
Abstract
MtDNA-based phylogeography has illuminated the impact of the Pleistocene Ice Age on species distribution dynamics and the build-up of genetic divergence. The well-known shortcomings of mtDNA in biogeographical inference can be compensated by integrating multilocus data and species distribution modelling into phylogeography. We re-visit the phylogeography of the Italian crested newt (Triturus carnifex), a species distributed in two of Europe’s main glacial refugia, the Balkan and Italian Peninsulas. While a new 51 nuclear DNA marker dataset supports the existence of three lineages previously suggested by mtDNA (Balkan, northern Italy and southern Italy), the nuclear DNA dataset also provides improved resolution where these lineages have obtained secondary contact. We observe geographically restricted admixture at the contact between the Balkan and northern Italy gene pools and identify a potential mtDNA ghost lineage here. At the contact between the northern and southern Italy gene pools we find admixture over a broader area, as well as asymmetric mtDNA introgression. Our species distribution model is in agreement with a distribution restricted to distinct refugia during Pleistocene glacial cycles and postglacial expansion with secondary contact. Our study supports: (1) the relevance of the north-western Balkan Peninsula as a discrete glacial refugium; (2) the importance of north-eastern Italy and the northern Apennine as suture zones; and (3) the applicability of a refugia-within-refugia scenario within the Italian Peninsula.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The glacial cycles of the Pleistocene Ice Age greatly influenced species distributions and are recognized as a major driver of intraspecific divergence in temperate species (Hewitt 2000; Hofreiter and Stewart 2009; Stewart et al. 2010). Geographical populations of a single species that were isolated in allopatric refugia during relatively long glacial periods, could repeatedly expand their ranges and establish hybrid zones upon secondary contact during relatively short interglacial periods (Hewitt 1988; Barton and Hewitt 1985). Yet, during the following glacial cycle, extinction in the interglacially colonized area would cause the ranges of the geographical populations to contract and become isolated again (Hewitt 2011a). This raises the question what the influence of such a temporary bridge for gene flow was on the genetic divergence of geographical populations within their respective glacial refugia – would genetic differentiation be negated or could it accumulate over multiple glacial cycles (Garrick et al. 2019)?
Phylogeographic surveys have mostly employed mtDNA (Riddle 2016; Beheregaray 2008; Avise 2000). Yet, mtDNA is generally more susceptible to geographical sub-structuring than the rest of the genome (Petit and Excoffier 2009), even in the face of uninterrupted gene flow (Irwin 2002), and the geographical structuring of mtDNA often deviates from that of nuclear DNA due to differential introgression (Toews and Brelsford 2012). While it is widely acknowledged that multiple unlinked nuclear markers are required to obtain an accurate estimate of the evolutionary independence of geographical populations (Edwards 2009), it has only relatively recently become feasible to consult many nuclear markers for a large number of individuals for non-model organisms (Ekblom and Galindo 2011; Garrick et al. 2015; McCormack et al. 2013). Additional insight into distribution dynamics can be obtained by the integration of species distribution modelling in phylogeography (Alvarado-Serrano and Knowles 2014; Svenning et al. 2011). This approach is regularly applied for the Last Glacial Maximum in particular, because climatic data layers are publicly available (Hijmans et al. 2005). By combining ‘next-generation phylogeography’ (Puritz et al. 2012) with species distribution modeling, mtDNA-based historical biogeographical hypotheses can be tested, identifying areas that acted as glacial refugia, and dissecting patterns of secondary admixture between lineages that originated in distinct refugia (e.g. Spinks et al. 2014; Dufresnes et al. 2020).
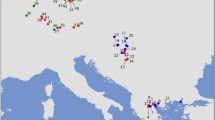
The range of the Italian crested newt Triturus carnifex encompasses two of Europe’s canonical glacial refugia, the Italian and the Balkan Peninsula (Wielstra et al. 2014b; Fig. 1). A previous mtDNA-based phylogeographic study revealed a basal split between the Italian and the Balkan populations, dated to the onset of the Pleistocene, and a further north-south division within the Italian Peninsula, in line with a refugia-within-refugia scenario (Canestrelli et al. 2012). Early studies consulting allozyme data suffered from incomplete sampling and yielded inconsistent results, preventing a proper understanding of the fate of nuclear genetic differentiation over multiple glacial-interglacial cycles (Scillitani and Picariello 2000; Arntzen 2001). We here collect multilocus nuclear DNA sequence data from populations across the entire range of T. carnifex, to determine the number of distinct gene pools present and the degree of gene flow between them. Additionally, we conduct species distribution modeling to assess glaciation-driven range reduction and fragmentation. We compare these independent estimates of distribution dynamics with each other and with the mtDNA-based hypothesis and discuss the impact of the Pleistocene climatic cycles on genetic divergence within T. carnifex.

Sampling scheme for Triturus carnifex and allocation of individuals to mtDNA clade and nuclear DNA gene pool. Numbered circles are populations, colored according to mtDNA clade (see Fig. 2), while the colored background approximates the distribution of nuclear DNA gene pools (“introgressed” refers to mtDNA derived from T. dobrogicus). Bar plots show allocation of individuals to mtDNA (mt) clade and nuclear DNA gene pool based on Structure under k =2 and k = 3 (details in Table S1). Populations 1–3 are excluded because they are affected by interspecific gene flow. Population numbers correspond to Table S1
Materials and Methods
Sampling and Sequencing
We sampled 85 individuals from 29 populations (Fig. 1; Table S1). Total genomic DNA was extracted using the DNeasy Tissue Kit (Qiagen). We obtained mtDNA data, an alignment comprising 635 bp of ND2 and 665 bp of ND4, from two phylogeographic studies (Canestrelli et al. 2012; Wielstra et al. 2013) and newly sequenced newts (13 for ND2 and 28 for ND4). We collected multilocus nuclear DNA sequence data, using an Ion Torrent next-generation sequencing protocol that is described in detail in Wielstra et al. (2014a). In brief, we sequenced 51 nuclear markers (a 52nd marker was removed as over 20% of individuals had less than 20 reads) of c. 140 bp in length (excluding primers), positioned in 3’ untranslated regions, in five multiplex PCRs. We pooled the multiplexes for each individual and ligated unique tags to be able to recognize the product belonging to each individual. We sequenced the amplicons on the Ion Torrent next-generation sequencing platform and processed the output with a bioinformatics pipeline that filters out poor quality reads, identifies alleles and converts data to a format directly usable for population genetic analysis. Mean coverage was 1157.6 reads (range 0–110648) per marker-individual combination and 98.0% of marker-individual combinations were considered successful (meaning they had at least 20 reads available). The range of the number of alleles per marker was 2–11 (Table S2). Data were phased and converted to a genotypic format that could directly be read in and converted with the software CREATE (Coombs et al. 2008).
