
Abstract
The study focuses on laboratory testing of carbon-dioxide adsorption. It compares three methods used to determine the capacity of the adsorbents proposed for CO2 capture. The tests have been performed on two samples, namely natural clinoptilolite and a 13X molecular sieve. Two methods were dynamic and one was static. The first dynamic method evaluated the capacity based on the change in CO2 volume fraction before and after a fixed-bed adsorber. For the same purpose, the second dynamic method used a gravimetric procedure. The static method detected a change in pressure during CO2 adsorption. The capacities determined for carbon-dioxide mole fractions of 5, 10, 13 and 20% at temperatures of 30 and 40 °C at atmospheric pressure were compared. The apparatus with a fixed-bed adsorber made it possible to perform tests also for overpressures of 2 and 5 bar and lower temperatures. The capacity obtained on the three apparatuses showed good agreement. The maximum capacity (3.32 mmol g−1) was determined for the 13X sample at 10 °C and a partial CO2 pressure of 120 kPa and, conversely, the lowest (0.12 mmol g−1) for clinoptilolite at 40 °C and a partial CO2 pressure of 5 kPa. There was no significant difference in capacity standard deviations between the apparatus (0.014–0.036 mmol g−1). The fixed-bed apparatus required one to three orders of magnitude more gas for the experiment and was more challenging to operate and evaluate the data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As part of the global effort to mitigate carbon-dioxide emissions, a large number of studies have been published on the adsorption of this greenhouse gas. However, the results discussed in them are based on different experimental and testing methods. Therefore, it is necessary to determine whether methods based on completely different principles are compatible.
To date, many materials have been proposed for the physical adsorption or chemisorption of carbon dioxide from flue gases or various waste gases. The literature most frequently discusses nine basic groups of adsorbents, namely activated carbonaceous adsorbents (mostly impregnated or chemically modified), natural zeolites and synthetic molecular sieves, silica gel, metal organic frameworks (MOFs), mesoporous silica, silica gel, γ-Al2O3 and hydrotalcite-like compounds (HTlcs). Advanced adsorbents usually have better adsorption characteristics balanced by higher cost. There are review studies on adsorption capacity, such as those published by Bonenfant et al. (2008), but these tend to compare the results obtained by several methods and under different conditions. Compared to older studies, chemically modified materials have been more frequently proposed for low-temperature CO2 sorption in recent years. For example, amine-impregnated silica gel has become very popular. Fujiki et al. (2017) demonstrated the achievement of higher capacities due to impregnation in a fixed-bed adsorber.
In view of the diversity of published experimental methods and types of sorbents, the text in the following does not comment on individual studies, but the data have been compiled in the form of Table 1. It provides examples of the adsorption capacity obtained with adsorbents from each of the above groups. The values given in the cited publications with other units have been converted to g kg−1. In the table, for clarity, the methods determining adsorption capacity have been divided into three categories, specifically: GR—gravimetric methods (thermogravimetry, magnetic suspension balance), CS—systems with an adsorption column and detection of the outlet CO2 content (IR analyzers, GC-TCD), and VA—static volumetric analyzers. Similarly, the adsorbents are divided in the table into: ZM—zeolites and molecular sieves, AC—activated carbonaceous sorbents, MOFs—metal organic frameworks, MPS—mesoporous silica, and ALU—alumina. The particular types and possible modifications of the adsorbents are also specified in Table 1.
From Table 1, it is evident that the adsorption capacity varies in a wide range, depending on experimental conditions, sample composition, etc. The published articles show that the laboratory techniques used for the determination of adsorption characteristics are dominated by the methods listed in Table 1, i.e., gravimetric, adsorption-column and CO2-detection, and finally volumetric (or manometric) methods. The question thus arises of whether the data obtained with the different laboratory methods are comparable. Nowadays, it is no longer merely a question of the state of scientific knowledge on the academic level, but of providing relevant data for the users of adsorption technologies. Thanks to international agreements and commitments on CO2 mitigation, the time has come when technologists need these data to implement CO2 capture on an industrial scale.
The largest number of publications on carbon-dioxide adsorption focuses on its post-combustion capture in power generation. Several review articles have been written on this subject, including those published by Samanta et al. (2012). CO2 capture from flue gas has in some cases been transferred from laboratory scale to pilot units. This was done mainly in states with a high proportion of energy production from fossil fuels. For example, the design and operational parameters of the vacuum pressure swing adsorption unit were addressed by Wawrzyńczak et al. (2019) and Majchrzak-Kucęba et al. (2021).
However, there is currently a relatively rapid shift away from coal-fired power plants in the EU and elsewhere in the world; therefore, there is no need to discuss this topic here. Just as post-combustion CO2 capture would work in fossil fuel-fired power plants, it can be implemented in biomass installations. Luberti et al. (2017) have developed a mathematical model describing the parameters of post-combustion CO2 capture by vacuum-pressure swing adsorption (VPSA) with 90% efficiency using 13X zeolite. The model simulates CO2 capture at an existing combined heat and power (CHP) plant in Gussing, Austria, which gasifies biomass and burns the resulting syngas in a gas engine.
Not only are the developed adsorption technologies applicable for the mentioned purpose, but CO2 adsorption can also be used in other industrial processes. The literature most frequently mentions hydrogen and biogas purification. Other applications include, for example, the treatment of metallurgical gases, the extraction of carbon dioxide from air, and the purification of end gas from cement production. Examples of these applications are given below.
Divekar et al. (2021) addressed the improvement of parameters for the recovery of pure H2 and CO2 from the natural gas steam-reforming process. For this purpose, they designed a PSA unit for hydrogen separation followed by a VSA unit producing CO2-rich gas. The adsorbers operated with a layered bed consisting of 13X zeolite and activated carbon. The same issue of the steam reforming of methane was also addressed by Liu et al. (2020). The proposed technology works according to the VPSA principle. Unlike the system above, carbon dioxide is recovered in the first stage and hydrogen in the second stage. A mathematical model developed in the study indicated that fixed-bed adsorbers would produce CO2 with a purity of up to 95.31%. The adsorbent proposed for the first stage was silica gel, and for the second stage, a combination of zeolite 5A and activated carbon was used.
CO2 separation from biogas was investigated by Vogtenhuber et al. (2018), who developed a mathematical model of a multistage process with fluidized bed operating in TSA mode. Polyethyleneimine-modified silica was selected as the sorbent. For the same purpose, Shen et al. (2018) proposed a VPSA process using pure silica gel in fixed-bed adsorbers. According to the simulation performed, it is thus possible to obtain CO2 with a purity of 96.74% and a yield of 97.58%. The PSA method was proposed for the separation of CO2 from biomethane by Abd et al. (2022). Using a mathematical model based on experimental data, they verified that spent coffee grounds can also be used for biogas purification in a fixed-bed column instead of a commercial sorbent. The only activation step in this case was degassing at 120 °C. The study showed, among other things, that a biogas-moisture content in the range of 3–10% improves the adsorption of CO2 in the adsorbent used.
Bjerge and Brevik (2014) published successful tests with CO2 adsorption in a Norcem cement plant. The installed fluidized-bed equipment operating in TSA mode was used to compare the process with an amine scrubber, membrane separation, and the regenerative calcium cycle, which were tested in parallel.
Saima et al. (2013) described the results of tests on a PSA pilot unit for the selective separation of CO and CO2 from blast-furnace gas. On a unit with the maximum daily capacity of 3 tons of CO2, it was possible to achieve the maximum CO2 purity of up to 99.5%. The system operated with a fixed bed of Zeolum F-9 molecular sieve.
Santori et al. (2018) designed an adsorption system using Zeolite 13X to recover highly concentrated CO2 from air. To achieve a mole fraction > 0.95, the system required five fixed-bed adsorbers arranged in serial order.
