
Abstract
CuO/ZrO2 (Cu/Zr) catalysts were synthesized via the co-precipitation method using Na2CO3 as the precipitating agent. The influence of CuO/ZrO2 ratio, aging time and calcination temperature of catalyst precursors on the physicochemical properties of Cu/Zr catalysts was studied. The catalysts were characterized by means of N2 adsorption, temperature-programmed reduction, N2O dissociative chemisorption, X-ray diffraction, inductively coupled plasma optical emission spectrometer and CO2 temperature-programmed desorption. It was found that only for samples calcined at 550 °C the ZrO2 was present as the tetragonal phase (t-ZrO2). In the case for samples which were aged for 1 h, metallic copper dispersion (DCu) does not depend on calcination. On the other hand, for non-aged catalysts a decrease in DCu is observed with higher temperature of calcination. The temperature corresponding to maximum reduction rate (Tmax) decreases for samples with higher DCu. The contribution of weak basic site is the highest for catalysts which were aged, whereas for non-aged catalysts mainly medium basic sites dominate on the surface.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cu/ZrO2 catalysts are commonly used in various catalytic reactions, e.g., methanol synthesis (Denise and Sneeden 1986; Samson et al. 2014), steam reforming of methanol (Oguchi et al. 2005; Ritzkopf et al. 2006), water gas shift reaction (Águila et al. 2008), selective catalytic reduction of NOx (Okamoto et al. 1998) and hydrogenation of levulinic acid to γ-valerolactone (GVL) (Ishikawa et al. 2017). The influence of zirconia polymorphism in Cu/ZrO2 catalysts plays an important role both in the physicochemical catalyst properties and catalyst activity. It has been shown that monoclinic (m-ZrO2) and tetragonal (t-ZrO2) ZrO2 exhibit different acid–base properties (Morterra et al. 1994; Bolis et al. 1998; Zhao et al. 2002) and surface hydroxyl group concentration (Pokrovski et al. 2001; Ardizzone and Bianchi 1999). The phase of ZrO2 being present in catalysts has influence on catalyst reducibility (Zhao et al. 2004) and CO2 adsorption capacity (Pokrovski et al. 2001). Additionally, ZrO2 prevents Cu particles from sintering during catalytic activity which enhances copper dispersion (Agrell et al. 2003; Shimokawabe et al. 1990).
Cu/ZrO2 catalysts containing monoclinic ZrO2 (m-ZrO2) exhibit higher selectivity to methanol than Cu catalysts deposited on tetragonal ZrO2 (t-ZrO2) (Jung and Bell 2002). The effect of ZrO2 phase on product selectivity is also visible in the ethanol dehydrogenation over Cu/ZrO2 catalysts. In this case, the highest rate of ethyl acetate formation is observed for Cu/m-ZrO2 catalyst and results from high oxygen mobility from the bulk m-ZrO2 phase to copper species. On the other hand, Cu deposition on t-ZrO2 leads to by-product formation (Sato et al. 2013). Maruya et al. (2000) came to the same conclusion that increased fraction of m-ZrO2 in the catalyst results in higher rate of isobutene formation from the H2/CO mixture. In methanol steam reforming, it has been stated that interaction of copper oxide with amorphous ZrO2 (am-ZrO2) is one of the factors responsible for high stability and activity of Cu/ZrO2 catalysts (Koeppel et al. 1992).
The objective of the conducted work was to evaluate the influence of synthesis conditions on the physicochemical properties of CuO/ZrO2 catalysts. Particularly, the influence of the composition of the catalyst, aging time and calcination temperature on catalyst basicity, reducibility and copper dispersion was determined.
Experimental
CuO/ZrO2 (Cu/Zr) catalysts were synthesized via the co-precipitation method at pH 7, using Na2CO3 as the precipitating agent with nominal composition (wt%) Cu/Zr = 70/30 (70Cu/Zr) and Cu/Zr = 50/50 (50Cu/Zr). Cations in the form of nitrates and Na2CO3 were simultaneously added dropwise into the beaker containing 100 mL of deionized water at 60 °C. During the precipitation, the mixture was vigorously stirred. For non-aged samples, the precipitates were subsequently washed by five times centrifugation and dried at 100 °C. For aged catalysts, prior to washing, the precipitates were kept in mother liquor for 1 h. After drying, samples were calcined at 300 °C, 350 °C and 550 °C. Catalysts which were aged for 1 h are denoted as 70Cu/Zr/X_1 h, while non-aged catalysts are denoted as 70Cu/Zr/X and 50Cu/Zr/X where X is the temperature of calcination.