Analysis of mtDNA
We obtained a dated mtDNA phylogeny with BEAST 2.6 (Bouckaert et al. 2019). As outgroup we used the sister species T. macedonicus (GenBank accession number NC015794). Introgressed mtDNA was excluded. The origin of the Adriatic Sea at 5.33 Ma, at the end of the Messinian Salinity Crisis, was interpreted as the vicariant event causing the split between T. carnifex and T. macedonicus (Arntzen et al. 2007) and appointed a normally distributed prior with a small standard deviation (0.001). The most appropriate models of sequence evolution were identified with jModelTest 2.1.10 (Darriba et al. 2012), based on the Bayesian Information Criterion (ND2: TN+I, ND4: TN+G). We applied the relaxed lognormal clock model and a Yule speciation model, with large uninformative priors for the Yule Birth Rate (gamma distribution, α = 0.001, β = 1000) and substitution rates (ucld.mean priors: exponential distribution, mean = 10). We conducted two independent runs of 150 million generation each, sampling parameters each 15,000 generations. We confirmed in Tracer 1.7 (Rambaut et al. 2018) that runs converged and effective sample sizes were at least 200. Trees from a stationary distribution (burn-in = 25%) were used to calculate an annotated Maximum Clade Credibility tree in TreeAnnotator 2.6. We calculated p-distances within and among the mtDNA clades identified (see Results) using MEGA X (Kumar et al. 2018).
Principal Component Analysis, Isolation by Distance and Allelic Richness of Nuclear DNA
We conducted a principal component analysis with the R package adegenet 2.1.3 (Jombart and Ahmed 2011), based on allele frequencies, and replacing missing data with the mean of the total dataset. We excluded those populations affected by interspecific gene flow (populations 1–3, see Results). We tested for isolation by distance using a Mantel test in the R package adegenet 2.1.3 (Jombart and Ahmed 2011) for the three groups identified in our Structure and PCA analyses (see Results). We excluded populations affected by interspecific (populations 1–3) or intraspecific (populations 8–9 and 14–16, see Results) gene flow. Allelic richness was determined using the R package DiveRsity 1.9.9 (Keenan et al. 2013). Again we excluded populations affected by interspecific (populations 1–3) or intraspecific (populations 8–9 and 14–16) gene flow, as well as population 4, because only a single individual was available.
Bayesian Genetic Clustering of Nuclear DNA
We analyzed multilocus nuclear DNA sequence data with Structure 2.3.3 (Pritchard et al. 2000) to assign individuals to distinct gene pools probabilistically under the admixture model, in combination with the correlated allele frequency model, with 100,000 iterations after 50,000 iterations of burn-in and ten replicates per run. We took a two-step approach in analyzing our dataset. First, considering that crested newt species hybridize at their contact zones (Arntzen et al. 2014), we aimed to exclude potentially confounding effects of interspecific gene flow. As T. carnifex is in parapatry with T. cristatus and T. dobrogicus, we tested for signs of interspecific gene flow with these two species, using reference data (three individuals from four populations for each species) available from Wielstra et al. (2014a). In a preliminary Structure run, we fixed the number of distinct gene pools k at 3, as we are dealing with three species. Yet, because the program had trouble allocating the T. cristatus and T. dobrogicus individuals to their respective species, presumably due to strong population structure within T. carnifex (see Results), we increased k to 4, under which T. cristatus and T. dobrogicus individuals were correctly allocated to their respective species. We considered T. carnifex individuals admixed if they were assigned to T. carnifex with an ancestry coefficient < 0.95.
Second, after excluding those populations affected by interspecific gene flow (populations 1–3), we explored population structure within T. carnifex. We tested for k over a range of 1–26 (with the upper limit determined by the total number of T. carnifex populations not affected by interspecific gene flow). We used CLUMPAK (Kopelman et al. 2015) to determine the optimum k value according to the Δ k criterion (Evanno et al. 2005). Individuals were assigned to a gene pool if the ancestry coefficient was ≥0.95 and otherwise were considered genetically admixed.
Nuclear DNA Phylogeny
We used Geneious Prime 2020.2.2 (https://www.geneious.com) to create a concatenated alignment with indels removed from the fasta files for the 51 nuclear loci (containing two alleles for each marker for each individual) produced by our bioinformatics pipeline (Wielstra et al. 2014a). A consensus sequence with polymorphic sites encoded by IUPAC codes was created for each individual using the SeqinR 3.6.1 package (Charif and Lobry 2007) in R (R-Development-Core-Team 2020). This resulted in a 7007 bp alignment. We excluded populations that showed signs of admixture with other crested newt species (populations 1–3) or among the three geographical groups delineated by the Structure and principal component analyses (populations 8–9 and 14–16). We calculated p-distances within and among the nuDNA clusters (see Results) using MEGA X (Kumar et al. 2018). We obtained a phylogeny with BEAST 2.6 (Bouckaert et al. 2019), using T. macedonicus as outgroup (ID 3248 taken from Wielstra et al. 2017). We considered heterozygote positions (useAmbiguities =”true”). We applied the strict clock model and a Yule speciation model. We conducted two independent runs of 30 million generations each, sampling parameters each 3,000 generations. Results were checked and summarized using the same procedure as described for the mtDNA phylogenetic analysis.