Experimental
Study objectives
From the literature cited in the introduction, it is clear that the state of knowledge in the field of the adsorption separation of carbon dioxide from flue gases and other gaseous mixtures is currently good. There is a wide range of promising adsorbents, and many of their key parameters can only be determined experimentally. The question arises, however, as to what extent the published data, particularly the adsorption capacity, are comparable. With the introduction of CO2 adsorption into practice, the need to have fast and reliable methods to verify the necessary properties of sorbents will grow as well.
Therefore, the study has four objectives, namely:
-
(a)
To determine the compatibility of the data on sorption properties obtained by three fundamentally different procedures
-
(b)
To compare the sorption properties of unmodified natural zeolite with a commercially available synthetic molecular sieve
-
(c)
To compare the three selected test methods in terms of user characteristics such as acquisition cost, operating cost, the complexity of operation, and the time demands of the experiment
-
(d)
To discuss the correlation of the experimental results with the Langmuir adsorption isotherm.
Experimental methods
Three different methods, each based on a different principle, were deliberately chosen to test the sorption behavior of the samples.
Two methods worked under dynamic conditions, where the gas flowed through the device during the experiment, and one method was static. The static method detected the pressure change associated with the transition of CO2 from the gas phase to the solid sample. The first dynamic method was based on detecting the change in the volume fraction of CO2 in the gas upstream and downstream of the adsorber. The second dynamic method was gravimetric and thus based on recording the mass change of the sample caused by CO2 adsorption.
Each of these methods used different equipment, with the first two being commercially available solid-sorbent analyzers and the third being a laboratory apparatus of our own design. Specifically, the instruments were an Autosorb iQ gas-sorption analyzer (hereafter called the Autosorb iQ), the DVS Advantage Surface Measurement System (hereafter called the DVS Advantage), and a laboratory flow PSA/TSA apparatus with a fixed bed (hereafter called the fixed bed). The main technical parameters, principles, and differences between the devices compared are summarized in Table 2.
Table 2 shows, among other things, the large difference in sample mass between commercially available analyzers and the apparatus of our own design with a fixed bed. The diametrically different adsorber size in the latter apparatus was deliberately chosen. The laboratory apparatus working with a sample mass two orders of magnitude larger than that of the adsorption analyzers made it possible to bring the experimental conditions closer to the parameters of real adsorption technology.
The samples tested
The selected test methods were applied to two porous inorganic materials that are commonly used as industrial and laboratory adsorbents.
The first sample was natural zeolite clinoptilolite mined at the site of Nižný Hrabovec in Slovakia (supplied by Zeocem, a.s., Slovakia). The material is currently supplied commercially for applications in agriculture, construction, waste- and service-water treatment, and flue-gas treatment. Since the material did not undergo any physicochemical modification prior to testing, it can be described as a low-cost sorbent.
In contrast, the second sample was the synthetic molecular sieve 13X (manufactured by Sigma-Aldrich, USA), which is one of the more expensive adsorbents supplied with certified composition and parameters.
The natural-zeolite sample was in the form of crushed irregularly shaped particles sieved to a fraction of 2–4 mm. On the other hand, the 13X molecular-sieve sample consisted of monodisperse spherical particles with a diameter of 2 mm. In this initial form, the samples were tested only on the flow apparatus of our own design. Before their testing on both commercial analyzers and also for all chemical analyses (except for bulk-density determination), the samples were ground in an agate mortar.
The principle of the Autosorb iQ analyzer
The Autosorb iQ instrument is an automatic analyzer designed to measure multiple parameters. It makes it possible to determine the specific surface area, the total pore volume, and pore-size distribution by measuring the nitrogen vapor adsorption at 77 K (− 196 °C) and by its subsequent evaluation using the BET (Brunauer–Emmett–Teller) and BJH (Barrett–Joyner–Halenda) equations. In addition, it is used to evaluate physical adsorption and chemisorption using different gases. It belongs to the group of so-called manometric (or also vacuum volumetric) analyzers. The method is based on the measurement of the adsorption isotherm under static conditions, where a known amount of gas (adsorbent) is sequentially dosed into a cell containing a sample of solid material. The quantity measured and mathematically evaluated during gas dosing is pressure. It is detected by means of electronic pressure transducers. Depending on the desired measurement temperature and the purpose of the analysis, the cuvette with the sample is placed in a Dewar vessel containing liquid refrigerant, in an electric resistance furnace, or in a thermostatic unit. Gas desorption even from micropores is ensured by an integrated turbo-molecular pump.
The instrument used had two degassing ports and one measuring station. One port was reserved for chemisorption. The apparatus included a blast furnace coupled with fan-assisted cooling. In the study presented here, the Autosorb iQ instrument was dedicated to a single purpose, namely the measurement of CO2 adsorption isotherms. The textural properties of the samples were determined on a different instrument.
The principle of the DVS Advantage analyzer
The DVS Advantage instrument belongs to the group of gravimetric sorption analyzers. It enables the measurement of adsorption and desorption isotherms over a wide range of humidity and temperature. The core of the apparatus is a microbalance module housed in a temperature-controlled box. The microbalance has two arms. In one of them, the quartz crucible contains a sample of the sorbent under test, whereas the other arm holds a reference (inert) material. Sorption and desorption characteristics are evaluated on the basis of the mass difference between the two materials. The measurements are taken under dynamic conditions where the gas flows through the weighing module over the sample. In addition to dry-gas measurements, the system is adapted to test the sorption of water vapor or the vapor of volatile organic compounds. For this purpose, it is equipped with a pair of saturators, in which the gas is enriched with a defined amount of the respective vapor. Constant steam partial pressure is achieved by circulating the gas in a closed loop. For this study, however, dry gas was used.
The principle of the fixed-bed flow apparatus
Measurements in the self-designed flow apparatus are taken under dynamic conditions, i.e., under a defined flow of a gaseous mixture containing a known concentration of CO2. By mathematically evaluating the measured breakthrough curves, it is possible to obtain the adsorption-capacity value as well as other data such as the sorption rate, separation efficiency under the given conditions, and specific sorbent consumption.
Adsorption and desorption are monitored by an online analysis of the gas flowing through the apparatus. The principle of the evaluation is to compare the content of CO2 in the gas entering the adsorber and in the gas leaving the adsorber. Adsorption can take place at atmospheric or elevated pressure, with the flow of dry or wet gas mixtures up to a maximum relative humidity of 90%. Desorption may be carried out by increasing the temperature at constant pressure, by decreasing the pressure at constant temperature, or by performing a combination of both. The interconnection of the components of the apparatus is shown in the scheme in Fig. 1.

Scheme of the fixed-bed apparatus: 1, 2—pressure cylinders with gas, 3—gas flow meter and controller, 4—total bypass three-way valve, 5—humidity meter display, 6—safety valve, 7—control manometer, 8—ball valves and needle valves of a humidifier, 9—humidifiers, 10—pressure capsule of the humidity probe, 11—heat exchanger, 12—adsorber bypass three-way valve, 13—adsorber, 14—climate chamber, 15—liquid chiller, 16—condensate collector, 17—back-pressure valve, 18—thermometer/datalogger, 19—PC for data acquisition, 20—IR analyzer, 21—IR analyzer bypass, 22—gas meter
With respect to the diverse principles and designs of the apparatus, the experimental procedures also differed. The specific conditions under which the tests were performed on all three devices are summarized in Table 3. More precise technical parameters of the fixed-bed flow apparatus as well as a more detailed description of the experimental procedure are summarized in appendix.
Supporting analytical methods
In addition to the measurement of their adsorption properties, the samples were subjected to further analyses. Characterization of the samples included the following parameters: elemental-composition determination, crystalline-phase determination, density determination, and textural analysis.
