
Abstract
The flame lily, Gloriosa superba L., is one of the two primary sources of the anti-inflammatory drug, colchicine. Previous studies have shown that a higher level of colchicine production occurs in the rhizomes than in leaves and roots. Earlier precursor feeding and transcriptome analysis of G. superba have provided a putative pathway and candidate genes involved in colchicine biosynthesis. Comparative analysis of expression levels of candidate pathway genes in different tissues of G. superba using quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) can reveal highly expressed genes in the rhizome compared to other tissues which could suggest roles of the gene products in colchicine biosynthesis. Normalization is an important step in effectively analyzing differential gene expression by qRT-PCR with broader applications. The current study selected candidate reference genes from the transcriptome datasets and analyzed them to determine the most stable genes for normalization of colchicine biosynthesis-related genes. Using RefFinder, one stable reference gene, UBC22, was selected to normalize gene expression levels of candidate methyltransferase (MT) genes in the leaves, roots, and rhizomes of G. superba. With UBC22 as reference gene, the methyltransferases, GsOMT1, GsOMT3, and GsOMT4 showed significantly higher expression levels in the rhizome of G. superba, while MT31794 was more highly expressed in the roots. In conclusion, the current results showed a viable reference gene expression analysis system that could help elucidate colchicine biosynthesis and its exploitation for increased production of the drug in G. superba.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The emergence of plant-extracted alkaloids in drug development is promising in human medicine due to their structural diversity and complex biological functions (Kishimoto et al. 2016; Mondal et al. 2019; Seca and Pinto 2018). Gloriosa superba L., a member of the Colchicaceae family, is a commercial source of pharmaceutical colchicine, an amino acid-derived compound that has anti-inflammatory functions and is approved by the US Food and Drug Administration for treatment of gout flares and familial Mediterranean fever (FMF) (Dubey et al. 2017; Leung et al. 2015; Sivakumar 2018). More recently, because of its known activity in reducing production of pro-inflammatory cytokines, colchicine’s use for COVID-19 treatment has been proposed (Perricone et al. 2020). Colchicine binds tubulin in mitotic cells, forming a complex that interrupts microtubule polymerization. Therefore, colchicine or its derivatives may also have promise as a possible treatment for cancer (Bhattacharya et al. 2016; Kumar et al. 2017; Pathak et al. 2022; Wang et al. 2016). However, as the demand for colchicine in medicine increases, the production of the natural isomer remains expensive, labor-intensive, and low-yielding (Kumar et al. 2017). Even with its prominent standing in human medicine, there is still not a clear understanding of the biosynthetic pathway of colchicine (Nett et al. 2020). To successfully establish alternative biomanufacturing platforms, the colchicine pathway genes and mechanism of regulation in G. superba must be clarified. One approach to identifying genes involved in metabolite biosynthesis is to identify genes that are differentially expressed in tissues that produce higher metabolite levels compared to other tissues that produce lower levels of the metabolite. In G. superba, it was shown that colchicine levels are higher in the rhizome than in the leaf and root (Nett et al. 2020). RNA Sequencing (RNA-Seq) data were used to identify methyltransferases (MT) and cytochrome P450-encoding genes that are upregulated in rhizome as compared to expression levels in leaves, roots and stems. One N-methyltransferase (GsNMT), four O-methyltransferases (GsOMT1-4), and two P450 genes were highly expressed in rhizomes. Combinations of the cloned genes were transformed using Agrobacterium tumefaciens into N. benthamiana Domin and transiently co-expressed, whereby their enzymatic roles in the biosynthesis of colchicine intermediates were confirmed.
A second set of RNA-Seq results and contigs from pooled tissues of G. superba and Colchicum autumnale L. is publicly available in https://medplantrnaseq.org/. Our preliminary RNA-Seq and BLAST2GO studies identified additional MTs, P450s, and other genes (N-acetyltransferase, NAT) that could be involved in additional or parallel steps in colchicine biosynthesis (S. Ganapathy, unpublished results). Quantitative real-time polymerase chain reaction (qRT-PCR) is the most effective method for gene expression analysis, and normalization using suitable reference genes is essential in obtaining reliable gene expression results (Jose et al. 2020; Li et al. 2017; Sahoo et al. 2019; Yuan et al. 2014). The selection of internal control genes is based on the consistency of expression across all tissue samples and under different growth conditions. Suitable control genes for this study would be housekeeping genes—constitutive genes essential in maintaining basic cellular functions (Yuan et al. 2014). This study evaluated ten commonly validated reference genes from Arabidopsis thaliana and X Doritaenopsis sp. (Table 1). Reference genes from A. thaliana and X Doritaenopsis sp. were compared using BLAST to G. superba cDNA contigs (https://medplantrnaseq.org/) to identify the G. superba orthologs. X Doritaenopsis belongs to the Orchidaceae family; Orchidaceae and Colchicaceae belong to subclass Petrosaviidae. Therefore, X Doritaenopsis sp. is more taxonomically related to G. superba, perhaps making it a better source for identifying reference genes. The specificity and stability of the reference genes were analyzed through qRT-PCR, and the RefFinder software package was used to determine the most stable reference gene to normalize qRT-PCR data of candidate OMT genes in G. superba.