Species Distribution Modelling
We created a species distribution model with Maxent 3.3.3k (Phillips et al. 2006) as this program is developed for presence-only data, outperforms other algorithms and can be used to reliably hindcast to past environments (Elith et al. 2006; Hijmans and Graham 2006). We restricted the feature type to hinge features as this produces a smoother model fit that emphasizes trends rather than idiosyncrasies of the data (Elith et al. 2010). We used a database of 127 T. carnifex localities presented in Wielstra et al. (2014b). For climate layers we used bioclimatic variables at 2.5 arcminute resolution (c. 5 x 5 km) available from the WorldClim database 1.4 (Hijmans et al. 2005). This resolution is appropriate to detect range-wide patterns, while still being biologically meaningful for Triturus newts, which have an estimated dispersal rate of 2 km per generation (Arntzen and Wallis 1991). Following Guisan and Thuiller (2005) and Peterson (2011) we selected a subset considered to reflect physiological limitations of the study species (in this case seasonality in temperature and precipitation) while showing little multicollinearity (a Pearson’s correlation of r < 0.7): bio10 = mean temperature of warmest quarter, bio11 = mean temperature of coldest quarter, bio15 = precipitation seasonality, bio16 = precipitation of wettest quarter, and bio17 = precipitation of driest quarter. Because the environmental range covered by pseudo-absence data, used by Maxent to discriminate presence data from the environmental background, should neither be too narrow nor too broad (Elith et al. 2011), we drew a 200 km radius buffer (following VanDerWal et al. 2009) around known Triturus localities (Wielstra et al. 2013). To determine whether our species distribution model performs better than random expectation, we tested its AUC value against a null model based on 99 models for random localities (Raes and ter Steege 2007). Random point data were created with ENMTools 1.3 (Warren et al. 2010). For hindcasting we used bioclimatic variables, available from the WorldClim database 1.4, for the Last Glacial Maximum (~ 21Ka), based on downscaled climate data from simulations of two global climate models: CCSM4 (Brady et al. 2013) and MIROC-ESM (Sueyoshi et al. 2013).
Results
We identified 43 T. carnifex mtDNA haplotypes (Table S1); mtDNA derived from T. dobrogicus was present in the two Austrian populations (populations 1–2 in Fig. 1). The mtDNA phylogeny strongly supports the presence of a distinct Balkan, northern Italian and southern Italian clade (Fig. 2; Table S3). A new, relatively distinct (Table S3) haplotype was identified from a geographically intermediate region (h09 in population 8; Fig. 1) that clusters with the Balkan clade (Fig. 2). The basal split in T. carnifex, dated to the late Pliocene (c. 3.2 Ma), is between the Balkan and the two Italian lineages. The subsequent split between the northern and southern Italian lineages is dated to the early Pleistocene (c. 2.3 Ma).

Time-calibrated mtDNA phylogeny for Triturus carnifex. The outgroup is not shown. The time axis (millions of years ago) is below the tree and grey bars represent the 95% highest posterior density interval of node ages for nodes with posterior probabilities ≥ 0.95 (labelled with an asterisk). Colors of mtDNA clades correspond to Fig. 1. Haplotype codes and population numbers (between parentheses) correspond to Table S1
The principal component analysis separated the Balkan and Italian populations (populations 4–7 versus 10–29, with 8–9 taking an intermediary position) along principal component 1 (explaining 26.5 % of the total variation) and the northern and southern Italian populations (populations 10–13 versus 17–29, with 14–16 taking an intermediary position) along principal component 2 (12.0%; Fig. 3). We found significant support for isolation by distance for the southern Italian cluster and not for the northern Italian of Balkan clusters. Allelic richness varied from 1.26–1.38, 1.15–1.39 and 1.11–1.31 for the Balkan, northern Italy and southern Italy clusters (Table S4).

Principal component analysis of nuclear DNA data for Triturus carnifex. Five clusters of populations have been given a unique color and population numbers correspond to Fig. 1
Structure identified nuclear admixture with T. cristatus in the two Austrian populations (populations 1–2 in Fig. 1; Table S1) and with T. dobrogicus in one of the Croatian populations (population 3). These populations, closely related to the other Balkan T. carnifex populations, were excluded from further analyses. The test for intraspecific structuring for T. carnifex in Structure suggested k = 2, separating the Balkan and Italian populations (populations 4–7 versus 10–29, with 8–9 showing admixture), as the optimal partitioning scheme, with k = 3, additionally separating northern and southern Italian populations, a close second (dividing Italian populations into 10–13 versus 17–29, with admixture in 14–16). The allocation of individuals to clusters under both k values, and the distribution of gene pools under k = 3, is shown in Fig. 1. Details are in Table S1 and results for additional k values are available from Dryad (see section ‘Data availability’). The BEAST phylogeny supports a basal bifurcation between a significantly supported Balkan and Italian clade. Within the latter clade, the northern and southern Italian populations do not form reciprocally monophyletic groups (Fig. S1). See Table S3 for p-distances within and among the nuDNA clusters.
The species distribution model (AUC value = 0.96) suggests that suitable conditions occur throughout the range of T. carnifex today (Fig. 4). Hence, the predicted ranges of the gene pools as delineated by nuclear and mitochondrial DNA are not geographically isolated at present, although the band of suitable habitat just west of the zone of admixture between the Balkan and Italian populations is narrow, squeezed between the Alps and the Po Delta. When projected on climate reconstructions for the Last Glacial Maximum, regardless of the global circulation models used, the species distribution model suggests suitable conditions were much reduced at the time (Fig. 4). Suitable area was predicted at the Last Glacial Maximum in the north-western Balkan Peninsula and in the Italian Peninsula. Within the Italian peninsula the range is predicted to have been highly fragmented, with patches of suitable habitat just south of the Alps and in the center and south of the Italian Peninsula. A pocket of suitable habitat remained in the area of current admixture between the Balkan and Italian groups.

