
Abstract
Pears (Pyrus spp.) are one of the most important fruit crops in temperate regions and are self-incompatible. Large numbers of interspecific hybrids occur naturally and, have been artificially produced in breeding programs. Also, many pear cultivars were generated from intraspecific crossing. Some varieties have been cultivated for a long time, but their breeding history remains unknown. The P. pyrifolia cv. Niitaka is one of key varieties in the Korean pear breeding program. Therefore, its complete chloroplast (cp) genome was determined, and its phylogenetic relationships with other Pyrus species and P. pyrifolia cultivars were addressed. The cp genome has a total of 133 genes, including 93 protein-coding genes, 32 tRNA genes, and eight rRNA genes. We found many SNPs in the ‘Niitaka’ cp genes when compared with that in another Korean cultivar ‘Wonwhang’ (BioSample SAMN05196235). The primer sets for six genes that had two or more SNPs in their sequence were used to amplify and sequence 27 Pyrus and one Malus cultivars. Of these, we found dramatic InDel polymorphisms in the ndhA and clpP genes. Phylogenetic relationships using the sequences of these two genes in 28 samples showed that they could mainly be classified into two groups of P. pyrifolia. Group I constitutes Niitaka and all cultivars that contain chloroplast maternally inherited from Niitaka, and group II constitutes the other cultivars of P. pyrifolia. We have developed a useful polymorphic molecular marker to confirm the maternal parent in the interspecific hybrids of Niitaka and previous mothers of Niitaka, (such as Amanogawa). Furthermore, these two genes could identify and greatly aid in understanding the subspecies classification in Pyrus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The chloroplast is a photosynthetic organelle used during the biosynthetic pathways of fatty acids, vitamins, pigments and amino acids (Bobik and Burch-Smith 2015; Rodríguez-Ezpeleta et al. 2005). The cp genome among land plants has a highly conserved genome structure with a large single copy region (LSC) and a small single copy region (SSC) separated by two inverted repeat regions (IR) (Chumley et al. 2006; Wicke et al. 2011). The complete cp genome sequences from tobacco and liverwort were reported in 1986 (Ohyama et al. 1986; Shinozaki et al. 1986). Since then, the number of fully sequenced cp genome has been increased rapidly by cost-effective next-generation sequencing technique. Over 800 complete cp genome sequences have been deposited in the National Center for Biotechnology Information (NCBI) organelle genome database including 300 entire cp genomes from crop and tree (Daniell et al. 2016). With the highly conserved sequence, compact size, lack of recombination, and maternal inheritance, the cp genomes have been used for generating genetic markers for phylogenetic classification (Hong et al. 2017), and DNA barcoding system for molecular identification (Dong et al. 2012).
Pears (Pyrus spp.), which belong to the family of Rosaceae, have been cultivated in East Asia, Europe, and North America for more than 3000 years, and are among the most important fruit crops in the temperate regions (Bell 1991). Pyrus has been divided into two groups, occidental, and oriental pears, based on their geographic distribution (Rubtsov 1944). According to their usage, Pyrus accessions are also classified into three groups. The first one comprises P. brestchenidri, P. pyrifolia, and P. ussuriensis which are commercially cultivated in Asia. And also, there is European pear (P. communis), the other rootstock group contains P. betulaefolia and P. calleryana (Yeo and Reed 1995). However, all species of Pyrus are likely to originate from natural interspecific hybridization events, which make it difficult to determine their exact genetic relationships. Understanding the domestication process of the cultivated pear and the evolutionary process of the pear species will be helpful in exploiting elite genetic resources in pears and aid in modern breeding.
In Korea, pear breeding began in the late 1920s at the National Institute of Horticultural and Herbal Science (NIHHS) of the Rural Development Administration (RDA). Asian pears have a sweet flavor, rich juice, and crisp flesh. Furthermore, they are considered to be of superior quality and more storage friendly compared to Occidental pears (Kim 2016). To improve fruit quality regarding fruit size, sugar content, flesh firmness, and storage quality, many breeders frequently used interspecific hybridization (Iketani et al. 2012). Consequently, the classification of pears is complicated because of the occurrence of both natural and artificial interspecific hybrids. Therefore, the genetic relationships and evolutionary history of Asian pears are unclear (Jiang et al. 2016). Studying the cp genome, which is maternally inherited, will enable us to gain a better understanding of the genetic relationships and evolutionary history of Asian pears (Zhang 2010).