Normalized qRT-PCR data of the colchicine pathway genes can be used for comparative analysis of gene expression in the leaf, root, and rhizome. These specific tissues are tested to obtain a context-specific understanding of metabolite production by comparing plant tissues that show differential expression of genes (Nett et al. 2020). Previous studies showed that rhizomes contain the highest concentration of colchicine (Nett et al. 2020), thereby proposing that candidate colchicine pathway genes will show a higher level of gene expression in rhizomes. The correlation of differential gene expression of methyltransferase genes by qRT-PCR from this study and the RNA-Seq results from (Nett et al. 2020) are presented.
Materials and methods
Plant cultures and growth conditions
Gloriosa superba tissue samples were excised from cultures in 2L balloon-type bubble bioreactors (Sivakumar 2018) and grown in small aseptic flask cultures with 50 ml of Murashige and Skoog (MS) media solution supplemented with 3% sucrose, 0.6 g/LKNO3, 1.5 g/L NH4NO3, and indole-3-butyric acid, and adjusted to a pH of 6.0. The flask cultures were placed on New Brunswick™ Innova® 2300 platform shaker set to 81 rpm for 24 h, 7 days a week at 25–27 °C for 6 weeks until rhizome germination, and shoots and roots formed. Tissue samples were collected from two separate G. superba plant cultures and the biological replicates designated as GS-1 and GS-2. One tissue sample was extracted from young leaf, root, and rhizome from each biological replicate. Each sample was weighed (80–120 mg), put in microfuge tubes, and flash-frozen in liquid nitrogen.
RNA extraction and cDNA synthesis
mRNA was extracted using the TRIzol Plus RNA purification system with PureLink RNA Mini Kit (ThermoFisher, USA). Frozen tissue samples in 1.5 ml microfuge tubes were homogenized in liquid nitrogen using RNase-free plastic pestles and passed repeatedly through an 18-gauge syringe needle before processing as per manufacturer’s instructions. 30 μl of RNase-free water was pipetted into the center of the spin cartridge and centrifuged to elute mRNA into the recovery tube. The concentration and purity of the isolated mRNAs were measured on a NanoDrop 2000 Spectrophotometer. RNA purity was measured with an OD260/280 value between 1.9 and 2.0 and mRNA concentrations ranged from 67 to 420 ng/μl. Quality of mRNA preparations were examined on 2% agarose gel. The mRNA was stored at − 80 °C until use. cDNA synthesis was performed using the reverse transcription protocol for SuperScript IV VILO Master Mix with ezDNase enzyme. A total of 500 ng mRNA per 20 μl reaction volume was reverse transcribed, diluted fivefold with 80 μl nuclease-free water, and used for qRT-PCR experiments.
qRT-PCR primer design and testing
A. thaliana and X Doritaenopsis sp. reference genes from NCBI were compared using BLAST to G. superba cDNA contigs obtained from https://medplantrnaseq.org/ to identify orthologs (Czechowski et al. 2005; Remans et al. 2008; Yuan et al. 2014). Coding sequences in G. superba contigs were identified and confirmed using BLASTX. Primers synthesized by ThermoFisher were 20–25 base pair long with 45–60% GC content, Tms of 58–63 °C, and amplicons ranging from 94 to 119 bp nucleotides with melting temperatures between 79.4 and 84.6 °C (Table 1). Master stocks of 100 mM were prepared from lyophilized forward and reverse primers. 10 mM working stocks were then prepared as a 1:10 dilution of the master stock for all qRT-PCR experiments.
Primer efficiency
All experiments were performed using PowerUp SYBR® Green Master Mix (Thermo Scientific, A25742) in 25 μl reaction containing cDNA corresponding to 5 ng total RNA, and no-template controls (water template) in 96-well plates (Applied Biosystems MicroAmp Cat. 43-067-37). qRT-PCR using Applied Biosystems QuantStudio 7 were run as follows: 96 °C denaturation for 6 min followed by 40 cycles of 30 s at 96 °C, 30 s at 55 °C, and 30 s at 72 °C. Fluorescence was read following each annealing and extension phase. All runs were followed by melting curve analysis. The linear range of template concentration to Ct value was determined by performing a series of two-fold dilutions using purified cDNA from three independent RNA extractions analyzed in three technical replicates. Primer efficiencies for all primer pairs were calculated using the standard curve method (Pfaffl 2001).