Elemental-composition analysis was performed by X-ray fluorescence spectrometry (XRF) using an XRF spectrometer ARL 9400 XP + (manufactured by Thermo Fisher Scientific, USA). In principle, the method is not capable of detecting the following elements: H, He, Be, B, C, N, O, and F. It was thus necessary to complement the XRF analysis with another method, making it possible to describe the mineralogical composition of the samples.
The method used was X-ray diffractometry (XRD), which is a key procedure for the analysis of the structure of crystalline substances. It was performed on an X´Pert PRO X-ray diffractometer (manufactured by PANalytical, the Netherlands). The crystalline phases present were identified by comparing the diffractograms with the PDF-4 + (Powder Diffraction File) database. The Rietveld method was used for quantification.
What is important for the practical use of sorbents is not only the bulk density of the material but also its real and apparent density. The real density was determined using an Ultrapyc 5000 helium pycnometer (manufactured by Anton Paar QuantaTec Inc., USA), working on the principle of measuring the He 6.0 pressure difference between the measuring and reference cells at constant temperature. The apparent density was established using a PoreMaster 60 mercury porosimeter (manufactured by Anton Paar GmbH, Austria). The bulk density was not determined by any instrumental analysis system, but the EN ISO 23145–2 (727,516) standard was followed. Since there is no specific standard for adsorbents, the selected procedure is considered relevant.
The specific surface area, total pore volume, and pore-size distribution were assessed by a Coulter SA 3100 automatic analyzer (manufactured by Beckman Coulter, Inc., USA). The system uses reversible nitrogen vapor adsorption at the boiling point of liquid nitrogen at atmospheric pressure (− 196 ºC). After the formation of a molecular monolayer on the sample surface, the specific surface area is calculated using the BET equation based on the knowledge of the area occupied by one N2 molecule and the number of adsorbed molecules. The next step is adsorption, which leads to the formation of multilayers, and pore-size distribution is calculated from the obtained adsorption isotherm by the BJH method. Once the pores are fully filled, the total pore volume is determined on the basis of the knowledge of the adsorbate density.
Mathematical evaluation
The adsorption-capacity values obtained independently by the three different instrumental procedures were used to calculate adsorption isotherms and to represent them graphically. Each of the known models is based on certain physicochemical prerequisites and uses a certain degree of simplification. In studies dealing with the adsorption of carbon dioxide on various materials, the authors most often fit the measured values with the Langmuir adsorption isotherm. The model is based on the assumptions that adsorption is only monolayer, all adsorption sites are equivalent, and adsorbed particles do not interact. Singh and Kumar (2016) used this model for CO2 adsorption in activated carbon at temperatures of 25–65 °C and relatively high pressures up to 45 bar. At lower pressures, the agreement with the Langmuir model was good, but at high pressures, the Dubinin–Astakhov model proved to be better in this case.
Liu et al. (2021) obtained very good agreement when applying the Langmuir model to the adsorption of CO2 in polyethyleneimine-impregnated essential oils at temperatures of 30, 50, and 70 °C and pressures up to about 32 kPa. The Langmuir model was also successfully applied by Yusuff et al. (2018) to test the adsorption of CO2 in alum sludge at 30 °C and an absolute pressure of 1 bar. Weishauptová et al. (2015) tested the adsorption of CO2 and CH4 on asphalt coal at pressures up to 15 MPa and a temperature of 45 °C. At such a high pressure, the Langmuir isotherm in standard form worked only for methane. Fitting the values for CO2 required the use of a modified equation supplemented with the density ratio of the gas phase to the adsorbed phase. Li et al. (2022) successfully used the Langmuir model to describe the adsorption of carbon dioxide in organic-rich shales. The data were obtained at 30 °C up to a maximum pressure of 5 MPa.
In papers published by Siqueira et al. (2017), Luberti et al. (2017), and Shen et al. (2018), an extended Langmuir model was recommended for fitting the measured capacity. In the first step, a calculation was performed for the adsorption of the pure components used to form the mixture. For the interpretation of the measured values for a pure component i, the Langmuir equation was used in a linearized form derived from its definition relation as follows:
where Θi is the degree of sorbent-surface coverage, ai is the amount of a component i actually adsorbed per unit mass of the adsorbent (mol g−1), aσ,i is the maximum amount of a component i adsorbed per unit mass of the adsorbent to form a saturated monolayer (mol g−1), p is the absolute pressure (Pa), and bi is the equilibrium constant of a component i. When expressed graphically, the dependence of p/ai on p represented a straight line with the slope 1/aσ,i and the segment 1/(aσ,ibi). The general form of the equation for an unlimited number of components was as follows:
where pi is the partial pressure of a gaseous component i, which can be expressed as the product of the molar fraction φi and the total pressure p, as reported by Siqueira et al. (2017), Luberti et al. (2017), and Shen et al. (2018).
Results and discussion
The basic characterization of the samples
For most laboratory analyses, the samples were prepared by grinding in an agate mortar. The only exception was the determination of the bulk density, for which samples were used in the initial granulometry. The basic parameters of the samples are summarized in Table 4.
The 13X molecular-sieve sample was formed by 95 wt. % of the crystalline phase of Faujasite-Na. The rest comprised a group of other related aluminosilicates. The elemental composition was consistent with the mineralogical composition described. Si, Na, and Al were determined as the major elements. Among the other elements, only Mg and Fe were significant. The other elements were either not detected at all or their mass fractions were below 0.1%.
The natural-zeolite sample was obviously more mineralogically diverse than the synthetic molecular sieve. In total, the crystalline phase detectable by XRD represented > 94%. This crystalline phase was dominated by clinoptilolite, but cristobalite, albite, quartz, and muscovite with an admixture of Mg also had significant mass fractions in the order of units of percent. In terms of elemental composition, Si, Al, K, Na, Ca, and Mg were predominant. The content of the other elements of the periodic system was either < 0.1%, or no elements were detected at all.
In comparison with natural zeolite, the molecular sieve had more than an order of magnitude larger specific surface area and almost three times the total pore volume. Therefore, it was expected to have adsorption capacity much better than that of zeolite.
When comparing the values from Table 4 with those mentioned by other authors, it is necessary to draw attention to their relatively wide range caused by different natural origin or sorbents production methods. For example, McEwen et al. (2013) reported the specific surface area of 488 m2 g−1 and the total pore volume of 0.27 cm3 g−1 for 13X. In contrast, Davarpanah et al. (2020) recorded the specific surface of 745 m2 g−1 and the total pore volume of 0.44 cm3 g−1 for the same adsorbent. The same author gives a specific surface area of 37 m2 g−1 and a total pore volume of 0.14 cm3 g−1 for clinoptilolite.
The pore-size distribution presented in the form of numerical data would not have been clear and was thus processed in the form of a bar chart in Fig. 2. Inorganic samples with such different parameters were deliberately chosen to enable a comparison of the applicability of the apparatus for testing high- and low-capacity adsorbents.

Pore-size distribution
Measured data and their processing
The DVS Advantage and Autosorb analyzers performed capacity evaluations automatically based on the total mass change of the sample and a change in the pressure in the measuring cell, respectively.
In contrast, the fixed-bed apparatus required the manual capacity calculation from breakthrough curves. An example of these curves obtained for a 13X sample exposed to 13% (mol.) of CO2 at 200 kPa overpressure is shown in Fig. 3 and at 500 kPa overpressure in Fig. 4. The graphs were supplemented with the blind-experiment curve, which played an integral role in the evaluation. The breakthrough curves in their initial form did not provide data on adsorption capacity, but it is possible to deduce some information from their course. The curves were similar in shape for both samples and for all measurements, with three recognizable phases. In the initial phase, the rate of sorption was such that CO2 was completely removed within the residence time of the gas in the sample layer. The second phase began with the breakthrough of CO2 through the sorbent. The time at which the first breakthrough was observed decreased with increasing experimental temperature.