X-ray diffraction patterns (XRD) were recorded on Bruker AXS D505 with CuKα radiation. The analysis was performed between 0.6° and 90° 2Θ range with accumulation time 5 s. The mean crystallite sizes of CuO and ZrO2 were calculated based on Rietveld refinement.
CO2 temperature-programmed desorption (CO2-TPD) measurements were carried out in quartz-fixed bed flow reactor connected online to a mass spectrometer (QMG 220 PRISMA PLUS). Signals: m/z = 2 (H2), m/z = 44 (CO2), m/z = 40 (Ar) were monitored during TPD. Prior to TPD run, the sample (100 mg) was reduced in 5% H2/Ar flow (30 mL/min) at 250 °C for 2.5 h. Next, the reactor was cooled down to room temperature (RT) and pulses (250 µL) of 5% CO2/Ar were introduced until saturation. Then, the sample was flashed with He flow (40 mL/min) for 0.5 h until obtaining stable CO2 line. TPD was done from RT to 400 °C with ∆T = 10°/min under He flow (40 mL/min).
N2O dissociative chemisorption (Shuyten et al. 2008) was performed using the same setup line as for CO2-TPD. The sample (100 mg) was reduced in the same manner as for CO2-TPD. The pulses (100 µL) of N2O were introduced at 90 °C under He flow (30 mL/min) until reoxidation was completed. Signals: m/z = 44 (N2O) and m/z = 28 (N2, N2O) were monitored. The metallic copper surface area (SCu) was calculated assuming atomic copper surface density of 1.47 × 1019 Cu atoms/m2, corresponding to 6.1 × 10−6 O2 mol/m2 for the stoichiometry Cu/O = 2. The Cu metal dispersion (DCu) was measured as percentage ratio of surface Cu moles from N2O chemisorption to the total moles of Cu present in the catalysts. Additionally, Cu° particle size (PCu) was determined using the following equation:
where d is the density of Cu metal (8.92 g/cm3) and wt% Cu loading is based on ICP. N2O chemisorption experiments were repeated few times for each sample resulting in relative standard deviation (RSD) below 1%.
Temperature-programmed reduction with hydrogen (H2-TPR) was carried out on Chembet-3000 (Quantachrome). The hydrogen consumption was monitored with TCD detector. For typical H2-TPR run, the sample (25 mg) was placed in quartz U-shaped tube reactor and conditioned at 100 °C under He flow (30 mL/min) for 1.5 h. Next, the sample was cooled down to RT under He flow and the H2-TPR experiment was performed in 5%H2/Ar (30 mL/min) in the temperature range RT − 850 °C (∆T = 10 °C/min). For quantitative H2-TPR studies, a four point calibration curve was prepared using appropriate amounts of CuO standard (Aldrich) mixed with Al2O3 in wt% ratio = 1:2, 1:4, 1:16, 1:12 (mechanical mixture). The calibration covered the hydrogen consumption range corresponding to TPR analysis. The H2-TPR and CO2-TPD profiles were deconvoluted into Gaussian peaks. Deconvolution was performed in Origin 2018 Pro software.
The BET surface area was measured with nitrogen adsorption at − 196 °C on Quantachrome Autosorb-1. Prior to measurements, samples were degassed for 18 h at 150 °C to remove adsorbed water and other surface impurities. The pore size distribution (PSD) profiles were obtained by the BJH method from the desorption branch.
The elemental analysis was performed by inductively coupled plasma optical emission spectrometer (ICP-OES). For ICP-OES analysis, 100 mg of sample was dissolved in a mixture of HNO3/H2O.
Results and discussion
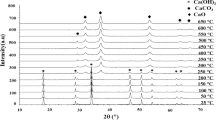
The XRD patterns of the CuO–ZrO2 precursors (Fig. 1) show diffraction peaks which are assigned only to the malachite phase. As can be seen from XRD patterns for samples after calcination, the XRD signals for the malachite phase disappeared and the precursors were fully decomposed to oxides. In the case of calcined samples, the CuO XRD peaks (signals at 2θ =32.5°, 35.5°, 38.7°, 48.9°, 53.4°, 58.3°, 61.6°, 66.4°, 68.1°, 72.5°, 75.0°, 82.9°) sharpened and their intensity increased with a calcination temperature for all samples, meaning that there was an increase in the samples’ crystallinity. The Rietveld refinement (Table 1) confirmed that the crystallite sizes of CuO got bigger for samples which were calcined at higher temperature. This is due to sintering which proceeds at higher temperatures. The estimated values of the CuO crystallite sizes for samples calcined at 550 °C are the biggest and are equal to 17.7 nm, 19.8 nm and 23.6 nm for 70Cu/Zr/550_1 h, 70Cu/Zr/550 and 50Cu/Zr/550, respectively. For samples calcined at 300 °C and 350 °C, only the CuO phase is visible. In this case, diffraction peaks for ZrO2 phase are not observed suggesting that the ZrO2 phase is present as the amorphous phase (Rhodes and Bell 2005) or its particles are too small to be detected by means of XRD (Figs. 2, 3, 4).

