
Abstract
Hydrogen is an alternative reductant to replace carbon for the production of metals. Reduction by hydrogen has advantages compared to carbothermic reduction, such as faster reaction rate and cleaner by-product (water vapor). This study investigated the application of hydrogen reduction for recycling and recovering cobalt and lithium from Li-ion battery cathode material (LiCoO2). The study consisted of thermodynamic simulations of the reactions and microstructure evolution analysis from experimental work to propose mechanisms of the reduction process. The thermodynamic assessment predicted that metallic Co could be generated from 400 °C and was stable up to 1200 °C, but strongly dependent on the molar amount of H2. The final experimental reduction products of lithium and cobalt were found to vary and consisted of Li2O, LiOH, Li2O2 and Co, CoO, Co3O4, respectively. The experimental work revealed that the overall reduction mechanism is uniquely characterized by the reduction temperature. The temperature range of 800 °C to 900 °C offered more benefit as Co could be generated as a larger mass indicating a more progressive reduction. The data and information obtained can help optimize the parameters in the recycling of Li-ion batteries.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Following the global trend on the electrification of transportation, car production is increasingly shifting to electric vehicles. In 2021, the sales of electric vehicles were 6.6 million which was four times higher than in 2019. It is projected by International Energy Agency (IEA) that the global electric vehicle stock will grow to 200 million by 2030 with the projection of 30 pct annual average growth. Global electric vehicle sales will reach 18 million units by 2025 and over 30 million by 2030. This means that by 2030, electric vehicle sales will represent 10 pct of global car sales.[1]
Due to the rapid growth in the production of electric vehicles, the demand for battery raw materials, particularly cobalt and lithium, is significantly increasing. Cobalt production in 2021 was 170 kt (1.5 times higher than 2017) and 80 kt for lithium (2 times higher than 2017). To meet the projected electric vehicle sales, a significant scaling up is needed for lithium and cobalt supply. Approximately 30 new lithium mines and 11 cobalt mines are needed by 2030 (assuming 8 kt lithium and 7 kt cobalt annual mine production capacity). Although battery-makers have been trying to decrease the cobalt content in batteries, the demand for electric vehicle batteries will keep increasing the cobalt demand at least for this decade.[1]
With the estimated battery lifespan of 8 to 12 years, a significant amount of spent batteries is expected in the near future.[2] It is forecast that by 2030, there will be nearly 1 million tons of battery scrap.[3] Based on the anticipated volume of spent batteries, it is essential to recover their valuable metal content. Thus, recycling spent Li-ion batteries is a good alternative to secure the raw material supply for the projected battery demand and to deal with the accumulated spent batteries.
Generally, the methods to recycle batteries are classified into two main routes: pyrometallurgical and hydrometallurgical. Pyrometallurgical route aims to process the spent batteries using thermal treatment. Hydrometallurgy encompasses pre-treatment to recover the cathode materials followed by leaching and subsequent purification and recovery techniques such as selective precipitation, ion exchange, and solvent extraction. Comparatively, hydrometallurgy routes typically require lower energy consumption and lower gas emission compared to pyrometallurgical routes.[4] However, there are some limitations on using the hydrometallurgical route, such as the complexity of its operation, generation of secondary pollutions and waste waters from the leaching process, long leaching time and low efficiency.[5] Alternatively, pyrometallurgical routes are capable of recovering the desired metals through physical and chemical transformations.[6]
Many metals can be produced by hydrogen reduction. Some commercial application of H2 as reducing agent has been applied on the production of platinum group metals, some rare metals like germanium and rhenium, and high-grade nickel and cobalt powder.[7,8] Reduction by H2 has been shown to have advantages compared to carbothermic reduction, such as mechanism of gas–solid reaction which can produce a very pure metal mostly in a form of powder. Moreover, only water vapor is present as the reaction by-product, and if the production of H2 is done by water electrolysis, no carbon dioxide will be produced during both hydrogen production and reduction in future.
Experiments on cobalt oxides reduction using H2 have been reported by many researchers.[9,10,11,12,13] An investigation of reduction by H2 was carried out on mixed powder of Co3O4 and CoO, heated isothermally in an electric furnace at 327 °C to 527 °C and H2 partial pressure of 0.25 to 0.6 atm (with N2 as the balance of 1 atm total pressure).[10] The degree of reaction was determined by H2 consumption measured from the change on heat capacities of the gas. The result showed that the reaction rate was directly proportional to H2 partial pressure. This linearity led to a conclusion that the reduction of Co3O4 and CoO was first-order reaction proportional to H2 concentration. The first stage of reduction was Co3O4 to CoO followed by reduction of CoO to Co with activation energy of 33.6 and 34.6 kJ/mol, respectively.