In our previous study, the complete cp and mitochondrial (mt) genome of a Korean pear (P. pyrifolia cv., Wonhwang), was de novo assembled using whole genome-sequencing data (Chung et al. 2017a, b). In this study, we sequenced the complete cp genome of another Korean variety (P. pyrifolia cv. Niitaka), and identified the deleted sequences in two cp genes of Niitaka and the maternal parent of the interspecific hybrids of Niitaka, despite the fact that species from the same genus of P. pyrifolia did not have these deletions. Many studies have employed the comparative genomic analysis using tandem repeats, InDels, simple sequence repeats (SSRs) polymorphism, and genetic diversity to identify valuable markers for DNA barcoding and phylogenetic analysis between species levels. Indel polymorphic markers within species in cp genome are rare, because they occur at a low evolutionary rate in most taxa. We reported the InDel polymorphisms within the ndhA and clpP gene as different species of Pyrus as well as within these genes in P. pyrifolia.
Materials and methods
Plant materials and DNA sample preparation
A total of 27 individuals in the Pyrus genus were cultivated in the Pear Research Station, NIHHS, RDA, Republic of Korea (latitude 35° 01′25.6″N, longitude 126° 44′38.4″E). The 16 cultivars of the P. pyrifolia, Niitaka, Whangkeumbae, Amanogawa, Gamro, Sunwhang, Shinhwa, Youngsanbae, Hanareum, Wonwhang, Imamuraaki, Geumchonjosaeng, Chuwangbae, Okusankichi, Seolwon, SuperGold, and Chanxixueli and two cultivars of P. ussuriensis, Doonggeullebae, and Cheongdangrori were used. Additionally, Yali and Dangshansuli of P. bretschneideri were used. We also used Danbae cultivar, an interspecific cross between P. pyrifolia and P. communis. As classifying by rootstock group, OPR125 and OPR195 of P. calleryana were included. One unclassified pear cultivar used with Kozo and a P. faurie cultivar Godang 5-1 were also used. We collected on two European pear cultivars of Bartlett and Max Red Bartlett. As an outgroup sequence cultivar with Malus, Fuji is listed in this analysis. These characteristics of 28 cultivars are listed in Supplement Table 1. Genomic DNA was extracted from young leaves using a DNeasy Plant Mini kit (Qiagen, CA, USA) according to the manufacturer’s instructions.
Chloroplast genome sequencing, assembly, and annotation
Whole genome sequencing was performed using an Illumina genome analyzer (Hiseq4000, Illumina, USA) at Macrogen (http://www.macrogen.com/) in Seoul, Republic of Korea. Genomic libraries with 350-bp inserts were prepared by following the paired-end standard protocol recommended by the manufacturer. Each sample was tagged separately with a different index. De novo assembly was performed using a CLC genome assembler (4.06 beta, CLC Inc., Aarhus, Denmark) with parameters of a minimum 200–600 bp autonomously controlled overlap size. The cp-coding contigs were identified by comparison to the entire cp genome of the reported P. pyrifolia (Terakami et al. 2012) and circularized. Gene annotation was conducted using CpGAVAS (http://www.herbalgenomics.org) and the cp map was drawn with the OrganellarGenomeDRAW (OGDRAW) program (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html).
