
Abstract
An inflammatory myofibroblastic tumor originating from the aorta is extremely rare. Here, we report a case involving a 41-year-old female patient with an aortic inflammatory myofibroblastic tumor. Although preoperative imaging showed a mediastinal mass indicative of invasive thymoma, surgical findings revealed that this mass originated from the aorta. The patient underwent partial resection of the aorta, including the mass with patch angioplasty. Based on the postoperative histological findings, the patient was diagnosed with an aortic inflammatory myofibroblastic tumor and is currently under outpatient follow-up.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammatory myofibroblastic tumor (IMT) is a rare neoplasm consisting of myofibroblastic spindle cells and various inflammatory cells. Although these tumors are usually considered benign, local recurrence and distant metastasis are commonly observed, suggesting the intermediate biological potential of these tumors. Since the first report of a pulmonary IMT, these tumors have been reported mostly in the lungs; however, extrapulmonary lesions, such as in the abdomen, retroperitoneal space, pelvis, and extremities, have also been reported [1].
Case
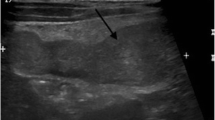
A 41-year-old female patient was admitted to the department of cardiology at our center, with the chief complaint of chest discomfort. Echocardiography revealed moderate pericardial effusion. Chest computed tomography (CT) revealed a 5.5-cm mass in the anterior mediastinum, along with pericardial effusion (Fig. 1a). The patient’s medical history was unremarkable, and routine preoperative examination findings were normal, except for slight elevation of the white blood cell count (11,900 cells/mm3). Cardiac magnetic resonance imaging (MRI) was performed, and while the boundaries of the mass were relatively clear and distinct, the possibility of its invasion into the aorta and pulmonary artery could not be completely ruled out (Fig. 1b).

a Chest CT scan (axial section) shows a relatively well-defined heterogeneously enhanced mass in the anterior mediastinum. b Cardiac MRI (sagittal section) shows a mass compressing the ascending aorta and proximal main pulmonary artery. CT computed tomography, MRI magnetic resonance imaging
Based on the preoperative imaging results, we suspected the patient of having invasive thymoma and planned to resect the mass by median sternotomy. However, contrary to our expectation, the mass was confirmed to be located in the intrapericardial space and not in the anterior mediastinal space. Following partial pericardiotomy, we could identify an irregular and solid mass in the upper region of the ascending aorta and proximal main pulmonary artery. The mass was carefully dissected from the right ventricular outflow tract, and we confirmed that it originated in the anterior wall of the aorta. We performed cardiopulmonary bypass through the femoral artery and right atrium to achieve complete resection. With sufficient safety margin (≥ 1 cm), partial resection of the aorta including the remnant mass and Dacron patch angioplasty was successfully performed. Total cardiopulmonary bypass time was 45 min (aortic cross-clamp time = 20 min).
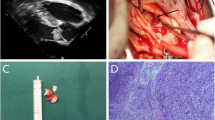
The patient was discharged on postoperative day 8, without any complications. Final pathology revealed that the tumor was composed of bland-appearing spindle cells and scattered inflammatory cells—mainly lymphocytes and plasma cells (Fig. 2a, b). On immunohistochemical staining, the tumor was positive for vimentin and anaplastic lymphoma receptor tyrosine kinase (ALK) and showed a positive signal pattern on ALK fluorescence in situ hybridization (FISH) test (Fig. 2c, d). Finally, IMT was diagnosed based on these findings. The resection margins and cytological examination of the pericardial fluid were negative for malignant cells. The patient did not undergo additional postoperative treatment, and no sign of recurrence was observed at the 6-months postoperatively. Regular follow-up imaging examinations have been planned for the patient.