Lin and Chen[13] proposed a mechanism of cobalt oxide reduction by H2 using temperature-programmed reduction with different heating rates. The consumption of H2 was measured by the change of H2 concentration through the exit gas. The study also concluded that the reduction occurred in two stages: Co3O4 reduction to CoO (Eq. [1]) followed by CoO reduction to Co (Eq. [2]) with the activation energy of 94.43 and 82.97 kJ/mol, respectively. The mechanism of reduction was reported to fit with two-dimensional nucleation model (Avrami–Erofeyev) for Co3O4 reduction and the unimolecular model for CoO reduction.
Bhandari and Dhawan[14] investigated H2 reduction of LiCoO2 at 400 °C to 900 °C for up to 120 minutes under pure H2 and low concentration of 10 pct H2, 90 pct Ar atmosphere. The thermodynamic assessment predicted that CoO can be obtained from 144 °C under H2 reduction. The pure H2 concentration caused a higher conversion to metallic Co compared to 10 pct H2 atmosphere. Additionally, the decomposition of LiCoO2 happened at slightly lower temperature (400 °C, as opposed to 500 °C). The effect of temperature was insignificant under pure H2 atmosphere, especially above 500 °C after full decomposition of LiCoO2. Temperature affected only the conversion of CoO to Co up to 600 °C before the full conversion to metallic Co was achieved. On the other hand, under 10 pct H2 atmosphere, the CoO conversion to metallic Co happened more gradual and would only achieve full conversion at 900 °C.
Pinegar[15] carried out a study of H2 reduction of LiCoO2 at limited temperature range of 500 °C to 600 °C. It was found that the reduction to metallic cobalt occurred after 60 minutes at 500 °C, and considerably faster at 600 °C, after 15 minutes. Cobalt was also recovered as CoO but existed fully as metallic Co after 120 minutes at 600 °C. Meanwhile, the lithium was recovered as LiOH, then converted to Li2O with longer reduction period of 90 minutes, and then achieved the full Li2O conversion after 120 min. They reported an activation energy of 82.8 to 92.6 kJ/mol and found that it varied over the extent of reaction (α) above 0.4 which indicated a multistep reduction process. However, the more detailed full mechanism was not investigated in those studies. Other studies of H2 reduction on different types of cathode materials are presented in Table I. This paper presents a systematic study of H2 reduction of LiCoO2 over a wider temperature range of 600 °C to 1000 °C through thermodynamic modeling and experimental works. Thermodynamic modeling was carried out using the thermochemical software FactSage™ (embedded with thermodynamic data of LiCoO2) to investigate the possible products of the reduction. An experimental study coupled with detailed characterization was conducted to verify the thermodynamic predictions and investigate the mechanisms and the microstructure evolution of the reduction.
Materials and Methodology
This study employed LiCoO2 with purity of 99.8 pct and particle size between 5 and 20 µm, supplied by Sigma Aldrich®. A pure material was used to investigate the fundamental phenomena occurring during reduction. However, it is expected that the results can be extended and applied for reduction of LiCoO2 cathode material. The experimental samples were prepared by pelletizing the powder through cold press in steel dies with load of 10 tons for 5 minutes, applied at a room temperature. The typical weight of each pellet was 3 g with a dimension of 8 mm in height and 13 mm in diameter.
Reduction Experiments
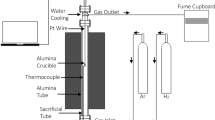
A vertical tube furnace (Nabertherm LT15/13/P330) connected with an analytical balance (FX-300i with 1 mg accuracy) was used. A sacrificial alumina tube of 47 mm diameter was used inside the main alumina tube. The pellet was placed inside an alumina crucible (18 mm diameter and 20 mm height), suspended, and connected to the balance on the top using a platinum wire, as shown in Figure 1. The temperature profile of the furnace and the hot zone were measured using K and R-type calibration thermocouples. Mass change during experiments was recorded by a balance via data acquisition program (DT80). A detailed study of the kinetics from the mass change data will be covered in a separate publication.

Schematic of the experimental setup used in the study
The experiments were carried out by first heating the furnace to the desired temperature. The sample was placed in the crucible and positioned inside the top water-cooled flange. The furnace was then sealed and purged with 0.5 L/min argon gas with a purity of 99.999 pct. After the inert atmosphere was stabilized, the sample was lowered to the hot zone using a lever mechanism. H2 at 0.3 L/min (15 pct H2/85 pct N2) was flowed to the furnace tube, and the sample was heated isothermally for 30 to 180 minutes. The low percentage of 15 pct H2 was used due to the safe laboratory procedure and to minimize the flammability of the gas mixture. At the end of the experiment, the sample was quenched by raising it to the upper water-cooled zone of the furnace. Once cooled, the H2 flow was stopped, and Ar was purged for 5 minutes. The top flange was opened, and the sample was recovered for characterization.