XRD patterns of catalyst precursors: 70Cu/Zr_1 h (1), 70Cu/Zr (2) and 50Cu/Zr (3)

XRD patterns of 70Cu/Zr/300_1 h (1), 70Cu/Zr/350_1 h (2) and 70Cu/Zr/550_1 h (3). Black diamond suit—CuO phase

XRD patterns of 70Cu/Zr/300 (1), 70Cu/Zr/350 (2) and 70Cu/Zr/550 (3). Black diamond suit—CuO phase
On the other hand, diffraction peaks ascribed to t-ZrO2 appear for samples calcined at 550 °C (2θ = 30.5°, 50.9° and 60.7°). For non-aged catalysts, which were calcined at 300 °C and 350 °C, the increase in the ZrO2 content had only minor influence on the CuO crystallite size. Nevertheless, the crystallite sizes of t-ZrO2 phase increase with ZrO2 content, reaching a value of 14.4 nm for 50Cu/Zr_550. Additionally, this catalyst possesses the biggest CuO crystallite size (23.6 nm) among all synthesized samples (Fig. 4).

XRD patterns of 50Cu/Zr/300 (1), 50Cu/Zr/350 (2) and 50Cu/Zr/550 (3). Black diamond suit—CuO phase
The N2 adsorption–desorption isotherms are presented in Figs. 5, 6 and 7 for aged 70/Cu/Zr samples, non-aged 70/Cu/Zr samples, and non-aged 50/Cu/Zr samples, respectively. All synthesized catalysts exhibit typical type V and type III isotherms with an H3-type hysteresis loop according to IUPAC classification (Thommes et al. 2015). The observed shape of the hysteresis loop is characteristic for samples possessing both mesoporous and macroporous (Basahel et al. 2016).

N2 adsorption–desorption isotherms for: A—70Cu/Zr/300_1 h, B—70Cu/Zr/350_1 h, C—70Cu/Zr/550_1 h

N2 adsorption–desorption isotherms for: A—70Cu/Zr/300, B—70Cu/Zr/350, C—70Cu/Zr/550