Species distribution model for Triturus carnifex. The species distribution model is projected on climate layers for the present (a) and for the Last Glacial Maximum based on the MIROC (b) and CCSM (C) climate models. Warmer colors refer to a higher predicted suitability
Discussion
Genome-enabled approaches to phylogeography allow us to test and extend biogeographical hypotheses proposed in studies employing mtDNA only (e.g. Spinks et al. 2014; Dufresnes et al. 2020). We here take a closer look at the Italian crested newt T. carnifex, a species whose range spans two canonical glacial refugia and for which an mtDNA-based phylogeographic study found deep geographical genetic structuring between and within these refugia (Canestrelli et al. 2012). We obtain a better understanding of the degree and extent of genetic admixture between populations derived from multiple glacial refugia.
Bayesian clustering analysis and principal component analysis confirm that, at the highest level of hierarchy, T. carnifex comprises two discrete gene pools. These gene pools occupy the Italian and the Balkan parts of the range (Figs. 1 and 3). Similarly, T. carnifex shows a basal split between an Italian and Balkan clade in both the mtDNA and nuclear DNA phylogeny (Figs. 2 and S1). The lack of isolation by distance in the Balkan and northern Italian (but not southern Italian) nuclear gene pools could reflect postglacial population expansion. Allelic richness for nuclear DNA shows no clear spatial signal. The species distribution model suggests that the Italian and Balkan groups were isolated in distinct refugia at the Last Glacial Maximum and are currently connected by a narrow band of suitable habitat (Fig. 4). These two groups presently meet in north-eastern Italy, in a region sandwiched between the Alps in the north and the Adriatic Sea in the south (Fig. 1). Here they show geographically restricted admixture. A comparison of intraspecific divergence in the genus Triturus (Table S2 in Wielstra et al. 2019) reveals that the degree of nuclear genetic divergence between the Balkan and Italian groups is unprecedented within crested newt species. The two lineages show apparently limited gene flow, despite hybridization upon secondary contact, suggesting the existence of two cryptic species in T. carnifex. A detailed hybrid zone analysis is required to test this hypothesis.
Within the Italian Peninsula, Bayesian clustering and principal component analysis suggests further sub-structuring into a northern and southern Italian group (Fig. 1), roughly in line with a northern and southern mtDNA clade (Fig. 2), whereas populations from northern and southern Italy are not reciprocally monophyletic in the nuclear DNA phylogeny (Fig. S1). The two Italian nuclear gene pools show admixture over a comparatively wide area along the northern Apennines. The species distribution model suggests that suitable conditions were much reduced at the Last Glacial Maximum, but remained just south of the Alps and, much further to the south, in central and southern Italy (Fig. 4).
While the phylogeographic signal from mtDNA in T. carnifex is generally concordant with nuclear DNA and supports the same three main lineages, it breaks down in areas of admixture (Figs. 1 and 2). First, we find mtDNA of a different crested newt species, T. dobrogicus, in T. carnifex populations north of the Alps (Fig.1). As T. dobrogicus is an obligatory lowland species (Arntzen et al. 1997), its mtDNA is unlikely to be native in this area. Yet, given that the same mtDNA haplotype still occurs throughout the range of T. dobrogicus (Wielstra et al. 2013), introgression must have occurred recently, upon postglacial secondary contact between the two species. Introgression into T. carnifex probably took place in the lowlands near the present day T. dobrogicus–T. carnifex hybrid zone and subsequently surfed (Klopfstein et al. 2006) beyond the T. dobrogicus range, in an expanding T. carnifex population (Mačát et al. 2019). Postglacial northwards expansion of T. carnifex around the Alps (from the Balkan population) is in line with our species distribution model (Fig. 4).
Second, admixture between the two Italian gene pools is relatively wider for the nuclear genome than for mtDNA. Also, mtDNA of the northern Italian clade is found to have introgressed into the southern Italian gene pool, well over a hundred kilometre southwards of the present day zone of admixture between the two gene pools (Fig. 1). We propose this mtDNA introgression could reflect expansion of the southern gene pool, at the expense of the northern one (Currat et al. 2008; Wielstra 2019) but it could also be explained by positive selection dragging the northern mtDNA into the southern gene pool (Bonnet et al. 2017).
Third, we identified a relatively distinct mtDNA haplotype (h09) of early Pleistocene origin (c. 1.6 Ma; Fig. 2) in the present day hybrid zone between the Italian and Balkan groups (Fig. 1). Given the geographically restricted distribution of the new mtDNA clade identified in T. carnifex it may have evolved in situ. The species distribution modelling agrees that suitable conditions remained here at the Last Glacial Maximum and presumably during previous glaciations as well (Hewitt 2011b). Under these circumstances, a population of T. carnifex could have survived the glaciations here, despite recurring secondary contact and admixture of the Italian and Balkan groups in north-eastern Italy. The origin of a mtDNA ‘ghost lineage’ that does not have precedent in nuclear DNA, could be explained by secondary contact of, and subsequent fusion of, populations that initially diverged in allopatry (Hogner et al. 2012; Dufresnes et al. 2020) or mtDNA capture from a population that has subsequently gone extinct (Mao et al. 2013; Zhang et al. 2019; Ottenburghs 2020). MtDNA ghost lineages are known from several Balkan newt taxa (Pabijan et al. 2015; Recuero et al. 2014; Wielstra and Arntzen 2020). Denser sampling east of the hybrid zone between the Balkan and northern Italian gene pool is required to test these hypotheses.