Characterization Methods
For X-ray Powder Diffraction (XRD) analysis, the samples were crushed in a mortar and pestle to form homogeneous powder. The analyses were carried out using a Bruker XRD (Bruker AXS GmbH) with Cu Kα radiation operated at 40 kV and 40 mA with 2θ angle from 15 to 140 deg at a scanning step and dwell time of 0.02 deg and 1 seconds, respectively. The XRD peaks of the phases present were identified by indexing and matching the diffraction patterns to known spectra from the International Centre for Diffraction Data Powder Diffraction File.
Scanning Electron Microscope (SEM) analyses were conducted using a ZEISS SUPRA model 40VP-25-38. The samples were prepared by either dispersing unconsolidated grains onto an aluminum stub or embedding in epoxy resin. The samples were coated using gold sputtering (Emitech K975X Turbo Evaporator). The SEM was operated using 276 mA emission current at a pressure of 2.8 × 10−6 Torr, accelerating voltage between 10 and 20 kV, and a working distance between 10 and 18 mm. In conjunction with the SEM imaging, Energy-Dispersive X-ray spectroscopy (EDX) analyses were conducted to obtain the elemental distribution maps and semi-quantitative elemental composition results.
High-Temperature XRD experiments were performed to investigate the decomposition of LiCoO2 upon heating under inert atmosphere. An INEL XRG3000 diffractometer was used, fitted with a Co tube operated at 40 kV and 35 mA. The diffraction patterns were collected using a CPS120 position sensitive detector, which collected 120 deg 2θ of diffraction data simultaneously. Datasets were collected in 60 seconds increments during heating. The samples were held in an Anton Paar HTK16N high-temperature chamber, which used a Pt strip resistance heater. The temperature and heating rate were controlled with a TCU 2000N temperature control unit. A slurry of the sample and acetone was prepared and placed within the sample well (20 × 7 × 0.2 mm) on the Pt strip heater. Flowing He was passed through the Anton Paar chamber during the measurements. Samples were heated at 10 °C min−1 from 25 °C up to the desired setpoint (900 °C or 1000 °C) and held for a period (up to 300 min).
Thermodynamic Analysis Method
The thermodynamic study was carried out using thermochemical software FactSage™ 8.2 (Thermfact/CRCT-Ecole Polytechnique de Montreal, QC, Canada). Two main databases of FactPS (database for pure substances) and FToxid (database for oxides) were applied for the thermodynamic calculations. Table II shows the phases considered in the calculations along with the databases and references. Thermodynamic data for LiCoO2 were not available in the built-in databases, thus a customized database was generated referring to available data in the literature. Table III shows the details of the selected values used.
The thermodynamic calculations covered an assessment of Gibbs free energy and equilibrium state calculations for relevant reactions, including the effect of H2 amount on the reduction of LiCoO2. The equilibrium calculations simulated the possible equilibrium products in a closed system corresponding to the main constraints of (1) the mass balance of the system components, and (2) the minimization of Gibbs energy for the selected products.
Results and Discussions
Thermodynamic Assessment
Gibbs free energy of key reactions
Figure 2 shows the standard Gibbs free energies of reactions (∆rG°) as a function of temperature by the relevant reactions associated with thermal decomposition and H2 reduction of LiCoO2, i.e., the reactions presented in Eqs. [3] through [6]. The Gibbs standard free energy for the thermal decomposition of LiCoO2 is shown by square data points and line at the top of the diagram. The products of LiCoO2 decomposition considered were Li2O, O2, and CoO, and were predicted to form at temperatures above 907 °C (Eq. [3]). In the presence of H2, reduction of LiCoO2 is thermodynamically possible through reactions (4) and (5) since the Gibbs free energy of these reactions is negative in the temperature range of interest. The end-products considered from these reactions were Co, Li2O, LiOH, and various gaseous components of H2O and the remaining H2. In the case where the intermediate product CoO, Eq. [6] (shown by inverse-triangle line) forms, H2 would reduce the CoO to Co. Overall, the stability of LiCoO2 decreases with increasing temperature, and reaction with H2 significantly decreases the ∆rG° of LiCoO2 reduction reactions.