The breakthrough curves of 13X measured at the overpressure of 2 bar

The breakthrough curves of 13X measured at the overpressure of 5 bar
In the second phase, the detected outlet CO2 content increased more or less linearly over time. With increasing temperature, there was always a change in the tangent slope (see Figs. 3 and 4). At temperatures of 30 and 40 °C, the CO2 content in the outlet gas after the breakthrough was observed to increase very steeply to 3/4 of the inlet concentration for both samples. On the contrary, at lower temperatures, the curve was more gradual. The third and last phase of adsorption was characterized by a very slow increase in the outlet concentration. Under the conditions used in the flow apparatus, the equilibrium state was reached approximately 1–3 h after the observed breakthrough. The time was again prolonged with decreasing temperature. These findings are valid for both samples tested. The 13X sample differed from the natural zeolite essentially only by a significantly longer first phase of the curve before the breakthrough occurred.
A study published by Nedoma et al. (2022) already proposed a mathematical model describing the breakthrough curves for the material and conditions tested here. The model fit well the experimental data measured using the fixed-bed apparatus described here.
The mathematical evaluation of the breakthrough curves was used to calculate the capacity and proceeded as follows. Data on temperature, gas flow rate, and outlet CO2 content were recorded synchronously at the frequency of 5 s. At each sub-interval, the adsorbed amount of CO2 was calculated from the flowed-gas volume at a given pressure and the difference between the input and outlet CO2 content. After equilibrium was reached, the adsorbed amounts of CO2 were summed in all five-second intervals. The resulting adsorption capacity was corrected by subtracting the value of the blank.
A comparison of the capacity determined by different methods
The equilibrium adsorption capacity determined by the three methods has been processed into bar graphs. Figure 5A compares the results for the 13X sample, and Fig. 5B shows the same for the natural-zeolite sample. The two comparative graphs include only those values that could be determined by all the apparatus used considering the technical limitations summarized in Table 2. The Autosorb has the minimum temperature limited by the ambient value in the laboratory, and the DVS Advantage is at risk of corrosion damage at higher CO2 concentrations. Therefore, the comparison involved only temperatures of 30 and 40 °C and the maximum volume fraction of 20% CO2 at 1 barabs pressure. For these reasons, each of the graphs compares the capacities determined using three apparatuses at four CO2 partial pressures (5, 10, 13, and 20 kPa) and two temperatures.

Adsorption capacity of 13X A and natural zeolite B measured by different methods
As can be seen in Figs. 5A and B, the results obtained on the fixed-bed apparatus and the DVS Advantage were in very good agreement. In contrast, the Autosorb gave slightly lower capacity values at all partial pressures of carbon dioxide and at both compared temperatures. Similar differences were also detected for other samples not included in this study. It is speculated that the reason for this could be the limited transport of gas molecules in the static atmosphere of the Autosorb apparatus in comparison with the dynamic gas flow in the fixed-bed and DVS Advantage. However, this assumption could not be verified.
The adsorption capacities in Fig. 5A and B are the arithmetic means of five measurements realized by cyclic repetitions of adsorption and desorption. Arithmetic means could be used because none of the samples showed a trend of decreasing (or increasing) capacity in individual cycles. Thus, there was no samples wear. The dependences of capacities on temperature and CO2 partial pressure are described in detail in separate chapters. However, trends can also be observed in Fig. 5. At 30 °C, for example, an increase in the partial pressure of CO2 from 5 to 20 kPa led to the following changes in the measured values. The capacities of the 13X measured by the fixed-bed apparatus increased from 0.48 to 1.32 mmol g−1, measured by the DVS from 0.53 to 1.30 mmol g−1 and measured by the Autosorb from 0.39 to 0.99 mmol g−1. The natural zeolite sample showed all capacities significantly lower than 13X. The change in the CO2 partial pressure from 5 to 20 kPa led to the increase in the capacity determined using the fixed-bed apparatus from 0.16 to 0.36 mmol g−1, using the DVS from 0.16 to 0.35 mmol g−1 and using the Autosorb from 0.11 to 0.30 mmol g−1.
Comparison of experimentally determined capacities with the literature is often difficult. The reason is both the different origin and composition of the sample and different experimental conditions. McEwen et al. (2013) pointed out a wide range of the capacities of zeolite 13 X obtained after the application of different outgassing temperatures. When comparing the capacities obtained by them with the values in the study presented here, a very good agreement can be noted. Davarpanah et al. (2020) report for zeolite 13X at 20 °C and a CO2 partial pressure of 10 kPa the capacity about 30% higher than the value measured here. Some discrepancies in published results demonstrate the difficulty of mutual comparing capacities, e.g., Najafi et al. (2023) report a capacity of zeolite 13X of 202.4 g kg−1 at a temperature of 25 °C and CO2 of 1 bar. This value is very high, as, e.g., Zhao et al. (2007) achieved only 214.2 g kg−1 under the same conditions even after alkaline modification.
Unlike natural zeolites, however, 13X compares slightly better because of its synthetic origin with more uniform properties. For example, in the study presented here, natural clinoptilolite of the given purity reached a capacity of 14.1 g kg−1 at 10 °C and CO2 partial pressure of 10 kPa. Davarpanah et al. (2020), however, report almost three times higher value for another clinoptilolite under the same conditions. Cavallo et al. (2023) determined at 25 °C the capacity of clinoptilolite to be even 88.0 g kg−1, i.e., significantly higher than the previous authors (for lower temperature).
All capacities shown in Figs. 5A and B were supplemented with error bars with the values − σ/ + σ, where σ represents the standard deviation. There was no significant difference between the standard deviations achieved by each device. In the case of the sample 13X, the standard deviation was in the range of 0.019–0.036 mmol g−1 for the fixed-bed apparatus, 0.014–0.035 mmol g−1 for the DVS Advantage, and 0.018–0.034 mmol g−1 for the Autosorb. In the case of natural zeolite, the standard deviation reached similar values, namely: 0.016–0.030 mmol g−1 for the fixed-bed apparatus, 0.018–0.033 mmol g−1 for the DVS Advantage, and 0.014–0.033 mmol g−1 for the Autosorb.
The standard deviations showed no dependence on absolute pressure, CO2 partial pressure, or temperature (see Figs. 5A and B). However, the situation was different in the case of the coefficient of variation, defined as the ratio of the standard deviation to the arithmetic mean and expressed as a percentage. As the sorption capacity increased with CO2 pressure and decreased with temperature, the value of the coefficient of variation also changed. The minimum coefficients of variation were always determined at the highest CO2 pressure and the lowest temperature, whereas the maximum values were determined under opposite conditions. The coefficients of variation of the capacity of the 13X sample were in the range of 1.4–8.5% for the fixed-bed apparatus, 1.3–8.2% for the DVS Advantage, and 2.3–10.3% for the Autosorb. For the reasons described, the coefficients of variation calculated for the natural zeolite were in the range of 6.0–25.9% for the fixed-bed apparatus, 5.3–28.5% for the DVS Advantage, and 4.5–41.8% for the Autosorb.
The temperature dependence of the capacity
The dependence of the adsorption capacity on temperature is shown in Figs. 6A and B. Six isobars obtained by measurements on a fixed-bed apparatus were plotted in the graph for each sample. Only for the sake of clarity, the graphs do not include the results for all tested CO2 partial pressures given by combinations of φCO2 and working pressures (see Table 3).