Histopathologic findings (hematoxylin and eosin staining) show a a proliferation of bland myofibroblastic spindle cells within the loose fibrous background and b the spindle cells containing small nuclei with vesicular chromatin mixed with inflammatory cells (mainly lymphocytes and plasma cells with occasionally eosinophils and neutrophils). Mild cytologic atypia and pleomorphism are observed. c Immunohistochemical staining shows that the spindle tumor cells are positive for ALK and d ALK break-apart FISH positive specimen shows splitting red and green signals (circles). ALK anaplastic lymphoma receptor tyrosine kinase, FISH fluorescence in situ hybridization
Discussion
To date, IMT has been ambiguously known as an inflammatory pseudotumor, fibromyxoid tumor, plasma cell granuloma, inflammatory fibrosarcoma, or pseudosarcomatous myofibroblastic tumor. However, Gleason et al. suggested that IMT should be clearly distinguished from various neoplastic and reactive lesions and that the unqualified designation of “inflammatory pseudotumor” should be avoided [2].
IMT mostly develops in children and young adults, with the main site of onset being the lungs and abdominopelvic cavity. Several studies have reported the onset of IMT in various somatic soft tissues. As described in this report, an IMT originating from the aorta is extremely rare, although less than 50 cardiac IMTs have been reported thus far [3].
The clinical symptoms of an IMT can vary depending on the infiltrated organ and mass effect. In 15–30% of the patients, general inflammatory symptoms, including fever, weight loss, and malaise, are commonly observed; however, asymptomatic development of the mass is also a distinct possibility [1, 4]. Cantera et al. reported that IMTs are often suspected to be malignant neoplasms, based on radiologic features, while they are not in reality. Their predominant radiologic presentation is as solid, irregular, and well-defined masses, although these findings are non-specific [5].
A definitive diagnosis of IMT is made by histologic examination. The characteristic feature of IMT is proliferation of myofibroblastic spindle cells in a myxoid to collagenous stroma with prominent inflammatory infiltrates, including plasma cells and lymphocytes and/or eosinophils. Immunohistochemical analysis can be helpful, and the tumor is often positive for smooth muscle actin, vimentin, and desmin. Approximately 50% of the IMTs exhibit positivity for ALK, and this is reported to affect the prognosis; however, there is no consensus on this association, and ALK positivity is not always necessary for diagnosing IMTs [6].
While there are various treatment options, the gold-standard treatment for IMT has not been clearly established. The majority of the previous studies have recommended complete surgical resection, if possible [7]. While local recurrence and distant metastasis rates of extrapulmonary IMTs vary based on their location and resectability, the overall rates are reported to be 25% and 5%, respectively. Although recurrence is often observed within a year from the initial surgery, recurrence after 9 years was observed in a patient, suggesting the need for long-term follow-up [8].
Conclusion
In this case report, we described a rare case of IMT originating from the ascending aorta in a female patient, which was successfully treated through surgery. Although this tumor originated from a vital organ, we expect a good prognosis after successful complete resection. Long-term follow-up is required for this patient.
References
Coffin CM, Watterson J, Priest JR, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol. 1995;19:859–72.
Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol. 2008;61:428–37.
Luo W, Teng P, Ni Y. A rare cardiac inflammatory myofibroblastic tumor involving aortic valve. J Cardiothorac Surg. 2017;12:13.
Cho MY, Min YK, Kim NR, Cho SJ, Kim HK, Lee KC, et al. Fever of unknown origin as a presentation of gastric inflammatory myofibroblastic tumor in a two-year-old boy. J Korean Med Sci. 2002;17:699–703.
Cantera JE, Alfaro MP, Rafart DC, Zalazar R, Muruzabal MM, Barquín PG, et al. Inflammatory myofibroblastic tumours: a pictorial review. Insights Imaging. 2015;6:85–96.
Coffin CM, Hornick JL, Fletcher CDM. Inflammatory myofibroblastic tumour: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31:509–20.
Tian J, Cheng L, Yung T. Multiple cardiac inflammatory myofibroblastic tumors in the right ventricle in an infant. Ann Thorac Surg. 2006;82:1531–5.
Morotti RA, Legman MD, Kerkar N, Pawel BR, Sanger WG, Coffin CM. Pediatric inflammatory myofibroblastic tumor with late metastasis to the lung: case report and review of the literature. Pediatr Dev Pathol. 2005;8:224–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yun, J.S., Song, S.Y., Na, K.J. et al. Inflammatory myofibroblastic tumor arising from the ascending aorta mimicking a thymoma. Gen Thorac Cardiovasc Surg 68, 1193–1195 (2020). https://doi.org/10.1007/s11748-019-01231-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11748-019-01231-2