N2 adsorption–desorption isotherms for: A—50Cu/Zr/300, B—50Cu/Zr/350, C—50Cu/Zr/550
The BJH pore size distribution (PSD) profiles and BJH cumulative pore volume are presented in Figs. 8, 9 and 10 for aged 70Cu/Zr samples, non-aged 70Cu/Zr samples, and non-aged 50Cu/Zr samples, respectively. PSD profiles for all synthesized catalysts show maxima in the 5–20 nm mesoporous range, meaning no microporosity. For aged catalysts, the BJH cumulative pore volume (Fig. 8B) is 0.18 cm3/g, 0.18 cm3/g, 0.17 cm3/g for 70Cu/Zr/300_1 h, 70Cu/Zr/350_1 h, and 70Cu/Zr/550_1 h, respectively, no matter the sample calcination temperature. On the other hand, the drop in BET specific surface area (Table 1) with increased temperature of calcination was observed for these samples. This is due to the changes in the PSD for aged samples which were calcined at different temperatures (Fig. 8A). As the temperature of calcination increases, the number of pores decreases but the diameter of pores become larger. For 70Cu/Zr/550_1 h the increase in the numbers of pores with diameter higher than 18 nm is visible which results in the value of the BJH cumulative pore volume being close to values for 70Cu/Zr/300_1 h and 70Cu/Zr/350_1 h. For both non-aged 70Cu/Zr/300 and 70Cu/Zr/350 samples, the BJH cumulative pore volume is the same and equal to 0.18 cm3/g. On the other hand, non-aged 70Cu/Zr/500 catalyst has slightly lower value of BJH cumulative pore volume (0.15 cm3/g). In this case, the visible maxima in PSD plots (Fig. 9A) above 18 nm are of lower intensity and there is no maximum in the PSD profile in the 3–5 nm range in comparison with 70Cu/Zr/550_1 h. Thus, this samples exhibits low BET specific surface area (18 m2/g).

The BJH pore size distribution (A) and BJH cumulative pore volume (B) for 70Cu/Zr/300_1 h (black square lines), 70Cu/Zr/350_1 h (red circle lines), and 70Cu/Zr/550_1 h (blue up-pointing triangle lines)

The BJH pore size distribution (A) and BJH cumulative pore volume (B) for 70Cu/Zr/300 (black square lines), 70Cu/Zr/350 (red circle lines), and 70Cu/Zr/550 (blue up-pointing triangle lines)

The BJH pore size distribution (A) and BJH cumulative pore volume (B) for 50Cu/Zr/300 (black square lines), 50Cu/Zr/350 (red circle lines), and 50Cu/Zr/550 (blue up-pointing triangle lines)
The increase in zirconia content for non-aged samples (Fig. 10) leads to lower cumulative pore volume (0.15 cm3/g, 0.12 cm3/g and 0.12 cm3/g for 50Cu/Zr_300, 50Cu/Zr_350 and 50Cu/Zr_550, respectively) and flattened pore size distribution in comparison with non-aged 70/Cu/Zr catalysts. The observed decrease in porosity results from catalysts sintering and corresponds with XRD analysis which shows an increase in crystal sizes of CuO with higher temperature of calcination.
The surface basicity of the catalysts was evaluated with CO2-TPD. The obtained results for three series of catalysts are presented in Figs. 11, 12 and 13. To confirm that the observed low-temperature desorption CO2 signals originate solely from the CO2 introduced via pulses prior to the desorption stage, the blank TPD test was performed for samples calcined at 300 °C (line 1 in Figs. 11, 12 and 13). For the blank TPD test, the sample was only reduced and later heated up in the same manner as during typical TPD, meaning no CO2 was introduced. In this case, a maximum of CO2 signal was observed above 350 °C, which was higher than the calcination temperature of the samples (i.e. 300 °C) used in blank TPD tests. This suggests that the visible CO2 signal from the precursor decomposition above 300 °C during TPD. The XRD analysis did not reveal any reflexes assigned to the malachite phase in the samples after calcination at 300 °C, meaning that the amount of undecomposed precursor, being still present in this catalysts, is below the limit of detection by the conventional X-ray diffraction method. For blank TPD tests (line 1 in Figs. 11, 12 and 13), the lowest amount of CO2 is released for the aged catalyst. On the other hand, it can been seen that for non-aged catalysts, the amount of CO2 being released upon heating is higher. A further increase of CO2 amount was observed for the catalysts with a higher content of ZrO2 (50Cu/Zr).