Conclusions
Triturus carnifex survived the Pleistocene Ice Age in three discrete glacial refugia, one in the north-western Balkan Peninsula (roughly the Istria region), one in the north of the Italian Peninsula, and one in the central/south of the Italian Peninsula—in line with a previous historical biogeographical scenario derived from mtDNA. Our study supports: (1) the relevance of the north-western Balkan Peninsula as a discrete glacial refugium (while traditionally the southern Balkans are considered most important; Poulakakis et al. 2015); (2) the importance of north-eastern Italy and the northern Apennine as suture zones (see also e.g. Dufresnes et al. 2014; Verardi et al. 2009; Schultze et al. 2020; Canestrelli et al. 2006; Canestrelli and Nascetti 2008); and (3) the applicability of a refugia-within-refugia scenario within the Italian Peninsula (Canestrelli et al. 2014; Canestrelli et al. 2010; Salvi et al. 2013; Maura et al. 2014).
While mtDNA overall is informative on the historical biogeography of T. carnifex, it does occasionally fall short. The deviation between mtDNA and our nuclear DNA dataset can be explained by well-understood gene flow upon secondary contact, resulting in asymmetric introgression, and ancient gene flow followed by genetic swamping might also account for the origin of a mtDNA ghost lineage. Here, next-generation phylogeography and species distribution modelling provide a more accurate and intricate picture. The Italian crested newt exemplifies how population fragmentation into multiple refugia, within the wider network of European glacial refugia, drove intraspecific divergence, despite repeated opportunities for merging during interglacial periods.
Data Availability
Sampling data are in Table S1, Supplementary Information. Raw and processed Ion Torrent sequence data and files associated with the analyses are available at the Dryad Digital Repository (https://doi.org/10.5061/dryad.g4f4qrfp5).
References
Alvarado-Serrano, D. F., & Knowles, L. L. (2014). Ecological niche models in phylogeographic studies: applications, advances, and precautions. Molecular Ecology Resources, 14(2), 233–248. https://doi.org/10.1111/1755-0998.12184
Arntzen, J. W. (2001). Genetic variation in the Italian crested newt, Triturus carnifex, and the origin of a non-native population north of the Alps. Biodiversity and Conservation, 10(6), 971–987. https://doi.org/10.1023/a:1016644814551
Arntzen, J. W., Bugter, R. J. F., Cogalniceanu, D., & Wallis, G. P. (1997). The distribution and conservation status of the Danube crested newt, Triturus dobrogicus. Amphibia-Reptilia, 18(2), 133–142. https://doi.org/10.1163/156853897x00026
Arntzen, J. W., Espregueira Themudo, G., & Wielstra, B. (2007). The phylogeny of crested newts (Triturus cristatus superspecies): nuclear and mitochondrial genetic characters suggest a hard polytomy, in line with the paleogeography of the centre of origin. Contributions to Zoology, 76(4), 261–278.
Arntzen, J. W., & Wallis, G. P. (1991). Restricted gene flow in a moving hybrid zone of the newts Triturus cristatus and T. marmoratus in western France. Evolution, 45(4), 805–826. https://doi.org/10.1111/j.1558-5646.1991.tb04352.x
Arntzen, J. W., Wielstra, B., & Wallis, G. P. (2014). The modality of nine Triturus newt hybrid zones, assessed with nuclear, mitochondrial and morphological data. Biological Journal of the Linnean Society, 113(2), 604–622. https://doi.org/10.1111/bij.12358
Avise, J. C. (2000). Phylogeography: The history and formation of species. Cambridge, Massachusetts: Harvard University Press.
Barton, N. H., & Hewitt, G. M. (1985). Analysis of hybrid zones. Annual Review of Ecology and Systematics, 16, 113–148. https://doi.org/10.1146/annurev.es.16.110185.000553
Beheregaray, L. B. (2008). Twenty years of phylogeography: the state of the field and the challenges for the Southern Hemisphere. Molecular Ecology, 17(17), 3754–3774. https://doi.org/10.1111/j.1365-294X.2008.03857.x
Bonnet, T., Leblois, R., Rousset, F., & Crochet, P.-A. (2017). A reassessment of explanations for discordant introgressions of mitochondrial and nuclear genomes. Evolution, 71(9), 2140–2158. https://doi.org/10.1111/evo.13296
Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duchêne, S., Fourment, M., Gavryushkina, A., et al. (2019). BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Computational Biology, 15(4), e1006650. https://doi.org/10.1371/journal.pcbi.1006650
Brady, E. C., Otto-Bliesner, B. L., Kay, J. E., & Rosenbloom, N. (2013). Sensitivity to glacial forcing in the CCSM4. Journal of Climate, 26(6), 1901–1925. https://doi.org/10.1175/JCLI-D-11-00416.1
Canestrelli, D., Aloise, G., Cecchetti, S., & Nascetti, G. (2010). Birth of a hotspot of intraspecific genetic diversity: notes from the underground. Molecular Ecology, 19(24), 5432–5451. https://doi.org/10.1111/j.1365-294X.2010.04900.x
Canestrelli, D., Bisconti, R., Sacco, F., & Nascetti, G. (2014). What triggers the rising of an intraspecific biodiversity hotspot? Hints from the agile frog. Scientific Reports, 4(5042). https://doi.org/10.1038/srep05042
Canestrelli, D., & Nascetti, G. (2008). Phylogeography of the pool frog Rana (Pelophylax) lessonae in the Italian peninsula and Sicily: multiple refugia, glacial expansions and nuclear–mitochondrial discordance. Journal of Biogeography, 35(10), 1923–1936. https://doi.org/10.1111/j.1365-2699.2008.01946.x
Canestrelli, D., Salvi, D., Maura, M., Bologna, M. A., & Nascetti, G. (2012). One Species, three Pleistocene evolutionary histories: phylogeography of the Italian crested newt, Triturus carnifex. PLoS One, 7(7), e41754. https://doi.org/10.1371/journal.pone.0041754
Canestrelli, D., Verardi, A., & Nascetti, G. (2006). Genetic differentiation and history of populations of the Italian treefrog Hyla intermedia: lack of concordance between mitochondrial and nuclear markers. Genetica, 130(3), 241. https://doi.org/10.1007/s10709-006-9102-9
Charif, D., & Lobry, J. R. (2007). SeqinR 1.0-2: a contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. In Structural approaches to sequence evolution (pp. 207-232): Springer.