The temperature dependence of 13X A and natural-zeolite B adsorption capacity with marked trend lines for different partial pressures of CO2
The capacity of the sample 13X decreased from the maximum value of 3.32 mmol g−1 (0.146 g g−1), reached at 10 °C and the CO2 partial pressure of 120 kPa, to 0.42 mmol g−1 (0.019 g g−1) at 40 °C and 5 kPa. The natural-zeolite sample also showed a significant decrease in the equilibrium capacity with increasing temperature and decreasing CO2 partial pressure. The maximum was 1.03 mmol g−1 (0.045 g g−1) at 10 °C and the CO2 partial pressure of 120 kPa, whereas the minimum of 0.12 mmol g−1 (0.005 g g−1) was determined at 40 °C and 5 kPa.
From the values for only four temperatures, it was not unambiguously possible to define the shape of the isobars; however, a simple quadratic function was suitable for fitting the points. In the case of the 13X sample, the trend lines were slightly convex in shape, whereas they were rather concave in the natural-zeolite sample. Nevertheless, because of the small number of points mentioned, it would be incorrect to draw conclusions from these findings regarding the physical or chemical nature of the sorption.
In principle, the practically (industrially) usable capacity is mostly lower than the equilibrium value. One possibility is to select a certain minimum sorption rate below which the capacity would no longer be counted. All three instruments allow the sorption rate to be determined.
The fixed-bed apparatus also offers the possibility of graphically estimating the usable capacity directly from the measured breakthrough curve. The procedure consists in interleaving the lines with two successive phases of the breakthrough curve as shown in Fig. 7. The first line linearizes the steep increase in the outlet volume fraction of CO2 after its breakthrough through the sample layer. The second line describes the linear trend of slow increase in CO2 content in the last phase of adsorption, when its molecules diffuse into the nearly saturated structure of the adsorbent. This phase of adsorption until equilibrium is reached is no longer of practical significance and should not be included in the capacity. The intersection of the two lines shows the transition time between these phases of the adsorption process. The intersections have been calculated from the slope shape of the lines of the dependence of the CO2 outlet volume fraction on time ((φCO2 = kt + q, where k and q are constants).

An example of the graphic determination of the practically usable capacity (20 °C, overpressure of 500 kPa, φCO2 = 20%)
For the sample 13X, the technically usable capacity was determined as described as 74.4–78.8% of the equilibrium capacity. The natural-zeolite sample had a significantly lower percentage of technically usable capacity, specifically 66.8–70.3% of the theoretical capacity. The determined ratios between the usable and theoretical capacity showed no dependence on temperature, CO2 partial pressure, or the total system pressure.
Breakthrough curves can also be used to calculate other characteristics such as breakthrough time or rate constant for a given pressure, temperature, and gas flow. An example is the Wheeler-Jonas equation, which was chosen based on the positive results obtained by Zhou et al. (2011) and others.
where tx is time (min), a is adsorbed amount (g g−1), m is weight of the adsorbent (g), φA,0 is the input volume fraction of the adsorbate, φA,x is the output volume fraction of the adsorbate in tx, Q is gas flow rate (m3 min−1), Kv is rate constant (min−1), and ρB is bulk density of the bed (g cm−3).
The following values determined for 20 °C, an overpressure of 5 bar, and an inlet volume fraction of CO2 of 20% are an example of the application of the equation. Kv of 0.033 min−1 for the sample 13X and 0.019 min−1 for natural zeolite was obtained. The calculated time to reach 50% and 95% input CO2 concentration was 22.78 and 62.43 min in the case of 13X. The difference between the experimentally determined and calculated time was 2.4% and 9.4%, respectively. In the case of natural zeolite, the calculated times were 6.89 and 22.12 min, which represented a difference compared to the measured data of 3.8 and 9.2%, respectively.
The correlation of experimental values with the Langmuir model
In agreement with the studies published by Siqueira et al. (2017), Luberti et al. (2017), and Shen et al. (2018), preference should be given to the extended Langmuir model. Nevertheless, the problem with all the test equipment used was the measurement of the isotherms of pure N2 and CO2. The fixed-bed apparatus with IR detection was fundamentally unable to determine the capacity for N2. The DVS Advantage and Autosorb systems, on the other hand, are able to measure pure components, but not in the required pressure range. Consequently, only control measurements of the adsorption capacity for N2 were taken at atmospheric pressure with the following results. At atmospheric pressure, the adsorption capacity for N2 in the sample 13X decreased from 0.0035 g g−1 at 10 °C to 0.0020 g g−1 at 40 °C. Therefore, these values are an order of magnitude lower than those recorded for CO2. In the case of natural zeolite, the situation was similar, as the capacity for N2 under the same conditions decreased from 0.0016 to 0.0015 g g−1. Based on the measurements described above, it appeared that the problem of parallel N2 sorption was not essential for the mathematical model. For that reason, the effect of N2 adsorption was neglected and the dependence of the adsorbed amount of CO2 on its partial pressure was calculated using the equation:
where the meaning of the symbols is the same as in Eq. (2).
Based on Eq. (4), linearization was performed for both samples, which provided the plots shown in Figs. 8 and 9, expressing the dependence φCO2p/a on φCO2p for CO2 partial pressures of 5, 10, 13, 15, 20, 30, 39, 60, 78, and 120 kPa. These corresponded to the respective combinations of the used CO2 molar fractions and the total pressures in the adsorber. The graphs were constructed using the capacity data obtained from the flow apparatus, because this instrument provided data over the widest range of pressure and temperature. For each temperature, the constants aσ and b were obtained by linearization (see Table 5).

Data for 13X linearized by the Langmuir model

Data for natural zeolite linearized by the Langmuir model
Subsequently, a back-calculation was performed to calculate the theoretical capacity values for the respective CO2 partial pressures and temperatures mentioned above using the Langmuir model. A comparison of the calculated adsorption isotherm curves with the actual measured capacity values is shown in Fig. 10 for the 13X sample and in Fig. 11 for the natural-zeolite sample. From the linearized plots in Figs. 8 and 9, and especially from Figs. 10 and 11, it is evident that the Langmuir model is very well applicable to both tested samples.

Correlation of the measured 13X adsorption capacity with the Langmuir model

Correlation of the measured adsorption capacity of natural zeolite with the Langmuir model
A comparison of the experimental apparatus and methods from the user’s perspective
All three devices have been found to be equivalent in terms of the comparability of results and variability. They differed significantly in some parameters, especially from the researcher/user perspective. The acquisition cost of the DVS Advantage and Autosorb sorption analyzers of the configurations used was similar—it was approximately 2.5 times the cost of the materials and parts for the construction of the fixed-bed apparatus.
Important data for research planning are among others: the total experiment time, including the machine time of the instrument and the time required for its operation and data evaluation, as well as gas, liquid, and energy consumption. Table 6 shows these data for an experiment consisting of five cycles of adsorption-capacity determination for one sample at one temperature, one pressure, and one mole fraction of carbon dioxide. The numbers refer to the whole experiment, i.e., its adsorption and desorption parts, and are long-term arithmetic means. The electricity-consumption figures were determined using a power meter and thus do not match the labelled power consumption of the device.
As Table 6 illustrates, the methods were similar in some parameters, especially in power consumption, the length of one measurement cycle, and the resulting total machine time of the five-cycle experiment. Nevertheless, there were large differences in gas consumption. The fixed-bed apparatus consumed three orders of magnitude more gas than the Autosorb apparatus and one order of magnitude more gas than the DVS Advantage to perform the same experiment.
There was a large difference in the difficulty of operation between the two commercially available analyzers and the fixed-bed adsorption apparatus. This apparatus required manual adjustment of flow rates and temperatures at each stage of the experiment. However, the most time-consuming part of the test was always the mathematical processing of the primary data in the calculation of adsorption capacity.