CO2-TPD profiles for blank TPD (1), 70Cu/Zr/300/_1 h (2), 70Cu/Zr/350_1 h (3) and 70/30-C/Z-550-1(4). Signal—black line, cumulative curve—red line, deconvoluted peaks used for quantification—blue line, additional deconvoluted peak—green line

CO2-TPD profiles for blank TPD (1), 70Cu/Zr/300 (2), 70Cu/Zr/350 (3) and 70Cu/Zr/550 (4). Signal—black line, cumulative curve—red line, deconvoluted peaks used for quantification—blue line, additional deconvoluted peak—green line

CO2-TPD profiles for blank TPD (1), 50Cu/Zr/300 (2), 50Cu/Zr/350 (3) and 50Cu/Zr/550 (4). Signal—black line, cumulative curve—red line, deconvoluted peaks used for quantification—blue line, additional deconvoluted peak—green line
For CO2-TPD runs, all catalysts show broad desorption profile ranging from RT − 400 °C. In the case of catalysts calcined at 300 °C and 350 °C, the signal from CO2 desorption overlaps with the signal assigned to CO2 which is released due to malachite phase decomposition in the temperature range above 300 °C. To have better insight into the desorption process, the TPD profiles were deconvoluted. The observed peak maxima were assigned accordingly to the areas related with CO2 desorption from weak (50–150 °C), medium (150–240 °C) and strong (> 240 °C) basic sites. It is known (Gao et al. 2013; Díez et al. 2000) that weak basic sites are related with the surface hydroxyl group, medium basic sites with Zr4+–O2− pairs, and the strong basic sites with the low-coordination oxygen anions.
The quantitative analysis of deconvoluted TPD profile shows (Table 2) that aged catalysts possess the lowest concentration of surface basic sites. Additionally, the highest contribution (C) of weak basic sites is observed for these catalysts. In the case of the catalysts which were aged for 1 h, the increase in the temperature of calcination influences the formation of medium basic sites and lowering the contribution of weak basic sites. For non-aged catalysts, medium basic sites dominate and in this case for catalysts calcined above 350 °C strong basic sites are also formed according to the CO2-TPD profiles.
For non-aged catalysts calcined at 550 °C, there is decreased amount of CO2 desorbed (Totdes) compared to the amount of CO2 introduced (Totads) via pulses during the adsorption stage. One can say that in this case, some fraction of CO2 is still bonded to the surface of these catalysts while heating up to 400 °C. This means that for aged catalysts calcined at 550 °C, the contribution of strong basic sites is higher than that calculated as well as total basicity. For other catalysts, the observed discrepancies between the amount of adsorbed CO2 and desorbed CO2 are due to experimental error related mainly with difficulties in integration of long-tailed CO2 pulse responses.
Average Cu° particle size (PCu) and metallic copper dispersion (DCu) obtained via N2O pulse chemisorption are presented in Table 1. The value for CuO crystal sizes based on Rietveld refinement is much lower than PCu for all synthesized catalysts. This means that during reduction at 250 °C prior to the introduction N2O pulses, the Cu particles sinter. Especially, surface Cu particles are more prone to agglomerate and sinter due to the low Hüttig temperature of copper (Twigg and Spencer 2001). According to Table 1, it is clear that the presence of small CuO crystallites in the catalyst leads to a lower value of PCu and higher DCu. On the other hand for aged catalysts, the increase of the CuO crystal size with temperature of calcination does not result in change in the values of PCu and DCu, whereas the loss of BET specific surface area for these catalysts is pronounced. According to Li et al. (2001), copper dispersion depends on two competitive phenomena. First is the segregation of CuO from the ZrO2 matrix, which is undergone when transformation of amorphous ZrO2 proceeds, leading to a higher copper surface area. The monoclinic to tetragonal phase transformation starts at 400 °C at the surface regions and then proceeds into the bulk as the temperature of calcination is increased. The XRD analysis for aged samples did not reveal any reflexes of m-ZrO2. Nevertheless, at the early stage of ZrO2 phase transformation, the concentration in the surface region of m-ZrO2 was low. Therefore, the absence of m-ZrO2 in the subsurface region cannot be ruled out, since crystallites of ZrO2 might be less than 4 nm in size which is below the detection limit of the XDR technique. The second factor is the increase in copper particle size with increased temperature of calcination, resulting in lowering the surface copper area. In the case of aged samples, both stabilization effect of ZrO2 and copper particle growth can take place simultaneously, leading to constant values of surface copper area and copper dispersion.
For catalysts which were not aged, the increase in PCu and decrease in DCu were observed with increase in the temperature of calcination. In this case, the surface copper stabilization is not observed. The addition of ZrO2 leads to catalysts of higher value of PCu which is in agreement with the CuO crystal sizes determined by XRD.
The reducibility of synthesized catalysts was investigated by means of temperature-programmed reduction with hydrogen (H2-TPR) and the results are presented in Figs. 14, 15 and 16. For all catalysts, the reduction stoichiometry (Cu/H2) is slightly above one, meaning that all CuO is reduced to metallic copper during TPR. The observed differences in H2 consumption (Table 3) are related with differences in concentration of CuO in the obtained catalysts. The ICP analysis shows that for aged 70Cu/Zr/, the CuO/ZrO2 ratio (wt%) is 7.3, while for non-aged 70Cu/Zr/550 and 50/Cu/Zr, the ratio (wt%) is 3.8 and 1.6, respectively. The observed differences are related with lower value of ZrO2 concentration in synthesized samples. The maximum reduction rate (Tmax) occurs at lower temperature for all catalysts than for standard CuO (Tmax = 338, not shown), resulting from copper–zirconia interaction. Based on the broad shape of the TPR profile, one can say that at least two copper species are present in the obtained catalysts. To distinguish between different copper species, the TPR signals were deconvoluted into two or three Gaussian peaks in all cases. The number of deconvoluted peaks can suggest that there are at least two or three types of CuO phases in the synthesized samples.