Gel extraction and sequencing
qRT-PCR amplification products were loaded in 2% agarose gel. The DNA bands that were confirmed to be the correct amplicon size were then excised from the agarose gel and stored at 20 °C before extraction with Zymoclean Gel DNA Recovery Kit (Cat. No. D4001). Sanger sequencing using ABI 3730xl DNA Analyzer confirmed the sequence identity and purity of all the amplified PCR products.
Quantitative real-time PCR of methyltransferase genes
Quantitative real-time PCR (qRT-PCR) reactions using PowerUp SYBR Green (Applied Biosystems Cat. A25741) were performed on the QuantStudio 7 Flex Real-Time PCR system with optical 96-well plates. The qRT-PCR reaction mixtures consisted of 1 μl of 1:5 diluted cDNA samples (with a concentration of approximately 5 ng mRNA/μl), 1.6 μl of pooled forward and reverse primers (10 mM), 10 μl PowerUp SYBR Green Master Mix (2 ×), and 7.4 μl of water for a 20 μl reaction. All samples had two biological replicates and three technical replicates with a no-template control (NTC). qRT-PCR was performed at a standard ramp speed and continuous melt curve ramp increment. The protocol consists of 15 s initial denaturation at 95 °C, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. The specificities of the primers were verified using melting curve analysis where the PCR products were heated from 60 to 95 °C (0.5 °C/5 s) with fluorescence measured within 20 min after the last qRT-PCR extension step. The raw Ct values were obtained with the default parameters in the QuantStudio system.
Results
Selection of reference genes
A total of ten candidate reference genes were evaluated to identify ideal reference genes for use in normalization and validation of gene expression for qRT-PCR analysis of candidate colchicine pathway genes. The specificity of amplification of the reference genes was tested by conventional PCR and qRT-PCR using a pool of cDNA samples from the leaf, root, and rhizome of G. superba. The specificity of each primer pair was confirmed with the presence of single peaks in the melt curve analysis and further validated with the amplification of single PCR products of the expected size using 2% agarose gel electrophoresis (not shown). At least three sets of forward and reverse primers were tested for each gene and one specific pair was chosen for each gene (Table 1). Additional testing involved Sanger sequencing of the gel-extracted PCR products to confirm the amplification of the correct gene of interest. A two-fold cDNA dilution series of mRNA was prepared to generate a standard curve for estimation of amplification efficiency [E = (10e(− 1/slope) − 1 × 100%] and correlation coefficient (R2), based on the slopes of the curve (Fig. 3).
The candidate reference genes were selected based on previous studies conducted in other plant transcriptomic analysis (Jose et al. 2020; Li et al. 2017; Sahoo et al. 2019; Yuan et al. 2014). They are traditional housekeeping genes consistently expressed in most living cells. However, the uniform expression of the genes across all sample tissue types must be evaluated to deduce the most stable gene/s to normalize qRT-PCR data in each new test organism. Previous studies tested eIF2, UBC22, RPL15, and PP2A orthologs of A. thaliana for qRT-PCR in I. indigotica (Li et al. 2017). Their study concluded that RPL15 had the highest stability in expression in varying tissue samples based on several factors, including the amplification specificity and efficiency (E = 1.05%), correlation coefficient (0.9920), and low experimental variation. The average expression stability value, calculated by the GeNorm software package, validated RPL15 as the most stable gene using stepwise exclusion (M = 0.06). Another similarly conducted study tested several actin genes (ACT1, ACT2, ACT3, ACT4, ACT5), TUA, TUB, and GADPH in Phalaenopsis cultivar Jiuhbao Red Rose, ranking ACT2 and ACT4 as the most stable genes based on amplification efficiencies of 0.99 and 1.00, correlation coefficients of 0.997 and 0.997, and M values less than 1.5 (Yuan et al. 2014).
Reference gene expression profiling
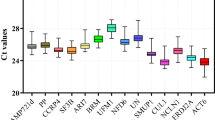
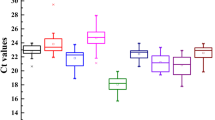
The Ct values of the ten candidate reference genes ranged from 19.42 to 34.98, with most values falling in the 22–28 range (Fig. 1). EF1, ACT7, and TUA showed the highest gene expression levels, while PP2A and RPL15 showed the lowest gene expression levels. The reference genes were tested using six different tissue samples isolated from the two biological replicates (GS-1, GS-2) with three technical replicates, and no-template controls (NTC) to analyze the variation of Ct values in the leaf, root, and rhizome in G. superba. Narrow Ct variations were observed for UBC22, EF1 and TUA, and wider Ct variations for PP2A, ACT3, ACT7, and GADPH. The two sets of primers for GADPH gave about the same range of Ct values.