Development of chloroplast InDel markers
The chloroplast sequence comparison of Niitaka vs. Wonwhang revealed many SNPs. From them, we selected six protein-coding genes that had more than two SNPs in this comparison. They are the ATP synthase subunit beta (atpB), ATP-dependent Clp protease (clpP), NADH dehydrogenase subunit 5 (ndhF), NADH dehydrogenase subunit 4 (ndhD), NADH dehydrogenase subunit 1 (ndhA), and yeast cadmium factor 1 (ycf1) genes. The primer set was designed using the Primer 3 program (http://bioinfo.ut.ee/primer3-0.4.0/). Six primers paired sequences were designed including the two SNPs with amplicon size of approximately 1 kb. Based on chloroplast genome (NC_015996 and KX904342), the polymorphic gene name, SNP location, primer pair sequences, annealing temperature, and expected product size are described in Table 1. PCR amplifications were carried out in 30 µl reaction containing 30 ng of genomic DNA template, 15 µl of 2X TOPsimpleTM DyeMIX-nTaq (Enzynomics, Korea), and 10 pM each of forward and reverse primers. The amplification was conducted by PCR machine (AB GeneAmp PCR system 9700) with a hot start of 94 °C for 5 min, 35 cycles of 30 s at 94 °C, 30 s at 60 °C, and 1 min at 72 °C, and a final extension at 72 °C for 10 min. Amplified fragments were analyzed by separation on agarose gels and sequencing. Then, the sequences were aligned with DNAstar software (DNASTAR Inc., Madison, Wisconsin USA) to compare each sequence.
Phylogenetic analysis
Phylogenetic analysis was conducted using the two InDel marker regions, ndhA, and clpP, shared by 27 Pyrus and one Malus. These Sanger sequences are shown in supplement file 5. Multiple sequence alignments were carried out using ClustalW, which was followed by phylogenetic tree generation using MEGA7 (Kumar et al. 2016) with 1000 bootstrap replicates. M. pumila cv. Fuji used as an outgroup. The maximum likelihood method was used, and the tree with the highest log likelihood was obtained.
Results
The complete chloroplast genome of Niitaka
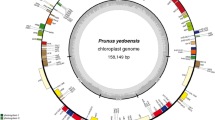
We sequenced, and de novo assembled the cp genome of P. pyrifolia cv. Niitaka. The entire cp genome sequences of Niitaka was submitted to GenBank under the accession number KX904342. The complete cp genome of Niitaka is 159,922 bp in length, with a pair of IR regions of 26,392 bp that separate an LSC region of 72,023 bp and an SSC region of 19,235 bp. A total of 133 genes were identified including 93 protein-coding genes, 32 tRNA genes, and 8 rRNA genes (Fig. 1). Twelve genes, atpF, ndhA, two ndhB, two rpl2, rpoC1, rpl22, two trnI-TAT and two ycf15 have one intron, and clpP and ycf3 gene have two introns. The SSC region contains 11 protein-coding, and 1 tRNA genes, and the respective IR regions contain 9 protein-coding genes, 5 tRNAs, and 4 rRNAs.

Gene map of the Niitaka chloroplast genome. The quadripartite structure includes two copies of an IR region (IRA and IRB) that separate the large single copy (LSC) and small single copy (SSC) regions. Genes on the outside of the map are transcribed in the counter-clockwise direction and genes on the inside of the map are transcribed in the clockwise direction. Differential functional gene groups are color-coded. The dashed area in the inner circle indicates the GC content of the chloroplast genome
Chloroplast InDel markers development
We determined the polymorphisms in the cp genomes between Niitaka and one of Asian pear cultivar with early ripening traits, ‘Wonwhang’ (Chung et al. 2017a, b). The sequence comparison of the two cp genomes identified many SNPs between Niitaka and Wonwhang. In particular, we found the 29 SNPs in 20 protein-coding genes that belonged to the diverse functional groups including house-keeping function and photosynthesis (Supplement Table 2). While most of the polymorphic genes had a single SNP, atpB for ATP synthase, three NADH dehydrogenase genes (ndhA, ndhD, and ndhF), psaA for photosystem I, and a hypothetical protein ycf1 had two SNPs (Supplement Table 2). ATP-dependent protease ClpP contained as many as four SNPs (Supplement Table 2). However, we did not observe any InDel between two cultivars in the protein-coding genes.