Coombs, J. A., Letcher, B. H., & Nislow, K. H. (2008). CREATE: a software to create input files from diploid genotypic data for 52 genetic software programs. Molecular Ecology Resources, 8(3), 578–580. https://doi.org/10.1111/j.1471-8286.2007.02036.x
Currat, M., Ruedi, M., Petit, R. J., & Excoffier, L. (2008). The hidden side of invasions: massive introgression by local genes. Evolution, 62(8), 1908–1920. https://doi.org/10.1111/j.1558-5646.2008.00413.x
Darriba, D., Taboada, G. L., Doallo, R., & Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat Meth, 9(8), 772–772. https://doi.org/10.1038/nmeth.2109
Dufresnes, C., Bonato, L., Novarini, N., Betto-Colliard, C., Perrin, N., & Stöck, M. (2014). Inferring the degree of incipient speciation in secondary contact zones of closely related lineages of Palearctic green toads (Bufo viridis subgroup). Heredity, 113(1), 9–20.
Dufresnes, C., Nicieza, A. G., Litvinchuk, S. N., Rodrigues, N., Jeffries, D. L., Vences, M., et al. (2020). Are glacial refugia hotspots of speciation and cytonuclear discordances? Answers from the genomic phylogeography of Spanish common frogs. Molecular Ecology, 29(5), 986–1000. https://doi.org/10.1111/mec.15368
Edwards, S. V. (2009). Is a new and general theory of molecular systematics emerging? Evolution, 63(1), 1–19. https://doi.org/10.1111/j.1558-5646.2008.00549.x
Ekblom, R., & Galindo, J. (2011). Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity, 107(1), 1–15.
Elith, J., Graham, C. H., Anderson, R., Dudík, M., Ferrier, S., Guisan, A., et al. (2006). Novel methods improve prediction of species’ distributions from occurrence data. Ecography, 29(2), 129–151. https://doi.org/10.1111/j.2006.0906-7590.04596.x
Elith, J., Kearney, M., & Phillips, S. (2010). The art of modelling range-shifting species. Methods in Ecology and Evolution, 1(4), 330–342. https://doi.org/10.1111/j.2041-210X.2010.00036.x
Elith, J., Phillips, S. J., Hastie, T., Dudík, M., Chee, Y. E., & Yates, C. J. (2011). A statistical explanation of MaxEnt for ecologists. Diversity and Distributions, 17(1), 43–57. https://doi.org/10.1111/j.1472-4642.2010.00725.x
Evanno, G., Regnaut, S., & Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: A simulation study. Molecular Ecology, 14(8), 2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Garrick, R. C., Banusiewicz, J. D., Burgess, S., Hyseni, C., & Symula, R. E. (2019). Extending phylogeography to account for lineage fusion. Journal of Biogeography, 46(2), 268–278. https://doi.org/10.1111/jbi.13503
Garrick, R. C., Bonatelli, I. A. S., Hyseni, C., Morales, A., Pelletier, T. A., Perez, M. F., et al. (2015). The evolution of phylogeographic data sets. Molecular Ecology, 24(6), 1164–1171. https://doi.org/10.1111/mec.13108
Guisan, A., & Thuiller, W. (2005). Predicting species distribution: offering more than simple habitat models. Ecology Letters, 8(9), 993–1009. https://doi.org/10.1111/j.1461-0248.2005.00792.x
Hewitt, G. (2000). The genetic legacy of the Quaternary ice ages. Nature, 405(6789), 907–913. https://doi.org/10.1038/35016000
Hewitt, G. (2011). Quaternary phylogeography: the roots of hybrid zones. Genetica, 139(5), 617–638. https://doi.org/10.1007/s10709-011-9547-3
Hewitt, G. M. (1988). Hybrid zones—natural laboratories for evolutionary studies. Trends in Ecology & Evolution, 3(7), 158–167. https://doi.org/10.1016/0169-5347(88)90033-x
Hewitt, G. M. (2011). Mediterranean peninsulas: the evolution of hotspots. In F. E. Zachos & J. C. Habel (Eds.), Biodiversity hotspots: distribution and protection of conservation priority areas (pp. 123–147). Berlin: Springer.
Hijmans, R. J., Cameron, S. E., Parra, J. L., Jones, P. G., & Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology, 25(15), 1965–1978. https://doi.org/10.1002/joc.1276
Hijmans, R. J., & Graham, C. H. (2006). The ability of climate envelope models to predict the effect of climate change on species distributions. Global Change Biology, 12(12), 2272–2281. https://doi.org/10.1111/j.1365-2486.2006.01256.x
Hofreiter, M., & Stewart, J. (2009). Ecological change, range fluctuations and population dynamics during the Pleistocene. Current Biology, 19(14), R584–R594. https://doi.org/10.1016/j.cub.2009.06.030
Hogner, S., Laskemoen, T., Lifjeld, J. T., Porkert, J., Kleven, O., Albayrak, T., et al. (2012). Deep sympatric mitochondrial divergence without reproductive isolation in the common redstart Phoenicurus phoenicurus. Ecology and Evolution, 2(12), 2974–2988. https://doi.org/10.1002/ece3.398
Irwin, D. E. (2002). Phylogeographic breaks without geographic barriers to gene flow. Evolution, 56(12), 2383–2394. https://doi.org/10.1111/j.0014-3820.2002.tb00164.x
Jombart, T., & Ahmed, I. (2011). adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics, 27(21), 3070–3071. https://doi.org/10.1093/bioinformatics/btr521
Keenan, K., McGinnity, P., Cross, T. F., Crozier, W. W., & Prodöhl, P. A. (2013). DiveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods in Ecology and Evolution, 4(8), 782–788. https://doi.org/10.1111/2041-210X.12067
Klopfstein, S., Currat, M., & Excoffier, L. (2006). The fate of mutations surfing on the wave of a range expansion. Molecular Biology and Evolution, 23(3), 482–490. https://doi.org/10.1093/molbev/msj057
Kopelman, N. M., Mayzel, J., Jakobsson, M., Rosenberg, N. A., & Mayrose, I. (2015). Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Molecular Ecology Resources, 15(5), 1179–1191. https://doi.org/10.1111/1755-0998.12387
Kumar, S., Stecher, G., Li, M., Knyaz, C., & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35(6), 1547–1549.