Box plot of ten candidate reference genes showing Ct distribution in leaf, root, and rhizome tissues of G. superba. Whiskers are the lowest and highest Ct values, boxes are the 25 and 75 percentiles, and the lines across each box are median values
RefFinder analysis
The expression stability of the ten selected reference genes was further analyzed using the RefFinder software package (Xie et al. 2012). RefFinder produces a comprehensive analysis of expression stabilities by using a combination of four different software packages, including BestKeeper (Pfaffl 2001), GeNorm (Vandesompele et al. 2002), NormFinder (Andersen et al. 2004), and Delta Ct (Silver et al. 2006). The geometric mean values of cycle quantification (Cq) from the four separate statistical algorithms are used to rank the reference genes from most stable to least stable genes in the final output analysis. Based on the RefFinder comprehensive data analysis of expression stability (Fig. 2), the ten reference genes were ranked as follows: UBC22 > TUA > TUB > eIF2 > RPL15 > EF1 > ACT7 > ACT3 > GADPH > PP2-A. UBC22 and TUA were consistently ranked highest across all expression analysis software packages. GADPH, which had been widely used to normalize many earlier qRT-PCR experiments, and PP2-A, consistently showed the lowest expression stabilities. BestKeeper generated a slightly modified ranking based on the standard deviation and Pearson’s correlation coefficient (Fig. 2). Genes with a standard deviation greater than one were considered unsuitable for normalization; UBC22 was the only gene that met all criteria. NormFinder also identified UBC22 as the most stable, while UBC22 came second to TUA and TUB using GeNorm. Using GeNorm, PP2-A and GADPH1 exceeded the M value cutoff of 1.5.

Analysis of stability of expression of ten candidate reference genes using RefFinder (Comprehensive Gene Stability), BestKeeper, NormFinder, and GeNorm
Amplification efficiency and specificity of six candidate reference genes
Based on the calculated correlation coefficients and computed stability ranking, six reference genes were chosen for further analysis of expression stability—UBC22, TUA, eIF2, RPL15, EF1, and ACT7; TUB was left out by mistake. The selected genes were tested by qRT-PCR using pooled leaf, root, and rhizome cDNA template and negative controls. The concentration of the templates was reduced by two-fold serial dilutions with 5–6 measurement points from a starting concentration of 10 ng mRNA/μl. Using the Ct data, a standard curve was generated to calculate the selected reference genes’ amplification efficiencies and correlation coefficients (Fig. 3). ACT7, RPL15, eIF2, UBC22, and TUA had R2 values > 0.98 and PCR efficiencies of 126–138%. Based on the cumulative analysis of expression stability of the candidate reference genes, UBC22 was selected to normalize expression data of candidate colchicine pathway genes.

Standard curve linear regression plot of Ct values of six candidate reference genes at different mRNA concentrations. Data are from three technical replicates from each of three biological replicates
Validation of UBC22 as reference gene using colchicine biosynthesis genes
Nett et al. (Nett et al. 2020) used sequences in https://medplantrnaseq.org/ and their newly generated RNA-Seq data to identify candidate colchicine pathway genes differentially expressed in rhizomes versus leaves and roots. They demonstrated that the transformation of some of those genes in Nicotiana tabacum leaves by Agrobacterium infiltration led to production of various colchicine intermediates. In this paper, the expression profiles of four methyltransferases studied by Nett et al. (2020), GsOMT1, GsOMT3, GsOMT4 and MT2864, were examined in three different plant tissues (Table 2) using qRT-PCR to see if the RNA-Seq results could be validated using UBC22 as reference gene for normalization. Using BLAST2GO, we identified a second, longer methyltransferase contig, MT31794 (Supplementary information), that aligns with MT2864 with 99% nucleotide sequence identity. Mismatches were due to ambiguous sequence reads in the 5′ end of MT2864. MT2864 is missing 117 bases from the start codon which are present in MT31794. BLASTX analysis of the longer MT31794 revealed a CDS that has 100% sequence coverage and 81.87% sequence identity with GsOMT4. MT31794 and MT2864 are likely sequences from the same gene and MT31794 was designated as representing MT2864 in this paper.