Next, we determined polymorphisms among Pyrus species. For this, the PCR primers were designed from six genes that showed two or more SNPs above (Table 1). The PCR and the subsequent Sanger sequencing with four protein-coding genes, ndhD, ndhF, ycf1, and atpB discovered SNPs but not Indels among 27 Pyrus and one Malus individuals (Supplement Table 2). However, two genes ndhA and clpP had both types of polymorphisms, SNPs and InDels. The nucleotide (nt) length of the ndhA amplicon ranged from 957 to 980 bp, and nt length of clpP amplicon ranged from 933 to 958 bp. The length of the InDel regions in ndhA, and clpP were 23 bp with ‘5-ATTAAGGATTAAAGTAACAAAGA-3’ and 10 bp with ‘5-T…CAAGATTTT-3’, respectively (Fig. 2). ClpP gene are located in 73,417–75,471 bp and ndhA gene which corresponds to 124,649–126,895 bp. ClpP gene has three exons and two introns and all of SNPs and InDel sequences from these two intron regions in our experiment. We found two SNPs of clpP gene in the first intron with 73,810 and 73,811 bp (Table 1) and this could be detected in variable deletions based on 11 bp less and more in the second intron (Supplement Figure 3). NdhA gene has two exons and an intron and SNPs and Indel was detected from an intron region (Supplement Figure 4). Deleted sequences were the same in the maternal parent of the interspecific hybrids of Niitaka and previous mothers of Niitaka, such as Amanogawa, as shown in Fig. 2, lanes 1–8. Danbae (lane 12), Seolwon (lane 15), Cheongdangrori (lane 18) and Chanxixueli (lane 24) had the same deleted sequences in this study. However P. calleryana, OPR125 and OPR195 (lanes 19 and 2), P. faurie, Godang 5-1 (lane 23), P. communis, Bartlett and Max Red Bartlett (lanes 26 and 27), and Malus, Fuji (lane 28) did not have a deleted sequences of clpP gene. The comparative SNPs and InDel sequences of ndhA and clpP amplicons in 28 cultivars from 166 to 230 bp and from 486 to 530 bp, respectively, are summarized in Tables 2 and 3.

Agarose gel electrophoresis of the amplicons of the ndhA and clpP chloroplast gene-specific primers (left photo). These chloroplast InDel markers were found in the 27 Pyrus and one Malus cultivars. M: DNA size marker (Enzynomics DM003, 10 µl of 150 µg/ml). The electrophoresis was performed on a 2.5% (ndhA) and 3% (clpP) agarose gel and run for 8 h at 100 V (ndhA) and 16 h at 80 V (clpP). The amplified products with indels were represented with asterisk, respectively. We assigned the accessions, intercross parentage, and species to each lane number

Maximum likelihood phylogenetic tree of 27 Pyrus species based on chloroplast InDel marker ndhA gene sequences. The numbers on the branches indicate the bootstrap values from 1000 replicates. Fuji was used as an outgroup

Maximum likelihood phylogenetic tree of 27 Pyrus species based on chloroplast InDel marker clpP gene sequences. The numbers on the branches indicate the bootstrap values from 1000 replicates. Fuji was used as an outgroup
Phylogenetic tree
Phylogenetic trees were constructed by conducting maximum likelihood analysis with MEGA7 program and each ndhA and clpP amplicon sequences of 27 Pyrus with Malus being as the outgroup (Figs. 3, 4). It was found that the occidental pears (Bartlett and max red Bartlett) were located outside of the oriental groups. The P. faurie cv. Godang 5-1 was located between the Asian and European pears. Whangkeumbae, Gamro, Sunwhang, Sinhwa, Youngsanbae, and Hanareum contained maternally inherited chloroplast from Niitaka according to the Korean Pear Breeding Program. Considering that Niitaka was generated from the crossing between Amanogawa (mother) and Imamuraaki (father) (Kim and Nou 2016). Geumchonjosaeng, Chuwhangbae, and SuperGold were generated as maternally inherited chloroplast from Imamuraaki. Most of Pyrus genus come together with the same subgroups, however, two P. ussuriensis cultivars, Cheongdangrori and Doonggeullebae were divided into the other groups.