H2-TPR profiles for 70Cu/Zr/300_1 h (1), 70CuZr/350_1 h (2) and 70Cu/Zr_550_1 h (3)

H2-TPR profiles for 70Cu/Zr/300 (1), 70Cu/Zr/350 (2) and 70Cu/Zr/550 (3)

H2-TPR profiles for 50Cu/Zr/300 (1), 50Cu/Zr/350 (2) and 50Cu/Zr/550 (3)
Only minor changes can be seen in TPR peaks for aged and non-aged catalysts which were calcined at 300 °C and 350 °C. In this case, the position of the deconvoluted peaks was similar. On the other hand, the increase of calcination temperature to 550 °C results in significant changes in the TPR profile shapes for all samples. The deconvoluted maxima shift toward higher reduction temperature. This effect is more pronounced for the non-aged catalyst which exhibits the deconvoluted peak at 294 °C and 307 °C for 70Cu/Zr/550 and 50Cu/Zr/550, respectively. Peaks which occur at higher temperature are attributed to the reduction of bulk CuO species, whereas the low-temperature peaks are due to dispersed and well-dispersed CuO species (Zhan et al. 2014). The observed changes in the deconvoluted peak numbers suggest that the ratio of dispersed and well-dispersed CuO species changes due to the synthesis condition.
The observed changes in the TPR profiles and in the value of Tmax result from copper dispersion. With increased metallic copper dispersion, there is pronounced decrease in the value of Tmax (Fig. 17). For aged catalysts, copper dispersion is the same, regardless of the temperature of calcination. This means that there is the same ratio of surface copper species to bulk copper species in these catalysts. Therefore, only minor changes in the value of Tmax are observed. The increased dispersion means that there is a higher ratio of surface copper species to bulk copper species. The surface copper species are reduced at lower temperature, while reduction of bulk copper species requires higher temperature. Considering this, the segregation of copper species and their stabilization on the surface of the catalysts facilitate the reduction.