Due to the greater sample weights, only the fixed-bed apparatus allowed their analysis after the end of the tests. Knowing whether cyclic sorptions/desorptions do not deteriorate the properties of materials is indeed important for industrial application. The samples were reanalyzed by XRD after their removal from the adsorber. In addition, their specific surface areas and pore-size distributions were measured. The hardness of adsorbents, determined by an automatic Kahl Pellet Hardness Tester (manufactured by Amandus Kahl GmbH& Co. KG, Germany), is another essential property for their industrial use. The pressures and temperatures used during the tests had no effect on the representation of the crystalline phases detected by XRD. The specific surface was also not affected. After all sorption tests, the 13X sample showed values of 510.8–513.0 m2 g‒1 and the natural zeolite 28.5–29.8 m2 g‒1. The relative representation of different pore diameters also remained unchanged, and the largest volume belonged to pores with a diameter of 20–80 nm. They still represented 51% for the sample 13X and 46% for the natural zeolite.
The hardness of the materials was also preserved. The average disintegration force of 100 randomly selected particles was 21 N for the 13X sample and 241 N for the natural zeolite. Considering the different origin of the samples, there was of course a greater variance of values for the natural zeolite, which coefficient of variation reached 18% (compared to 7% for 13X).
Conclusions
Among the findings verified by the experiments performed, it is necessary to emphasize the following. The three devices, each operating on a different principle, gave well-comparable standard deviations of the results. It has been verified that the fixed-bed apparatus achieves good agreement of results with the dynamic sorption analyzer (DVS Advantage). In contrast, the static volumetric analyzer provided capacity values about 25% lower than those of both dynamic devices in all tests performed. The cause was probably a worse transport of gas molecules into the samples.
The main drawback of the automatic sorption analyzers was the high demands on sample homogeneity, resulting from charges. In addition, the fixed-bed flow apparatus enabled operation over the widest range of test conditions closer to that of a real adsorption unit. For example, at an overpressure of 5 bar, a temperature of 20 °C and a CO2 volume fraction of 13%, which are the conditions expected in the treatment of flue gases from solid fuels including biomass, the samples showed the following results. The sample 13X had a capacity of 0.132 g g−1 (3.00 mmol g−1), whereas the sample of untreated natural zeolite had a capacity of 0.039 g g−1 (0.89 mmol g−1), which is less than 30% of the capacity of the sample 13X.
Primarily thanks to fixed-bed apparatus, the dependences of adsorption capacities on temperature (in the range 10–40 °C) and CO2 partial pressure (in the range 5–120 kPa) were described. The capacity data obtained for different temperatures and pressures were used to calculate the constants of the Langmuir model. In agreement with the model, the largest capacities were achieved at the maximum partial pressure of CO2 (120 kPa) and the minimum temperature (10 °C). Under these conditions, the equilibrium capacity of the sample 13X was 0.146 g g−1(3.32 mmol g−1) and of natural zeolite 0.045 g g−1 (1.03 mmol g−1). Increasing the temperature to 40 °C caused a decrease in the capacity of 13X by 35% and natural zeolite by 41% compared to the values mentioned above. On the contrary, while keeping the temperature at 10 °C, the reduction of the CO2 partial pressure to 5 kPa was manifested in the capacity decrease by 74% for the 13X sample and by 80% in the case of natural zeolite.
The agreement between the experimentally determined values and the Langmuir model was very good. The average difference between the measured and calculated capacity values was 3.8% in the case of 13X and 5.1% in the case of natural zeolite. Only at the lowest tested CO2 partial pressure was the difference somewhat higher, but a maximum of 14%.
By cyclical repeating of five adsorption and desorption steps with the same sample batch, it was verified that none of the samples underwent degradation under the given conditions. No changes were detected in the structure of the samples, specifically in the representation of crystalline phases detected by the XRD method, in the specific surface area, or in the pore-size distribution. There were also no changes in the hardness of the material particles and, above all, in the determined adsorption capacities.
The disadvantage of using a fixed-bed apparatus was the high consumption of industrial gases and especially the ten times longer time required for operation and mathematical data evaluation.
Since automatic sorption analyzers generally have a significantly limited range of operating conditions and sample weights, the following can be recommended. Only for routine measurements of the adsorption capacity of a large number of samples at different temperatures, it is preferable to use an automatic sorption analyzer. For laboratory simulations of the conditions under which the majority of the industrial units are operated (i.e., PSA, TSA, etc.), the pressure adsorption fixed-bed apparatus presented here is the optimal solution.
Abbreviations
- a i :
-
Adsorbed amount of a component i (mol g−1)
- a σ ,i :
-
Maximum adsorbed amount of a component i (mol g−1)
- b i :
-
Equilibrium constant of a component i
- K v :
-
Adsorption rate constant (min−1)
- k :
-
Slope of a straight line
- M :
-
Weight of a packed bed (g)
- Q :
-
Volumetric flow rate (m3 min−1)
- p :
-
Pressure (Pa)
- q :
-
Y-axis intercept of a straight line
- φ i :
-
Mole fraction of a component i
- ρ B :
-
Bulk density of the packed bed (g cm−3)
- Θ:
-
Fractional occupancy of the adsorption sites
- σ :
-
Standard deviation
References
Abd AA, Othman MR, Shabbani HJK, Helwani Z (2022) Biomethane upgrading to transportation fuel quality using spent coffee for carbon dioxide capture in pressure swing adsorption. J Environ Chem Eng 10(2):107169. https://doi.org/10.1016/j.jece.2022.107169
Agbaje TA, Singh S, Reddy SK, Polychronopoulou K, Vega LF, Khaleel M, Wang K, Karanikolos GN (2021) Salt-free synthesis of Cu-BTC metal-organic framework exhibiting mesoporosity and enhanced carbon dioxide adsorption. Microporous Mesoporous Mater 324:111265. https://doi.org/10.1016/j.micromeso.2021.111265
Auta M, Hameed BH (2014) Adsorption of carbon dioxide by diethanolamine activated alumina beads in a fixed bed. Chem Eng J 253:350–355. https://doi.org/10.1016/j.cej.2014.05.018
Auta M, Darbis NDA, Din ATM, Hameed BH (2013) Fixed-bed column adsorption of carbon dioxide by sodium hydroxide modified activated alumina. Chem Eng J 233:80–87. https://doi.org/10.1016/j.cej.2013.08.012
Bjerge LM, Brevik P (2014) CO2 capture in the cement industry, Norcem CO2 capture project (Norway). Energy Procedia 63:6455–6463. https://doi.org/10.1016/j.egypro.2014.11.680
Bonenfant D, Kharoune M, Niquette P, Mimeault M, Hausler R (2008) Advances in principal factors influencing carbon dioxide adsorption on zeolites. Sci Technol Adv Mater 9(1):013007. https://doi.org/10.1088/1468-6996/9/1/013007
Cavallo M, Dosa M, Porcaro NG, Bonino F, Piumetti M, Crocellá V (2023) Shaped natural and synthetic zeolites for CO2 capture in a wide temperature range. J CO2 Util 67: 102335. https://doi.org/10.1016/j.jcou.2022.102335
Chen C, Kim J, Yang D, Ahn W (2011) Carbon dioxide adsorption over zeolite-like metal organic frameworks (ZMOFs) having a sod topology: structure and ion-exchange effect. Chem Eng J 168(3):1134–1139. https://doi.org/10.1016/j.cej.2011.01.096