Discussion
Chloroplast sequences evolve relatively slowly and are highly conserved between species within a genus (Dong et al. 2014). Therefore, chloroplast sequences are ideal for species identification, phylogenetic research, and genetic diversity research, etc. With the decreasing cost of next-generation sequencing, sequencing of cp genome and development of cp-based DNA markers have attracted attention (Davey et al. 2011). In this study, we utilized Illumina sequencing to provide the genomic resource and ultimately the whole cp sequences of an Asian pear cultivar, from which we have expected to develop cp-based molecular markers. The obtained cp genome showed the canonical feature of cp genome: the size of higher plants ranging from 120 to 160 kb and a quadripartite structure consisted of a pair of inverted repeats (IRs), LSC and SSC regions.
While most of sequences were conserved, we found many SNPs in the protein-coding genes from the cp sequences comparison between the two cultivars, Niitaka and Wonwhang, which served as a fundamental resource for marker development, species identification, phylogenetic study, etc. Within the cp genome, the matK, rbcL, rpoB, rpoC1 and ycf1 genes are the main DNA makers used in identifying genetic diversity in land plants (Dong et al. 2015; Group et al. 2009). In this study, we also observed that matK and ycf1 was polymorphic (Supplement Table 2). Besides, ndhA and clpP genes were identified to be highly polymorphic and could be used for marker development. SNPs, as well as InDels, were discovered in ndhA and clpP genes at the level of Pyrus species and cultivars. Recently, InDel markers in cp have been used for genetic identification in many crops such as potato, quinoa, ginseng, onion, and soybean (Hong et al. 2017; Joh et al. 2017; Lee et al. 2017; Sohn et al. 2017; Cho et al. 2016). It is easy and inexpensive to use (Qiao et al. 2016). Our primer sets designed for two genes could easily detect InDels among subspecies of P. pyrifolia or species of Pyrus on the agarose gel (Fig. 2). Niitaka is an intercross cultivar of Amanogawa and Imamuraaki and is also used as maternal plants for Whankeumbae, Gamro, Sunwhang, Youngsanbae, and Hanareum. The breeding history of Niitaka-related plants was consistent with the genotyping result with the primer pairs for ndhA and clpP genes, as revealed by the same deletion events of 23 bp as ndhA (5′-ATTAAGGATTAAAGTAACAAAGA-3′) and 10 bp as clpP (5′-T…CAAGATTTT-3′) (Fig. 2). Danbae, Seolwon, and Cangxixuel of P. pyrifolia, Cheongdangrori of P. ussuriensis had the 23 bp, and 11 bp deleted sequences in their amplicons of ndhA and clpP, respectively. OPR125 and OPR195 of P. calleryana, Godang 5-1 of P. faurie, and Bartlett and Max red Bartlett of P. communis had the deletion in ndhA but not in clpP. The species P. pyrifolia and P. ussuriensis did not share the deleted sequences with similar species, P. calleryana, P. faurie, and P. communis, which had a different InDel. These ndhA and clpP genes could assist in classifying species and even subspecies.
The genetic relationships among the Pyrus species were identified. As expected, the European pears were located outside of the Asian pears. P. faurie was present between the Asian and European pears. Unclassified of Pyrus cultivar, Kozo was relevant to P. calleryana, P. bretschneideri and P.pyrifolia as maternally inherited chloroplast from Imamuraaki. We could confirm that it contained deleted sequences or not in these cultivars, and the deletion of ndhA gene occurred first, followed by deletion of clpP gene. A molecular population genetic analysis was previously performed with Japanese population of P. ussuriensis, and of cultivated pears. They cited the relationships among cultivars and wild populations and used true natives of P. ussuriensis that originated in Japan or on the Asian continent (Iketani et al. 2012). In our study, two P. ussuriensis cultivars, Cheongdangrori had the deletion of two genes together, but Doonggeullebae did not have the deletion. Cheongdangrori and Doonggeullebae were showed the different sub-group as phylogenetic relationship. Cheongdangrori belonged to maternally inherited chloroplast from Niitaka, while Doonggeullebae clustered into the other cultivars of P. pyrifolia, which included P. calleryana, and P. bretschneideri. The polymorphisms used for phylogenetic analysis were found in the short sequences of two cp genes, which was not enough to fully distinguish the species. With the completion of genomic sequencing and cp-genome assembly, the phylogenetic analysis will be repeated to confirm this present result and further understand the genetic relationship with the higher resolution.