The specificity of amplification of the methyltransferase genes was initially tested by conventional PCR and qRT-PCR using a pool of cDNA samples and further validated through the presence of a single band in agarose gel, followed by DNA sequencing. Gene expression was measured using two biological replicates (GS-1 and GS-2), three technical replicates, and no-template controls (NTC) for three tissue samples; leaf, root, and rhizome. The raw Ct values, obtained by qRT-PCR, were analyzed through the comparative ΔΔCt method (Pfaffl 2001). The method normalized the values of the target gene against the values of the reference gene. The relative expression of the colchicine pathway genes in the different tissue samples was calculated using the following formula:
The relative expression fold change (FC) of rhizome and root sample Ct values in comparison with the Ct values obtained from the leaf samples were calculated as follows:
Melt curve analysis after qRT-PCR provided additional validation of the presence of single amplified PCR products (Online Resource, Fig. S1).
qRT-PCR analysis of O-methyltransferase genes
Nett et al. (Nett et al. 2020) transiently expressed 11 putative OMT genes by Agrobacterium tumefaciens infiltration of N. benthamiana leaves and showed by liquid chromatography–mass spectrometry (LC–MS) analysis that five OMTs (GsNMT, GsOMT1, GsOMT2, GsOMT3 and GSOMT4) were involved in the biosynthesis of the colchicine precursor, N-formyldemecolcine. In contrast, six other OMTs (including MT2864) did not seem to be involved. Nett et al. (2020) also showed high accumulation of colchicine in the rhizomes of G. superba relative to other tissues, which correlated with the differential expression levels of GsOMTs 1–4, therefore, lending additional support for their possible role in the colchicine pathway.
To validate the suitability of UBC22 as reference gene for qRT-PCR for looking at differential expression of colchicine biosynthetic genes in rhizomes versus leaves and roots, expression of GsOMT1, GsOMT3, GsOMT4, and MT31794 (= MT2864) was studied. qRT-PCR was performed using GS-1 and GS-2 and produced melt curves showing single amplification products, which were further confirmed by Sanger sequencing. GsOMT1 was significantly upregulated in the rhizome of G. superba (Fig. 4). There was an average of 1416-fold change (FC) in gene expression in the rhizome compared to the root and leaf (FC = 1.53 and 1, respectively). Ct values of GsOMT3 also showed a consistently high level of expression in the rhizome of the G. superba plants (Ct ~ 26) compared to the root and leaf (Ct ~ 33) (Fig. 4). The high average FC (FC = 76.25) observed in the rhizome and smaller FC observed in the root and leaf (FC = 0.27 and 0.24, respectively) indicate the high expression of GsOMT3 in the rhizome. Normalization and comparative analysis of GsOMT4 also resulted in a significantly high level of gene expression in the rhizome of G. superba. The average expression fold change in the rhizome for the two biological replicates is 41.11, while FC values for root and leaf are 1.79 and 1.0, respectively.

Comparative ∆∆Ct analysis of methyltransferase genes in G. superba tissues and melting curves of qRT-PCR amplicons. PCR products were heated from 60 to 95 °C (0.5 °C/5 s) and fluorescence measured within 20 min after the last qRT-PCR extension step. 5 ng/μl mRNA used per reaction
MT31794 was included in this study, because the equivalent in Nett et al. (2020), MT2864, did not show differential tissue expression in their work based on FPKMs (fragments per kilobase million) and had a poor correlation with GsOMT1 expression (r = 0.56). Unlike the other OMTs, MT2864 failed to catalyze the synthesis of colchicine precursors upon transformation into N. benthamiana. Thus, it would be a desirable gene to use as negative control to contrast with the rhizome-specific expression of GsOMT1, GsOMT3, and GsOMT4. In this study, MT31794 was not highly expressed in the rhizomes as compared to that in roots and in leaves. Instead, MT31794 had the highest level of expression occurring in roots and the lowest expression in rhizomes (Fig. 4). The average FC in the rhizome, 0.15, was considerably low compared to the roots’ 2.85. It is important to note that fold increases determined by qRT-PCR and RNA-seq are not always 100% correlated (R2 = 0.83 in Marriott et al. 2016), with the fold change sometimes higher for qRT-PCR than from RNA-Seq. RNA was also collected from plants grown in solid substrate in (Nett et al. 2020), while the present study used plants grown in liquid media with shaking.