Influence of metallic copper dispersion (DCu) on maximum reduction rate (Tmax)
Conclusions
The influence of aging time, temperature of precursor calcination and catalysts’ composition on ZrO2 polymorphism, catalyst reducibility, basicity and copper dispersion was investigated. It can be stated that the above-mentioned catalyst parameters are closely related with the synthesis method. There is an observed stabilization effect of surface copper species for aged CuO/ZrO2 catalysts, which is due to the subsurface ZrO2 transformation from tetragonal to monoclinic. For non-aged catalysts, this effect is not present and a decrease in copper dispersion is observed with increased temperature of calcination. This is in agreement with the TPR profile. The value of Tmax practically does not change for aged catalysts, no matter the temperature of calcination. This mean that aged catalysts calcined at 300 °C, 350 °C and 550 °C possess the same surface copper species concentration. In the case of non-aged catalysts, the increased content of ZrO2 results in increased temperature of reduction. This is due to lower copper dispersion, meaning that a bulk of the copper species dominates. One can say that increased temperature of precursor calcination leads to higher basicity of catalyst surface, which can play a crucial role in catalytic reactions involving CO2.
References
Agrell J, Birgersson H, Boutonnet M, Melián-Cabrera I, Navarro RM, Fierro JLG (2003) Production of hydrogen from methanol over Cu/ZnO catalysts promoted by ZrO2 and Al2O3. J Catal 219:389–403. https://doi.org/10.1016/S0021-9517(03)00221-5
Águila G, Guerrero S, Araya P (2008) Influence of the crystalline structure of ZrO2 on the activity of Cu/ZrO2 catalysts on the water gas shift reaction. Catal Commun 9:2550–2554. https://doi.org/10.1016/j.catcom.2008.07.011
Ardizzone S, Bianchi CL (1999) Electrochemical features of zirconia polymorphs. The interplay between structure and surface OH species. J Electroanal Chem 465:136–141. https://doi.org/10.1016/S0022-0728(99)00069-8
Basahel S, Mokhtar M, Alsharaeh E, Ali T, Mahmoud H, Narasimharao K (2016) Physico-chemical and catalytic properties of mesoporous CuO–ZrO2 catalysts. Catalysts 6:57–77. https://doi.org/10.3390/catal604005
Bolis V, Cerrato G, Magnacca G, Morterra C (1998) Surface acidity of metal oxides. Combined microcalorimetric and IR-spectroscopic studies of variously dehydrated systems. Thermochim Acta 312:63–77. https://doi.org/10.1016/S0040-6031(97)00440-1
Denise B, Sneeden RPA (1986) Oxide-Supported copper catalysts prepared from copper formate: differences in behavior in methanol synthesis from CO/H2 and CO2/H2 mixtures. Appl Catal 28:235–239
Díez VK, Apesteguía CR, Di Cosimo JI (2000) Acid–base properties and active site requirements for elimination reactions on alkali-promoted MgO catalysts. Catal Today 63:53–62. https://doi.org/10.1016/S0920-5861(00)00445-4
Gao P, Li F, Zhan HJ, Zhao N, Xiao FK, Wei W, Zhong LS, Wang H, Sun YH (2013) Influence of Zr on the performance of Cu/Zn/Al/Zr catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol. J Catal 298:51–60. https://doi.org/10.1016/j.jcat.2012.10.030
Ishikawa Satoshi, Jones DR, Iqbal Sarwat, Reece C, Morgan DJ, Willock DJ, Miedziak PJ, Bartley JK, Edwards JK, Murayama T, Uedaa W, Hutchings GJ (2017) Identification of the catalytically active component of Cu–Zr–O catalyst for the hydrogenation of levulinic acid to γ-valerolactone. Green Chem 19:225–236. https://doi.org/10.1039/C6GC02598F
Jung KT, Bell AT (2002) Effects of zirconia phase on the synthesis of methanol over zirconia-supported copper. Catal Lett 80:63. https://doi.org/10.1023/A:1015326726898
Koeppel RA, Baiker A, Wokaun A (1992) Copper/zirconia catalysts for the synthesis of methanol from carbon dioxide. Appl Catal A Gen 84(1):77–102. https://doi.org/10.1016/0926-860X(92)80340-I
Li M, Feng Z, Xiong G, Ying P, Xin Q, Li C (2001) Phase transformation in the surface region of zirconia detected by UV Raman spectroscopy. J Phys Chem B 105:8107–8111. https://doi.org/10.1021/jp010526l