Mačát, Z., Rulík, M., Jablonski, D., Reiter, A., Jeřábková, L., Rada, S., et al. (2019). Species-specific habitat preferences do not shape the structure of a crested newt hybrid zone (Triturus cristatus x T. carnifex). Ecology and Evolution, 9(22), 12446–12458. https://doi.org/10.1002/ece3.5683
Mao, X., He, G., Hua, P., Jones, G., Zhang, S., & Rossiter, S. J. (2013). Historical introgression and the persistence of ghost alleles in the intermediate horseshoe bat (Rhinolophus affinis). Molecular Ecology, 22(4), 1035–1050. https://doi.org/10.1111/mec.12154
Maura, M., Salvi, D., Bologna, M. A., Nascetti, G., & Canestrelli, D. (2014). Northern richness and cryptic refugia: phylogeography of the Italian smooth newt Lissotriton vulgaris meridionalis. Biological Journal of the Linnean Society, 113(2), 590–603. https://doi.org/10.1111/bij.12360
McCormack, J. E., Hird, S. M., Zellmer, A. J., Carstens, B. C., & Brumfield, R. T. (2013). Applications of next-generation sequencing to phylogeography and phylogenetics. Molecular Phylogenetics and Evolution, 66(2), 526–538. https://doi.org/10.1016/j.ympev.2011.12.007
Ottenburghs, J. (2020). Ghost introgression: Spooky gene flow in the distant past. Bioessays, 42(6), 2000012. https://doi.org/10.1002/bies.202000012
Pabijan, M., Zieliński, P., Dudek, K., Chloupek, M., Sotiropoulos, K., Liana, M., et al. (2015). The dissection of a Pleistocene refugium: phylogeography of the smooth newt, Lissotriton vulgaris, in the Balkans. Journal of Biogeography, 42(4), 671–683. https://doi.org/10.1111/jbi.12449
Peterson, A. T. (2011). Ecological niche conservatism: a time-structured review of evidence. Journal of Biogeography, 38(5), 817–827. https://doi.org/10.1111/j.1365-2699.2010.02456.x
Petit, R. J., & Excoffier, L. (2009). Gene flow and species delimitation. Trends in Ecology & Evolution, 24(7), 386–393. https://doi.org/10.1016/j.tree.2009.02.011
Phillips, S. J., Anderson, R. P., & Schapire, R. E. (2006). Maximum entropy modeling of species geographic distributions. Ecological Modelling, 190(3–4), 231–259. https://doi.org/10.1016/j.ecolmodel.2005.03.026
Poulakakis, N., Kapli, P., Lymberakis, P., Trichas, A., Vardinoyiannis, K., Sfenthourakis, S., et al. (2015). A review of phylogeographic analyses of animal taxa from the Aegean and surrounding regions. Journal of Zoological Systematics and Evolutionary Research, 53(1), 18–32. https://doi.org/10.1111/jzs.12071
Pritchard, J. K., Stephens, M., & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959.
Puritz, J. B., Addison, J. A., & Toonen, R. J. (2012). Next-generation phylogeography: A targeted approach for multilocus sequencing of non-model organisms. PLoS One, 7(3), e34241. https://doi.org/10.1371/journal.pone.0034241
R-Development-Core-Team. (2020). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Raes, N., & ter Steege, H. (2007). A null-model for significance testing of presence-only species distribution models. Ecography, 30(5), 727–736. https://doi.org/10.1111/j.2007.0906-7590.05041.x
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., & Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67(5), 901–904. https://doi.org/10.1093/sysbio/syy032
Recuero, E., Buckley, D., García-París, M., Arntzen, J. W., Cogălniceanu, D., & Martínez-Solano, I. (2014). Evolutionary history of Ichthyosaura alpestris (Caudata, Salamandridae) inferred from the combined analysis of nuclear and mitochondrial markers. Molecular Phylogenetics and Evolution, 81, 207–220. https://doi.org/10.1016/j.ympev.2014.09.014
Riddle, B. R. (2016). Comparative phylogeography clarifies the complexity and problems of continental distribution that drove A R Wallace to favor islands. Proceedings of the National Academy of Sciences, 113(29), 7970–7977. https://doi.org/10.1073/pnas.1601072113
Salvi, D., Harris, D. J., Kaliontzopoulou, A., Carretero, M. A., & Pinho, C. (2013). Persistence across Pleistocene ice ages in Mediterranean and extra-Mediterranean refugia: phylogeographic insights from the common wall lizard. BMC Evolutionary Biology, 13(1), 147. https://doi.org/10.1186/1471-2148-13-147
Schultze, N., Spitzweg, C., Corti, C., Delaugerre, M., Di Nicola, M. R., Geniez, P., et al. (2020). Mitochondrial ghost lineages blur phylogeography and taxonomy of Natrix helvetica and N. natrix in Italy and Corsica. Zoologica Scripta, 49(4), 395–411. https://doi.org/10.1111/zsc.12417
Scillitani, G., & Picariello, O. (2000). Genetic variation and its causes in the crested newt, Triturus carnifex (Laurenti, 1768), from Italy (Caudata: Salamandridae). Herpetologica, 56(1), 119–130.