Discussion
Inclusion of a reference gene as internal control is a widespread practice when using qRT-PCR for examining differential gene expression in plants (You et al. 2021; Sharma et al. 2021; Mafra et al. 2012) and non-plants (Silver et al. 2006; Cankorur-Cetinkaya et al. 2012; Hu et al. 2016). Reference gene readings are used to normalize Ct data for possible analyst and other experimental errors especially those that can skew the starting mRNA amounts (Chapman and Waldenström 2015). An ideal reference gene should be expressed stably in a broad range of treatment conditions and/or tissues. In the present study, four algorithms (geNorm, NormFinder, BestKeeper, and Delta Ct), followed by RefFinder, were used to evaluate the stability of ten G. superba housekeeping genes. With the exception of geNorm, the other four programs identified UBC22 as the highest scoring of the ten candidates, followed by TUA, TUB and eIF2. UBC22 was second best after TUA and TUB using geNorm. UBC homologues worked best among tested reference genes, at least in certain conditions, in qRT-PCR studies done in many plants (Ye et al. 2018; Niu et al. 2015; Taylor et al. 2016; Li et al. 2016; Chen et al. 2019), but not in others (Czechowski et al. 2005; Qu et al. 2019; Li et al. 2019).
UBC22 was validated as a reference gene for examining differential gene expression of four G. superba methyltransferases. GsOMT1, GsOMT3 and GsOMT4 were highly expressed, while MT31794 was poorly expressed in colchicine-overproducing rhizomes compared to leaves and roots. The results were consistent with the RNA-Seq and heterologous colchicine pathway reconstitution studies done in N. benthamiana using those same four genes (Nett et al. 2020). Using BLAST2GO on cDNA contigs, we have identified additional candidate MT and cytochrome P450 genes in G. superba (S. Ganapathy, unpublished) that were not studied in Nett et al. (2020). A qRT-PCR approach using UBC22 as reference gene would be used to test rhizome overexpression of those remaining candidate genes. Genes that will show increased expression in the rhizomes of G. superba, versus leaves or roots, would be reasonable candidates for prioritizing functionality studies in the colchicine biosynthetic pathway. Therefore, this study is one step towards complete elucidation of the biosynthetic pathway in G. superba. Once gene expression is evaluated and confirmed for the entire colchicine pathway, additional studies to validate the exact biochemical contributions of the enzyme products are necessary for laying a foundation for synthetic biotechnology and possible large-scale production of colchicine and its novel derivatives (Ghawanmeh et al. 2020) in heterologous systems like yeast. Plant biosynthetic genes have been successfully engineered in yeast to produce pharmaceutical drugs or their precursors (Cravens et al. 2019; Ro et al. 2006; Galanie et al. 2015; Srinivasan and Smolke 2020; Luo et al. 2019). In addition, overexpression of pathway genes in transgenic G. superba or C. autumnale can further increase colchicine yield. In Papaver somniferum L., increased production of the alkaloids, morphine and codeine, was achieved by constitutively expressing just one protein in the pathway, codeinone reductase (Larkin et al. 2007). In Panax ginseng C.A.Mey, over-expression of squalene synthase increased levels of ginsenoside and phytosterols (Lee et al. 2004). In Macleaya cordata (Willd.) R.Br., overexpression of berberine bridge enzyme increased production of benzylisoquinoline alkaloids (Huang et al. 2018). More recently, the production of anticholinergic and arrhythmia alkaloid, hyoscyamine, was increased in Atropa belladonna L. by using CRISPR/CAS9 to inactivate a dioxygenase which converts hyoscyamine to an unwanted derivative (Zeng et al. 2021).
While using UBC22 for normalizing qRT-PCR readouts of methyl transferase genes successfully produced results consistent with differential FPKM patterns from RNA-Seq data (Nett et al. 2020), more studies are needed to determine if UBC22 could serve as an ideal reference for qRT-PCR studies in G. superba beyond colchicine biosynthetic genes.
Data availability
G. superba cDNA contig sequences are available at https://medplantrnaseq.org/. Coding sequences used in this study are provided in the supplementary information.
