, Ikebuchi K, Jimi E, Komori T, Maruki Y, Matsuoka M, Miyazono K, Nakayama K, Nanba A, Tomoda H, Okazaki Y, Ohtake A, Oda H, Owan I, Yoda T, Haga N, Furuya H, Katagiri T (2008) A unique mutation of ALK2, G356D, found in a patient with fibrodysplasia ossificans progressiva is a moderately activated BMP type I receptor. Biochem Biophys Res Commun 377:905–909
Song G‑A, Kim H‑J, Woo K‑M, Baek J‑H, Kim G‑S, Choi J‑Y, Ryoo H‑M (2010) Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem 285:22542–22553
Hatsell SJ, Idone V, Wolken DMA, Huang L, Kim HJ, Wang L, Wen X, Nannuru KC, Jimenez J, Xie L, Das N, Makhoul G, Chernomorsky R, D’Ambrosio D, Corpina RA, Schoenherr CJ, Feeley K, Yu PB, Yancopoulos GD, Murphy AJ, Economides AN (2015) ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med 7:303ra137
Wang H, Shore EM, Pignolo RJ, Kaplan FS (2018) Activin A amplifies dysregulated BMP signaling and induces chondro-osseous differentiation of primary connective tissue progenitor cells in patients with fibrodysplasia ossificans progressiva (FOP). Bone 109:218–224
Hino K, Horigome K, Nishio M, Komura S, Nagata S, Zhao C, Jin Y, Kawakami K, Yamada Y, Ohta A, Toguchida J, Ikeya M (2017) Activin‑A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J Clin Invest 127:3339–3352
Valer JA, Sanchez-de-Diego C, Gamez B, Mishina Y, Rosa JL, Ventura F (2019) Inhibition of phosphatidylinositol 3‑kinase α (PI3Kα) prevents heterotopic ossification. EMBO Mol Med 11(9):e10567. https://doi.org/10.15252/emmm.201910567
Kaplan J, Kaplan FS, Shore EM (2012) Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele-specific targeting. Gene Ther 19:786–790
Shi S, Cai J, de Gorter DJ, Sanchez-Duffhues G, Kemaladewi DU, Hoogaars WM, Aartsma-Rus A, ’t Hoen PA, ten Dijke P (2013) Antisense-oligonucleotide mediated exon skipping in activin-receptor-like kinase 2: inhibiting the receptor that is overactive in fibrodysplasia ossificans progressiva. PLoS One 8:e69096
Fukuda T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, Noguchi Y, Iwakiri K, Kondo T, Kurose J, Endo K, Awakura T, Fukushi J, Nakashima Y, Chiyonobu T, Kawara A, Nishida Y, Wada I, Akita M, Komori T, Nakayama K, Nanba A, Maruki Y, Yoda T, Tomoda H, Yu PB, Shore EM, Kaplan FS, Miyazono K, Matsuoka M, Ikebuchi K, Ohtake A, Oda H, Jimi E, Owan I, Okazaki Y, Katagiri T (2009) Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem 284:7149–7156
Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD (2008) BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med 14:1363–1369
Sako D, Grinberg AV, Liu J, Davies MV, Castonguay R, Maniatis S, Andreucci AJ, Pobre EG, Tomkinson KN, Monnell TE, Ucran JA, Martinez-Hackert E, Pearsall RS, Underwood KW, Seehra J, Kumar R (2010) Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J Biol Chem 285:21037–21048
Rauner M, Baschant U, Roetto A, Pellegrino RM, Rother S, Salbach-Hirsch J, Weidner H, Hintze V, Campbell G, Petzold A, Lemaitre R, Henry I, Bellido T, Theurl I, Altamura S, Colucci S, Muckenthaler MU, Schett G, Komla Ebri D, Bassett JHD, Williams GR, Platzbecker U, Hofbauer LC (2019) Transferrin receptor 2 controls bone mass and pathological bone formation via BMP and Wnt signaling. Nat Metab 1:111–124
Shimono K, Tung WE, Macolino C, Chi AH, Didizian JH, Mundy C, Chandraratna RA, Mishina Y, Enomoto-Iwamoto M, Pacifici M, Iwamoto M (2011) Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists. Nat Med 17:454–460
Chakkalakal SA, Uchibe K, Convente MR, Zhang D, Economides AN, Kaplan FS, Pacifici M, Iwamoto M, Shore EM (2016) Palovarotene inhibits heterotopic ossification and maintains limb mobility and growth in mice with the human ACVR1(R206H) fibrodysplasia ossificans progressiva (FOP) mutation. J Bone Miner Res 31:1666–1675
Lees-Shepard JB, Yamamoto M, Biswas AA, Stoessel SJ, Nicholas SE, Cogswell CA, Devarakonda PM, Schneider MJ Jr., Cummins SM, Legendre NP, Yamamoto S, Kaartinen V, Hunter JW, Goldhamer DJ (2018) Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat Commun 9:471
Matsumoto Y, Hayashi Y, Schlieve CR, Ikeya M, Kim H, Nguyen TD, Sami S, Baba S, Barruet E, Nasu A, Asaka I, Otsuka T, Yamanaka S, Conklin BR, Toguchida J, Hsiao EC (2013) Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet J Rare Dis 8:190
Dai Q, Xu Z, Ma X, Niu N, Zhou S, Xie F, Jiang L, Wang J, Zou W (2017) mTOR/Raptor signaling is critical for skeletogenesis in mice through the regulation of Runx2 expression. Cell Death Differ 24:1886–1899
Barruet E, Morales BM, Cain CJ, Ton AN, Wentworth KL, Chan TV, Moody TA, Haks MC, Ottenhoff TH, Hellman J, Nakamura MC, Hsiao EC (2018) NF-κB/MAPK activation underlies ACVR1-mediated inflammation in human heterotopic ossification. JCI Insight 3(22):122958. https://doi.org/10.1172/jci.insight.122958
Convente MR, Chakkalakal SA, Yang E, Caron RJ, Zhang D, Kambayashi T, Kaplan FS, Shore EM (2018) Depletion of mast cells and macrophages impairs heterotopic ossification in an Acvr1(R206H) mouse model of fibrodysplasia ossificans progressiva. J Bone Miner Res 33:269–282
Kaplan FS, Andolina JR, Adamson PC, Teachey DT, Finklestein JZ, Ebb DH, Whitehead B, Jacobs B, Siegel DM, Keen R, Hsiao E, Pignolo RJ (2018) Early clinical observations on the use of imatinib mesylate in FOP: a report of seven cases. Bone 109:276–280
Williams E, Bullock AN (2018) Structural basis for the potent and selective binding of LDN-212854 to the BMP receptor kinase ALK2. Bone 109:251–258
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
M. Rauner, L. Seefried and E. Shore declare that they have no competing interests.
For this article no studies with human participants or animals were performed by any of the authors. All studies performed were in accordance with the ethical standards indicated in each case.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rauner, M., Seefried, L. & Shore, E. Genetics and future therapy prospects of fibrodysplasia ossificans progressiva. medgen 31, 391–396 (2019). https://doi.org/10.1007/s11825-019-00279-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11825-019-00279-y