Spinks, P. Q., Thomson, R. C., & Shaffer, H. B. (2014). The advantages of going large: genome-wide SNPs clarify the complex population history and systematics of the threatened western pond turtle. Molecular Ecology, 23(9), 2228–2241. https://doi.org/10.1111/mec.12736
Stewart, J. R., Lister, A. M., Barnes, I., & Dalen, L. (2010). Refugia revisited: individualistic responses of species in space and time. Proceedings of the Royal Society B-Biological Sciences, 277(1682), 661–671. https://doi.org/10.1098/rspb.2009.1272
Sueyoshi, T., Ohgaito, R., Yamamoto, A., Chikamoto, M. O., Hajima, T., Okajima, H., et al. (2013). Set-up of the PMIP3 paleoclimate experiments conducted using an Earth system model MIROC-ESM. Geosci. Model Dev, 6(3), 819–836. https://doi.org/10.5194/gmd-6-819-2013
Svenning, J.-C., Fløjgaard, C., Marske, K. A., Nógues-Bravo, D., & Normand, S. (2011). Applications of species distribution modeling to paleobiology. Quaternary Science Reviews, 30(21–22), 2930–2947. https://doi.org/10.1016/j.quascirev.2011.06.012
Toews, D. P. L., & Brelsford, A. (2012). The biogeography of mitochondrial and nuclear discordance in animals. Molecular Ecology, 21(16), 3907–3930. https://doi.org/10.1111/j.1365-294X.2012.05664.x
VanDerWal, J., Shoo, L. P., Graham, C., & Williams, S. E. (2009). Selecting pseudo-absence data for presence-only distribution modeling: How far should you stray from what you know? Ecological Modelling, 220(4), 589–594. https://doi.org/10.1016/j.ecolmodel.2008.11.010
Verardi, A., Canestrelli, D., & Nascetti, G. (2009). Nuclear and mitochondrial patterns of introgression between the parapatric European treefrogs Hyla arborea and H. intermedia. Annales Zoologici Fennici, 46(4), 247–258. https://doi.org/10.5735/086.046.0402
Warren, D. L., Glor, R. E., & Turelli, M. (2010). ENMTools: a toolbox for comparative studies of environmental niche models. Ecography, 33(3), 607–611. https://doi.org/10.1111/j.1600-0587.2009.06142.x
Wielstra, B. (2019). Historical hybrid zone movement: more pervasive than appreciated. Journal of Biogeography, 46(7), 1300–1305. https://doi.org/10.1111/jbi.13600
Wielstra, B., & Arntzen, J. W. (2020). Extensive cytonuclear discordance in a crested newt from the Balkan Peninsula glacial refugium. Biological Journal of the Linnean Society, 130(3), 578–585. https://doi.org/10.1093/biolinnean/blaa062
Wielstra, B., Burke, T., Butlin, R. K., & Arntzen, J. W. (2017). A signature of dynamic biogeography: Enclaves indicate past species replacement. Proceedings of the Royal Society of London B: Biological Sciences, 284(1868), 20172014. https://doi.org/10.1098/rspb.2017.2014
Wielstra, B., Crnobrnja-Isailović, J., Litvinchuk, S. N., Reijnen, B. T., Skidmore, A. K., Sotiropoulis, K., et al. (2013). Tracing glacial refugia of Triturus newts based on mitochondrial DNA phylogeography and species distribution modeling. Frontiers in Zoology, 10, 13. https://doi.org/10.1186/1742-9994-10-13
Wielstra, B., Duijm, E., Lagler, P., Lammers, Y., Meilink, W. R. M., Ziermann, J. M., et al. (2014). Parallel tagged amplicon sequencing of transcriptome-based genetic markers for Triturus newts with the Ion Torrent next-generation sequencing platform. Molecular Ecology Resources, 14(5), 1080–1089. https://doi.org/10.1111/1755-0998.12242
Wielstra, B., McCartney-Melstad, E., Arntzen, J. W., Butlin, R. K., & Shaffer, H. B. (2019). Phylogenomics of the adaptive radiation of Triturus newts supports gradual ecological niche expansion towards an incrementally aquatic lifestyle. Molecular Phylogenetics and Evolution, 133, 120–127. https://doi.org/10.1016/j.ympev.2018.12.032
Wielstra, B., Sillero, N., Vörös, J., & Arntzen, J. W. (2014). The distribution of the crested and marbled newt species (Amphibia: Salamandridae: Triturus) – an addition to the New Atlas of Amphibians and Reptiles of Europe. Amphibia-Reptilia, 35(3), 376–381. https://doi.org/10.1163/15685381-00002960
Zhang, D., Tang, L., Cheng, Y., Hao, Y., Xiong, Y., Song, G., et al. (2019). “Ghost introgression” as a cause of deep mitochondrial divergence in a bird species complex. Molecular Biology and Evolution, 36(11), 2375–2386. https://doi.org/10.1093/molbev/msz170
Acknowledgements
Michela Maura (Roma Tre University) helped during field and laboratory work. Marco A. Bologna (Roma Tre University) and G. Nascetti (Tuscia University) supported in sample collection. Wiesław Babik and Dick Groenenberg provided advice on data analysis.
Funding
None
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethics approval
Newts were captured and handled under permits from the Italian Ministry of Environment (DPN/2D/2003/2267).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wielstra, B., Salvi, D. & Canestrelli, D. Genetic Divergence Across Glacial Refugia Despite Interglacial Gene Flow in a Crested Newt. Evol Biol 48, 17–26 (2021). https://doi.org/10.1007/s11692-020-09519-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11692-020-09519-5