References
Andersen CL, Jensen JL, Ørntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5250
Bhattacharya S, Das A, Datta S, Ganguli A, Chakrabarti G (2016) Colchicine induces autophagy and senescence in lung cancer cells at clinically admissible concentration: potential use of colchicine in combination with autophagy inhibitor in cancer therapy. Tumor Biol 37:10653–10664
Cankorur-Cetinkaya A, Dereli E, Eraslan S, Karabekmez E, Dikicioglu D, Kirdar B (2012) A novel strategy for selection and validation of reference genes in dynamic multidimensional experimental design in yeast. PLoS One 7(6):e38351
Chapman JR, Waldenström J (2015) With reference to reference genes: a systematic review of endogenous controls in gene expression studies. PLoS One 10(11):e0141853
Chen C, Wu J, Hua Q, Tel-Zur N, Xie F, Zhang Z, Chen J, Zhang R, Hu G, Zhao J, Qin Y (2019) Identification of reliable reference genes for quantitative real-time PCR normalization in pitaya. Plant Methods 15:70
Cravens A, Payne J, Smolke CD (2019) Synthetic biology strategies for microbial biosynthesis of plant natural products. Nat Commun 10:1–12
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible W-R (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139:5–17
Dubey KK, Kumar P, Labrou NE, Shukla P (2017) Biotherapeutic potential and mechanisms of action of colchicine. Crit Rev Biotechnol 37:1038–1047
Galanie S, Thodey K, Trenchard IJ, Filsinger Interrante M, Smolke CD (2015) Complete biosynthesis of opioids in yeast. Science 349:1095–1100
Ghawanmeh AA, Al-Bajalan HM, Mackeen MM, Alali FQ, Chong KF (2020) Recent developments on (−)-colchicine derivatives: synthesis and structure-activity relationship. Eur J Med Chem 185:111788
Hu Y, Xie S, Yao J (2016) Identification of novel reference genes suitable for qRT-PCR normalization with respect to the zebrafish developmental stage. PLoS One 11(2):e0149277
Huang P, Liu W, Xu M, Jiang R, Xia L, Wang P, Li H, Tang Z, Zheng Q, Zeng J (2018) Modulation of benzylisoquinoline alkaloid biosynthesis by overexpression berberine bridge enzyme in Macleaya cordata. Sci Rep 8:1–9
Jose S, Abbey J, Jaakola L, Percival D (2020) Selection and validation of reliable reference genes for gene expression studies from Monilinia vaccinii-corymbosi infected wild blueberry phenotypes. Sci Rep 10:1–10
Kishimoto S, Sato M, Tsunematsu Y, Watanabe K (2016) Evaluation of biosynthetic pathway and engineered biosynthesis of alkaloids. Molecules 21:1078
Kumar A, Sharma PR, Mondhe DM (2017) Potential anticancer role of colchicine-based derivatives: an overview. Anticancer Drugs 28:250–262
Larkin PJ, Miller JA, Allen RS, Chitty JA, Gerlach WL, Frick S, Kutchan TM, Fist AJ (2007) Increasing morphinan alkaloid production by over-expressing codeinone reductase in transgenic Papaver somniferum. Plant Biotechnol J 5(1):26–37
Lee M-H, Jeong J-H, Seo J-W, Shin C-G, Kim Y-S, In J-G, Yang D-C, Yi J-S, Choi Y-E (2004) Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant Cell Physiol 45:976–984
Leung YY, Hui LLY, Kraus VB (2015) Colchicine—update on mechanisms of action and therapeutic uses. Seminars in arthritis and rheumatism, vol 45. Elsevier, Amsterdam, pp 341–350
Li J, Han J, Hu Y, Yang J (2016) Selection of reference genes for quantitative real-time PCR during flower development in tree peony (Paeonia suffruticosa Andr.). Front Plant Sci 7:516
Li T, Wang J, Lu M, Zhang T, Qu X, Wang Z (2017) Selection and validation of appropriate reference genes for qRT-PCR analysis in Isatis indigotica Fort. Front Plant Sci 8:1139
Li Z, Lu H, He Z, Wang C, Wang Y, Ji X (2019) Selection of appropriate reference genes for quantitative real-time reverse transcription PCR in Betula platyphylla under salt and osmotic stress conditions. PLoS One 14(12):e0225926
Luo X, Reiter MA, d’Espaux L, Wong J, Denby CM, Lechner A, Zhang Y, Grzybowski AT, Harth S, Lin W (2019) Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 567:123–126
Mafra V, Kubo KS, Alves-Ferreira M, Ribeiro-Alves M, Stuart RM, Boava LP, Rodrigues CM, Machado MA (2012) Reference genes for accurate transcript normalization in citrus genotypes under different experimental conditions. PLoS One 7(2):e3126337
Marriott AS, Vasieva O, Fang Y, Copeland NA, McLennan AG, Jones NJ (2016) NUDT2 disruption elevates diadenosine tetraphosphate (Ap4A) and down-regulates immune response and cancer promotion genes. PLoS One 11:e0154674
Mondal A, Gandhi A, Fimognari C, Atanasov AG, Bishayee A (2019) Alkaloids for cancer prevention and therapy: current progress and future perspectives. Eur J Pharmacol 858:172472