Maternal origins of the Asian pears are still unclear because of frequent hybrid events, fast radial evolution, and lack of informative data. Pyrus pyrifolia cv. Niitaka is the leading cultivar accounting for over 80% of pear production in the Republic of Korea and has been used as a maternal source to develop new cultivars such as Sinhwa, Whangkeumbae, Sunwhang, Hanareum, and Youngsanbae. This study confirmed the breeding history of Niitaka-related plants as well as another important cultivar Imamuraaki-related group. Moreover, Danbae and Seolwon seemed to be generated from the Niitaka, but not their originally recorded maternal plant, and manpungbae, respectively. Admittedly, our results need to be confirmed with more genetic resources and molecular markers later.
The cp genomes are maternally inherited and have markers that are highly conserved within species, i.e., they show a lack of polymorphism. Here, we reported useful polymorphic genetic markers that can be used to examine plant genome evolution and differentiation in Rosaceae.
References
Bell RL (1991) Pears (Pyrus). Acta Hortic 290:657–700
Bobik K, Burch-Smith TM (2015) Chloroplast signaling within, between and beyond cells. Front Plant Sci 6:781
Cho K-S, Cheon K-S, Hong S-Y, Cho J-H, Im J-S, Mekapogu M, Yu Y-S, Park T-H (2016) Complete chloroplast genome sequences of Solanum commersonii and its application to chloroplast genotype in somatic hybrids with Solanum tuberosum. Plant Cell Rep 35(10):2113–2123
Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, Jansen RK (2006) The complete chloroplast genome sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol 23(11):2175–2190
Chung HY, Lee T-H, Kim Y-K, Kim JS (2017a) Complete chloroplast genome sequences of Wonwhang (Pyrus pyrifolia) and its phylogenetic analysis. Mitochondrial DNA Part B 2(1):325–326
Chung HY, Won SY, Kang S-H, Sohn S-H, Kim JS (2017b) The complete mitochondrial genome of Wonwhang (Pyrus pyrifolia). Mitochondrial DNA Part B 2(2):902–903
Daniell H, Lin C-S, Yu M, Chang W-J (2016) Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol 17(1):134
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12(7):499–510
Dong W, Liu J, Yu J, Wang L, Zhou S (2012) Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One 7(4):e35071
Dong W, Liu H, Xu C, Zuo Y, Chen Z, Zhou S (2014) A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs. BMC Genet 15(1):138
Dong W, Xu C, Li C, Sun J, Zuo Y, Shi S, Cheng T, Guo J, Zhou S (2015) ycf1, the most promising plastid DNA barcode of land plants. Sci Rep 5:8348
Group CPW, Hollingsworth PM, Forrest LL, Spouge JL, Hajibabaei M, Ratnasingham S, van der Bank M, Chase MW, Cowan RS, Erickson DL (2009) A DNA barcode for land plants. Proc Natl Acad Sci 106(31):12794–12797
Hong S-Y, Cheon K-S, Yoo K-O, Lee H-O, Cho K-S, Suh J-T, Kim S-J, Nam J-H, Sohn H-B, Kim Y-H (2017) Complete chloroplast genome sequences and comparative analysis of Chenopodium quinoa and C. album. Front Plant Sci 8:1696
Iketani H, Katayama H, Uematsu C, Mase N, Sato Y, Yamamoto T (2012) Genetic structure of East Asian cultivated pears (Pyrus spp.) and their reclassification in accordance with the nomenclature of cultivated plants. Plant Syst Evol 298(9):1689–1700
Jiang S, Zheng X, Yu P, Yue X, Ahmed M, Cai D, Teng Y (2016) Primitive genepools of Asian pears and their complex hybrid origins inferred from fluorescent sequence-specific amplification polymorphism (SSAP) markers based on LTR retrotransposons. PLoS One 11(2):e0149192
Joh HJ, Kim N-H, Jayakodi M, Jang W, Park JY, Kim YC, In J-G, Yang T-J (2017) Authentication of golden-berry P. ginseng cultivar ‘Gumpoong’ from a landrace ‘Hwangsook’ based on pooling method using chloroplast-derived markers. Plant Breed Biotechnol 5(1):16–24