Maruya K, Komiya T, Hayakawa T, Lu L, Yashima M (2000) Active sites on ZrO2 for the formation of isobutene from CO and H2. J Mol Catal A Chem 159:97–102. https://doi.org/10.1016/S1381-1169(00)00176-X
Morterra C, Cerrato G, Ferronia L, Montanaro L (1994) Surface characterization of yttria-stabilized tetragonal ZrO2 Part 1. Structural, morphological, and surface hydration features. Mater Chem Phys 37:243–257. https://doi.org/10.1016/0254-0584(94)90160-0
Oguchi H, Kanai H, Utani K, Matsumura Y, Imamura S (2005) Cu2O as active species in the steam reforming of methanol by CuO/ZrO2 catalysts. Appl Catal A Gen 293:64–70. https://doi.org/10.1016/j.apcata.2005.07.010
Okamoto Y, Kubota T, Gotoh H, Ohto Y, Aritani H, Tanaka T, Yoshida S (1998) XAFS study of zirconia-supported copper catalysts for the NO–CO reaction: deactivation, rejuvenation and stabilization of Cu species. J Chem Soc Faraday Trans 94:3743–3752
Pokrovski K, Jung KT, Bell AT (2001) Investigation of CO and CO2 adsorption on tetragonal and monoclinic zirconia. Langmuir 17:4297–4303. https://doi.org/10.1021/la001723z
Rhodes MD, Bell AT (2005) The effects of zirconia morphology on methanol synthesis from CO and H2 over Cu/ZrO2 catalysts Part I. Steady-state studies. J Catal 233:198–209. https://doi.org/10.1016/j.jcat.2005.04.026
Ritzkopf I, Vukojevic S, Weidenthaler C, Grunwaldt JD, Schüth F (2006) Decreased CO production in methanol steam reforming over Cu/ZrO2 catalysts prepared by the microemulsion technique. Appl Catal A Gen 302:215–223. https://doi.org/10.1016/j.apcata.2006.01.014
Samson K, Śliwa M, Socha RP, Góra-Marek K, Mucha D, Rutkowska-Zbik D, Paul JF, Ruggiero-Mikołajczyk M, Grabowski R, Słoczyński J (2014) Influence of ZrO2 structure and copper electronic state on activity of Cu/ZrO2 catalysts in methanol synthesis from CO2. ACS Catal 4:3730–3741. https://doi.org/10.1021/cs500979c
Sato AG, Volanti DP, Meira DM, Damyanova S, Longo E, Bueno JMC (2013) Effect of the ZrO2 phase on the structure and behavior of supported Cu catalysts for ethanol conversion. J Catal 307:1–17. https://doi.org/10.1016/j.jcat.2013.06.022
Shimokawabe M, Asakawa H, Takezawa N (1990) Characterization of copper/zirconia catalysts prepared by an impregnation metod. Appl Catal 59:45–58
Shuyten S, Dinka P, Mukasyan A, Wolf E (2008) A novel combustion synthesis preparation of oxidative hydrogen production from methanol. Catal Lett 121:189–198. https://doi.org/10.1007/s10562-007-9336-x
Thommes M, Kaneko K, Neimark AV, Olivier JP, Reinoso FR, Rouquerol J, Sing KSW (2015) Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC technical report). Pure Appl Chem 87(9–10):1051–1069. https://doi.org/10.1515/pac-2014-1117
Twigg MV, Spencer MS (2001) Deactivation of supported copper metal catalysts for hydrogenation reactions. Appl Catal A 212:161–174. https://doi.org/10.1016/S0926-860X(00)00854-1
Zhan H, Li F, Gao N, Zhao N, Xiao F, Wei W, Zhong L, Sun Y (2014) Methanol synthesis from CO2 hydrogenation over La–M–Cu–Zn–O (M=Y, Ce, Mg, Zr) catalysts derived from perovskite-type precursors. J Powers Sources 251:113–121. https://doi.org/10.1016/j.powsour.2013.11.037
Zhao Y, Li W, Zhang M, Tao K (2002) A comparison of surface acidic features between tetragonal and monoclinic nanostructured zirconia. Catal Commun 3:239–245. https://doi.org/10.1016/S1566-7367(02)00089-4
Zhao Y, Tao K, Wan HL (2004) Effect of zirconia phase on the reduction behaviour of highly dispersed zirconia-supported copper oxide. Catal Commun 5:249–252. https://doi.org/10.1016/j.catcom.2004.02.011
Acknowledgements
This work has been financially supported by the National Science Centre, Poland (SONATA Grant: 2016/23/D/ST4/02492, 2017–2020).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all the authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Śliwa, M., Samson, K. Influence of synthesis parameters on physicochemical properties of CuO/ZrO2 catalysts. Chem. Pap. 73, 2793–2802 (2019). https://doi.org/10.1007/s11696-019-00832-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-019-00832-8