Nett RS, Lau W, Sattely ES (2020) Discovery and engineering of colchicine alkaloid biosynthesis (published correction appears in Nature. 2020 Jul 30). Nature 584:148–153
Niu X, Qi J, Zhang G, Xu J, Tao A, Fang P, Su J (2015) Selection of reliable reference genes for quantitative real-time PCR gene expression analysis in Jute (Corchorus capsularis) under stress treatments. Front Plant Sci 6:848
Pathak S, Gupta R, Parkar H, Joshi N, Nagotu S, Kale A (2022) The role of colchicine on actin polymerization dynamics: as a potent anti-angiogenic factor. J Biomol Struct Dyn 40:11729–11743
Perricone C, Bartoloni E, Gerli R (2020) Colchicine, an anti-rheumatic agent, as a potential compound for the treatment of COVID-19. Reumatologia 58:261
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res 29:e45
Qu R, Miao Y, Cui Y, Cao Y, Zhou Y, Tang X, Yang J, Wang F (2019) Selection of reference genes for the quantitative real-time PCR normalization of gene expression in Isatis indigotica fortune. BMC Mol Biol 20(1):9
Remans T, Smeets K, Opdenakker K, Mathijsen D, Vangronsveld J, Cuypers A (2008) Normalisation of real-time RT-PCR gene expression measurements in Arabidopsis thaliana exposed to increased metal concentrations. Planta 227:1343–1349
Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MC, Withers ST, Shiba Y, Sarpong R, Keasling JD (2006) Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440(7086):940–943
Sahoo PK, Parida S, Mohapatra A, Mohanty J (2019) Selection of candidate reference genes for RT-qPCR analysis in Argulus siamensis and their validation through screening of drugs and drug targets. Sci Rep 9:1–11
Seca AM, Pinto DC (2018) Plant secondary metabolites as anticancer agents: successes in clinical trials and therapeutic application. Int J Mol Sci 19:263
Sharma S, Vengavasi K, Kumar MN, Yadav SK, Pandey R (2021) Expression of potential reference genes in response to macronutrient stress in rice and soybean. Gene 792:145742
Silver N, Best S, Jiang J, Thein SL (2006) Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol Biol 7:1–9
Sivakumar G (2018) Upstream biomanufacturing of pharmaceutical colchicine. Crit Rev Biotechnol 38:83–92
Srinivasan P, Smolke CD (2020) Biosynthesis of medicinal tropane alkaloids in yeast. Nature 585:614–619
Taylor CM, Jost R, Erskine W, Nelson MN (2016) Identifying stable reference genes for qRT-PCR normalisation in gene expression studies of narrow-leafed lupin (Lupinus angustifolius L.). PLoS One 11(2):e0148300
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:1–12
Wang Y, Zhang H, Gigant B, Yu Y, Wu Y, Chen X, Lai Q, Yang Z, Chen Q, Yang J (2016) Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J 283:102–111
Xie F, Xiao P, Chen D, Xu L, Zhang B (2012) miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol 80:75–84
Ye J, Jin CF, Li N, Liu MH, Fei ZX, Dong LZ, Li L, Li ZQ (2018) Selection of suitable reference genes for qRT-PCR normalisation under different experimental conditions in Eucommia ulmoides Oliv. Sci Rep 8(1):15043
You S, Cao K, Chen C, Li Y, Wu J, Zhu G, Fang W, Wang X, Wang L (2021) Selection and validation reference genes for qRT-PCR normalization in different cultivars during fruit ripening and softening of peach (Prunus persica). Sci Rep 11(1):7302
Yuan X-Y, Jiang S-H, Wang M-F, Ma J, Zhang X-Y, Cui B (2014) Evaluation of internal control for gene expression in Phalaenopsis by quantitative real-time PCR. Appl Biochem Biotechnol 173:1431–1445
Zeng L, Zhang Q, Jiang C, Zheng Y, Zuo Y, Qin J, Liao Z, Deng H (2021) Development of Atropa belladonna L. plants with high-yield hyoscyamine and without its derivatives using the CRISPR/Cas9 system. Int J Mol Sci 22:1731
Acknowledgements
This manuscript is dedicated to the memory of Sivakumar Ganapathy and his lifetime work on Gloriosa superba.
Funding
The research was supported by an internal grant from the University of Houston to Sivakumar Ganapathy and United States National Institutes of Health Grant 5RC2GM092561-02 to Toni M. Kutchan.
Author information
Authors and Affiliations
Contributions
NJ and DRD performed the qRT-PCR experiments and analyses and contributed equally to this work. TMK generated transcriptomes and JSB carried out BLAST2GO. SG and ABF conceptualized and directed this research. ALK helped with writing and data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Johnson, N., Rodriguez Diaz, D., Ganapathy, S. et al. Evaluation of reference genes for qRT-PCR studies in the colchicine producing Gloriosa superba L.. Plant Biotechnol Rep 17, 561–571 (2023). https://doi.org/10.1007/s11816-023-00840-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-023-00840-x