Kim Y-K (2016) Diversity of Pyrus germplasm and breeding of red skin colored and scab resistant pear by using interspecific hybridization. Thesis, Chonnam National University, Jeollanam-do
Kim H-T, Nou I-S (2016) Confirmation of parentage of the pear cultivar ‘Niitaka’ (Pyrus pyrifolia) based on self-incompatibility haplotypes and genotyping with SSR markers. Korean J Hortic Sci Technol 34(3):453–460
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874
Lee J, Chon J, Lim J, Kim E-K, Nah G (2017) Characterization of complete chloroplast genome of Allium victorialis and its application for barcode markers. Plant Breed Biotechnol 5(3):221–227
Ohyama K, Fukuzawa H, Kohchi T, Shirai H, Sano T, Sano S, Umesono K, Shiki Y, Takeuchi M, Chang Z (1986) Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 322(6079):572–574
Qiao J, Cai M, Yan G, Wang N, Li F, Chen B, Gao G, Xu K, Li J, Wu X (2016) High-throughput multiplex cpDNA resequencing clarifies the genetic diversity and genetic relationships among Brassica napus, Brassica rapa and Brassica oleracea. Plant Biotechnol J 14(1):409–418
Rodríguez-Ezpeleta N, Brinkmann H, Burey SC, Roure B, Burger G, Löffelhardt W, Bohnert HJ, Philippe H, Lang BF (2005) Monophyly of primary photosynthetic eukaryotes: green plants, red algae, and glaucophytes. Curr Biol 15(14):1325–1330
Rubtsov G (1944) Geographical distribution of the genus Pyrus and trends and factors in its evolution. Am Nat 78(777):358–366
Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J 5(9):2043
Sohn H-B, Kim S-J, Hwang T-Y, Park H-M, Lee Y-Y, Markkandan K, Lee D, Lee S, Hong S-Y, Song Y-H (2017) Barcode system for genetic identification of soybean [Glycine max (L.) Merrill] cultivars using InDel markers specific to dense variation blocks. Front Plant Sci 8:520
Terakami S, Matsumura Y, Kurita K, Kanamori H, Katayose Y, Yamamoto T, Katayama H (2012) Complete sequence of the chloroplast genome from pear (Pyrus pyrifolia): genome structure and comparative analysis. Tree Genet Genomes 8(4):841–854
Wicke S, Schneeweiss GM, Müller KF, Quandt D (2011) The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol 76(3–5):273–297
Yeo DY, Reed BM (1995) Micropropagation of three Pyrus rootstocks. HortScience 30(3):620–623
Zhang Q (2010) Why does biparental plastid inheritance revive in angiosperms? J Plant Res 123(2):201–206
Acknowledgements
This work was carried out with the support of the Cooperative Research Program for Agriculture Science and Technology Development (PJ0133812018, PJ0104502017).
Author information
Authors and Affiliations
Contributions
JSK conceived and designed all experiments. JSK performed the experiments. HYC and SYW analyzed the data and wrote the paper. Y-KK contributed the materials. SYW and JSK revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that have no conflict of interest.
Ethical standards
The authors declare that the experiments comply with current laws of the countries in which the experiments were performed.
Data archiving statement
The chloroplast genome sequences of Pyrus has been deposited in the National Center Biotechnology Information (NCBI) under the accession number K904342 (Pyrus pyrifolia_cv. Niitaka), KX450876 (Pyrus pyrifolia_cv. Wonwhang), KX450877 (Pyrus pyrifolia_cv. Whangkeumbae), KX450884 (intercross cultivar of Pyrus pyrifolia and P. communis, Greensis), KX450880 (P. bretschneideri cv. Dangshansuli), KX450881 (P. bretschneideri cv. Yali), KX450883 (P. ussuriensis cv. Cheongshilli), and KX450882 (P. ussuriensis cv. Xioxiangshui).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chung, H.Y., Won, S.Y., Kim, YK. et al. Development of the chloroplast genome-based InDel markers in Niitaka (Pyrus pyrifolia) and its application. Plant Biotechnol Rep 13, 51–61 (2019). https://doi.org/10.1007/s11816-018-00513-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-018-00513-0