Davarpanah E, Armandi M, Hernández S, Fino D, Arletti R, Bensaid S, Piumetti M (2020) CO2 capture on natural zeolite clinoptilolite: effect of temperature and role of the adsorption sites. J Environ Manag 275:111229. https://doi.org/10.1016/j.jenvman.2020.111229
Divekar S, Arya A, Hanif A, Chauhan R, Gupta P, Nanoti A, Dasgupta S (2021) Recovery of hydrogen and carbon dioxide from hydrogen PSA tail gas by vacuum swing adsorption. Sep Purif Technol 254:117113. https://doi.org/10.1016/j.seppur.2020.117113
Esteves LM, Ferreira TJ, Keba A, Esperanca JMSS, Esteves IAAC (2023) Carbon materials derived from cyano-based IL@ZIF-8 composites for CO2 sorption separation systems. Mater Today Sustain 22:100353. https://doi.org/10.1016/j.mtsust.2023.100353
Fujiki J, Chowdhury FA, Yamada H, Yogo K (2017) Highly efficient post-combustion CO2 capture by low-temperature steam-aided vacuum swing adsorption using a novel polyamine-based solid sorbent. Chem Eng J 307:273–282. https://doi.org/10.1016/j.cej.2016.08.071
Hauchhum L, Mahanta P (2014) Carbon dioxide adsorption on zeolites and activated carbon by pressure swing adsorption in a fixed bed. Int J Energy Environ Eng 5:349–356. https://doi.org/10.1007/s40095-014-0131-3
Lei L, Cheng Y, Chen C, Kosari M, Jiang Z, He C (2022) Taming structure and modulating carbon dioxide (CO2) adsorption isosteric heat of nickel-based metal organic framework (MOF-74(Ni)) for remarkable CO2 capture. J Colloid Interface Sci 612:132–145. https://doi.org/10.1016/j.jcis.2021.12.163
Li Z, Zhang J, Mo X, Xu X, Zhang Y, Wang D, Tong Z, Tang X (2022) Adsorption behaviors of near-critical carbon dioxide on organic-rich shales: Modeling, multifractality, and kinetics. Chem Eng J 428:132526. https://doi.org/10.1016/j.cej.2021.132526
Liu B, Yu X, Shi W, Shen Y, Zhang D, Tang Z (2020) Two-stage VSA/PSA for capturing carbon dioxide (CO2) and producing hydrogen (H2) from steam-methane reforming gas. Int J Hydrog Energy 45(46):24870–24882. https://doi.org/10.1016/j.ijhydene.2020.06.264
Liu L, Jin S, Park Y, Kim KM, Lee CH (2021) Sorption equilibria, kinetics, and temperature-swing adsorption performance of polyethyleneimine-impregnated silica for post-combustion carbon dioxide capture. Sep Purif Technol 266:118582. https://doi.org/10.1016/j.seppur.2021.118582
Luberti M, Oreggioni GD, Ahn H (2017) Design of a rapid vacuum pressure swing adsorption (RVPSA) process for post-combustion CO2 capture from a biomass-fuelled CHP plant. J Environ Chem Eng 5(4):3973–3982. https://doi.org/10.1016/j.jece.2017.07.029
Majchrzak-Kucęba I, Wawrzyńczak D (2021) Treatment of flue gas in a CO2 capture pilot plant for a commercial CFB boiler. Energies 14(9):2458. https://doi.org/10.3390/en14092458
McEwen J, Hayman JD, Yazaydin AO (2013) A comparative study of CO2, CH4 and N2 adsorption in ZIF-8, Zeolite-13X and BPL activated carbon. Chem Phys 412:72–76. https://doi.org/10.1016/j.chemphys.2012.12.012
Najafi AM, Soltanali S, Khorashe F, Ghassabzadeh H (2023) Effect of binder on CO2, CH4, and N2 adsorption behavior, structural properties, and diffusion coefficients on the extruded zeolite 13X. Chemosphere 324:138275. https://doi.org/10.1016/j.chemosphere.2023.138275
Nedoma M, Staf M, Hrdlička J (2022) Experimental and simulation study of CO2 breakthrough curves in a fixed-bed adsorption process. Acta Polytechnica 62(3):370–385. https://doi.org/10.14311/AP.2022.62.0370
Saima WH, Mogi Y, Haraoka T (2013) Development of PSA system for the recovery of carbon dioxide and carbon monoxide from blast furnace gas in steel works. Energy Procedia 37:7152–7159. https://doi.org/10.1016/j.egypro.2013.06.652
Samanta A, Zhao A, Shimizu GKH, Sarkar P, Gupta R (2012) Post-combustion CO2 capture using solid sorbents: a review. Ind Eng Chem Res 51(4):1438–1463. https://doi.org/10.1021/ie200686q
Santori G, Charalambous C, Ferrari M, Brandani S (2018) Adsorption artificial tree for atmospheric carbon dioxide capture, purification and compression. Energy 162:1158–1168. https://doi.org/10.1016/j.energy.2018.08.090
Shafeeyan MS, Daud WMAW, Houshmand A, Arami-Niya A (2011) Ammonia modification of activated carbon to enhance carbon dioxide adsorption: effect of pre-oxidation. Appl Surf Sci 257(9):3936–3942. https://doi.org/10.1016/j.apsusc.2010.11.127
Shen Y, Shi W, Zhang D, Na P, Fu B (2018) The removal and capture of CO2 from biogas by vacuum pressure swing process using silica gel. J CO2 Util 27: 259–271. https://doi.org/10.1016/j.jcou.2018.08.001
Singh VK, Kumar EA (2016) Measurement and analysis of adsorption isotherms of CO2 on activated carbon. Appl Therm Eng 97:77–86. https://doi.org/10.1016/j.applthermaleng.2015.10.052
Siqueira RM, Freitas GR, Peixoto HR, Do Nascimento JF, Musse AP, Torres AE, Azevedo DC, Bastos-Neto M (2017) Carbon dioxide capture by pressure swing adsorption. Energy Procedia 1(114):2182–2192. https://doi.org/10.1016/j.egypro.2017.03.1355
Thote JA, Chatti RV, Iyer KS, Kumar V, Valechha AN, Labhsetwar NK, Biniwale RB, Yenkie MKN, Rayalu SS (2012) N-doped mesoporous alumina for adsorption of carbon dioxide. J Environ Sci 24(11):1979–1984. https://doi.org/10.1016/S1001-0742(11)61022-X
Vogtenhuber H, Hofmann R, Helminger F, Schöny G (2018) Process simulation of an efficient temperature swing adsorption concept for biogas upgrading. Energy 162:200–209. https://doi.org/10.1016/j.energy.2018.07.193
Wawrzyńczak D, Majchrzak-Kucęba I, Srokosz K, Kozak M, Nowak W, Zdeb J, Smółka W, Zajchowski A (2019) The pilot dual-reflux vacuum pressure swing adsorption unit for CO2 capture from flue gas. Sep Purif Technol 209:560–570. https://doi.org/10.1016/j.seppur.2018.07.079
Weishauptová Z, Přibyl O, Sýkorová I, Machovič V (2015) Effect of bituminous coal properties on carbon dioxide and methane high pressure sorption. Fuel 139:115–124. https://doi.org/10.1016/j.fuel.2014.08.030
Widiastuti N, Susanti I (2019) Activation of zeolite-Y templated carbon with KOH to enhance the CO2 adsorption capacity. Mal J Fund Appl Sci 15(2):249–253
Yaumi AL, Abu Bakar MZ, Hameed BH (2017) Reusable nitrogen-doped mesoporous carbon adsorbent for carbon dioxide adsorption in fixed-bed. Energy 138:776–784. https://doi.org/10.1016/j.energy.2017.07.130
Yusuff SM, Khim OK, Yunus WMZM, Fitrianto A, Ahmad M, Ibrahim N, Ros FC, Teoh C (2018) Carbon dioxide sorption isotherm and kinetics by alum sludge. Mater Today Proc 5(10–2):21948–21955. https://doi.org/10.1016/j.matpr.2018.07.055
Zhao Z, Cui X, Ma J, Li R (2007) Adsorption of carbon dioxide on alkali-modified zeolite 13X adsorbents. Int J Greenh Gas Control 1(3):355–359. https://doi.org/10.1016/S1750-5836(07)00072-2
Zhao G, Aziz B, Hedin N (2010) Carbon dioxide adsorption on mesoporous silica surfaces containing amine-like motifs. Appl Energy 87(9):2907–2913. https://doi.org/10.1016/j.apenergy.2009.06.008
Zhao Y, Shen Y, Bai L (2012) Effect of chemical modification on carbon dioxide adsorption property of mesoporous silica. J Colloid Interface Sci 379(1):94–100. https://doi.org/10.1016/j.jcis.2012.04.064
Zhou C, Feng S, Zhou G, Jin Y, Liang J, Xu J (2011) A simple method for calculating the overall adsorption rate constant in the Wheeler–Jonas equation. Adsorpt Sci Technol 29(1):71–82. https://doi.org/10.1260/0263-6174.29.1.71
Zukal A, Čejka PJ, J, (2013) MgO-modified mesoporous silicas impregnated by potassium carbonate for carbon dioxide adsorption. Microporous Mesoporous Mater 167:44–50. https://doi.org/10.1016/j.micromeso.2012.05.026
Acknowledgements
This work was carried out as part of the project TK03030167 “Low-Emission Technologies of Energy Conversion of Biomass and Alternative Fuels,” supported by the Technology Agency of the Czech Republic (TACR), which is gratefully acknowledged.
Funding
Open access publishing supported by the National Technical Library in Prague. This study was funded by Technologická Agentura České Republiky,TK03030167
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
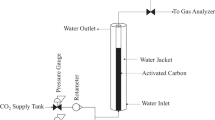
Appendix
Appendix
The description of the flow apparatus can be supplemented with the following detailed information. The source of the gaseous mixture simulating dedusted flue gas is a pressure cylinder. Downstream of the flow meter/controller, the gas is split into two streams. Alternatively, the gas can be fed to the adsorber containing the sample or through a bypass at the same pressure to the analyzer. This ensures the evaluation of the adsorption capacity. The apparatus also allows the testing of gas enriched with water vapor in a controlled manner, but this procedure has not been applied in this study. The core of the apparatus is a vertical steel fixed-bed adsorber, installed in a common frame together with a humidifier and a humidity probe. The adsorber of length 260 mm and the inner diameter 40 mm is fitted with a standard DN40/PN6 flange for sample filling and removal. The gas is introduced at the head of the adsorber and exits it through its bottom. After leaving the climate chamber, the gas pressure is reduced to atmospheric level. The gas enters the IC analyzer and subsequently the drum gas meter, displaying the total volume of gas that has flown through the device in a particular phase of the experiment. A general view of the apparatus in operation is shown in Fig.

Photographs of the fixed-bed apparatus (left: 1—climate chamber, 2—adsorber assembly in the closed chamber, 3—PC, 4—lateral passage of pipes and cables, 5—liquid chiller, 6—thermometer/datalogger, 7—IR analyzer, 8—gas flow meter/controller, 9—safety valve, 10—gas meter, 11—condensate collector; right: adsorber assembly in the open chamber)
12.
The high-pressure part of the system is made of DIN 1.4401 (AISI 316) stainless steel. Connections are made with A-LOK dismountable fittings (manufactured by Parker-Hannifin Corp., USA). The gas flow rate at the inlet of the apparatus is measured and adjusted in the range 0.4–20 dm3 min−1 (normal conditions) by means of an EL-FLOW Prestige FG-111BP thermal mass flow meter with an electromagnetic controller (manufactured by Bronkhorst High-Tech B.V., the Netherlands). The temperature in the layer of the adsorbent under investigation is measured by a K-type thermocouple connected to a Greisinger GMH 3250 thermometer/datalogger (manufactured by GHM Messtechnik GmbH, Germany).
If necessary, gas humidity is measured using a HUMICAP probe HMP8 with an Indigo520 transmitter (manufactured by Vaisala Oyj, Finland). The pressure in the apparatus is adjusted by a manual BP300 diaphragm-sensed back-pressure regulator (manufactured by Pressure Tech Ltd, UK). The temperature during both adsorption and desorption is maintained by placing the adsorber in an MKT 240 E3.1 climate chamber (manufactured by Binder GmbH, Germany). The CO2 content in the gas is measured using an Ultramat 23 infrared analyzer (manufactured by Siemens AG, Germany) with the measurable volume fraction range of 0–100% and a sensitivity of 0.05%.
A specific method was developed for each of the measuring devices. Measurement of adsorption capacity on the Autosorb iQ instrument consisted of the following sequence of steps. The sample was weighed into the crucible of a glass measuring cell with a volume of ca 2 ml, and the cell was placed in the measuring port of the device. The sample was degassed directly in the Autosorb iQ instrument at the temperature of 120 °C and at atmospheric pressure for 120 min. After being cooled to laboratory temperature, the sample in the crucible was weighed outside the device and then returned to the measuring port. Subsequently, the system was heated to the selected operating temperature at the rate of 20 °C min−1. Nitrogen was circulated through the system at atmospheric pressure until the temperature stabilized. All tests were carried out under isothermal conditions at 30 and 40 °C. The gas mixture containing the required volume fraction of CO2 was fed into the measuring cell via a solenoid valve, with the dosing stopped when the pressure reached 800 Torr (106.7 kPa). This pressure was automatically indicated by the system as reaching equilibrium. The primary output of the experiment was an adsorption isotherm curve constructed from ten points of the dependence of volume (cm3 g−1) on absolute pressure. Volume conversion using state variables provided the corresponding adsorption capacity, expressed as the amount of CO2 adsorbed per unit mass of the sample at the time equilibrium, was reached.
Measurement using the DVS Advantage analyzer was taken as follows. The sample was weighed into a 0.1-ml crucible and placed in the device. The sample was degassed at the temperature of 120 °C in a stream of nitrogen until a constant mass was reached. The inert gas was then exchanged for the selected model mixture of CO2, and the measurement was taken under isothermal conditions until a constant mass was obtained. The achievement equilibrium was followed by desorption in a nitrogen stream at 120 °C, and the sample was prepared for the next cycle. As with the other apparatus used, the capacity calculation always included five cycles of adsorption.
For illustrative purposes, the measurement procedure using the fixed-bed flow apparatus is described for a single-cycle experiment. In real experiments, the alternation of the adsorption and desorption phases was repeated a total of five times to verify reproducibility.
A sample dried externally at 120 °C to a constant mass was initially loaded into the adsorber. All measurement phases were carried out at the same gas flow rate of 2.0 dm3 min−1 (normal conditions). After the introduction of inert gas into the adsorber, the climate chamber at 20 °C min−1 began to be heated at atmospheric pressure until the desorption temperature of 120 °C was reached in the adsorber. After it was verified that the sample did not release any CO2, the temperature was lowered and the pressure in the adsorber was increased to the desired level during adsorption. Once the conditions in the adsorber stabilized, nitrogen was replaced by a mixture with CO2 to start the adsorption. The adsorption phase of the experiment ended when the IR analyzer displayed the equilibration of the inlet and outlet CO2 concentrations. The described procedure yielded breakthrough curves, whose evaluation provided the data discussed below. This was followed by depressurization and performance of the above-described desorption under N2 flow.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Staf, M., Kyselová, V. Searching for a cost-effective method of determining sorption properties for the industrial use of CO2 capture. Chem. Pap. 77, 6217–6233 (2023). https://doi.org/10.1007/s11696-023-02933-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-023-02933-x