ΔrG° vs temperature of LiCoO2 reduction by H2
Equilibrium calculations
Figure 3 presents the predicted equilibrium amount of the reduction of LiCoO2 by H2 with molar ratio of LiCoO2/H2 equal to 1 at temperatures 400 °C to 1200 °C. It shows that H2 has strong reducing influence on LiCoO2. Between 400 °C and 1200 °C, metallic Co was predicted to be stable. Along with metallic Co, CoO was formed as a decomposition product. As for lithium, LiOH was stable up to 1035 °C, after which it gradually transformed into Li2O. The intermediate product CoO existed with lower amounts of H2. It is also observed from Figure 3 that above 1000 °C, there is an increase of CoO while metallic Co decreases. This happens during the conversion of LiOH to Li2O which boosts the formation of water vapor. Due to the release of water vapor, the H2/H2O ratio will significantly decrease thus creating a pH2/H2O that is lower than the equilibrium value (relatively less hydrogen to water) which may cause Co to be oxidized to CoO.

Predicted equilibrium amount of phases after hydrothermic reduction of 1 mol LiCoO2 by1 mol of H2 at 400 °C to 1200 °C
Effect of different amount of H2
Another set of calculations investigating the effect of different amounts of H2 on the reduction of LiCoO2 was carried out. For optimizing the yield of metallic Co as the final product (rather than cobalt oxides), it is important to identify the minimum molar ratio of H2 to LiCoO2 that can produce 100 pct conversion to metallic Co.
Figure 4(a) through (d) presents the predicted cobalt phases in equilibrium at different molar amounts of H2 (0 to 1.6 mol) at temperatures 400 °C to 1200 °C, and 1 atm. At low H2 (below 0.5 mol), CoO and Co3O4 are predicted to form. With no H2, Co3O4 is predicted to form at 810 °C before it converts to CoO at 950 °C (Figures 4(a) and (c)). Reaction with 0.1 mol H2 does not affect the results significantly, only lowering the decomposition temperature by 20 °C. The reducing effect of H2 is prominent above 0.2 mol, which is seen by the conversion to Co3O4 and CoO at the much lower temperature of 400 °C. At 0.5 mol H2, CoO is predicted as the only cobalt phase at all temperatures (Figure 4(a)). Starting from 0.6 mol H2, metallic Co was predicted to form, and the Co conversion percentage was increasing with increasing amount of H2. Simultaneously, the amount of CoO decreases (Figure 4(b)) until reaching full conversion to metallic Co at 1.6 mol of H2 (Figure 4(d)). More H2 over 1.6 mol was not predicted to affect the cobalt conversion.

The predicted cobalt phases from LiCoO2 hydrogen reduction under 0 to 1.5 mol H2 per 1 mol LiCoO2: (a) CoO(s) with 0 to 0.5 mol H2, (b) CoO(s) with 0.6 to 1.5 mol H2, (c) Co3O4(s), (d) Co(s)
Figure 5(a) through (c) presents the predicted lithium phases at equilibrium from reaction with H2 amounts from 0 to 0.5 mol (the trend of 0.5 to 1.5 mol H2 is identical). Unlike for cobalt products, the lithium phases are more dependent on the temperature rather than H2 amount. The effect of H2 is significant at amounts below 0.5 mol. In this range, LiOH is predicted to form rather than Li2O along with the increase of H2. The more H2 in the system, the higher is the conversion temperature from LiOH to Li2O. The amount of H2 between 0.5 and 1.5 mol is predicted to not affect the formation of lithium products. The lines for the H2 above 0.5 mol are identical; thus, they are not shown in Figure 5(c). In this range, the temperature was predicted to solely control the variation of lithium products where solid LiOH was predicted to present up to 470 °C before transforming to its liquid phase. The LiOH was predicted to be stable up to 1035 °C (also refer to Figure 3) before transforming to Li2O. Reactions with 1.6 to 2 mol H2 (Figures 6(a) and (b)) are predicted to not significantly affect the lithium products, only decreasing the conversion temperature of LiOH to Li2O as the amount of H2 is increased.

The lithium phases products from LiCoO2 hydrothermic reduction under 0 to 0.5 mol H2 per 1 mol of LiCoO2: (a) LiOH(s), (b) LiOH(l), (c) Li2O(s), the trend of 0.5 to 1.5 mol H2 is identical

The lithium phases products from LiCoO2 hydrothermic reduction under 1.6 to 2 mol H2 per 1 mol of LiCoO2 (a) LiOH(l), (b) Li2O(s)
The amount of LiCoO2 remaining in the system unreacted is presented in Figure 7(a). LiCoO2 is predicted to be fully consumed in reaction with 0.1 mol H2. The generated by-products are water vapor and/or O2. Oxygen is generated only in reactions with low amount of H2 (below 0.5 mol, Figure 7(b)), while higher H2 amounts favor the generation of water vapor (Figures 7(c), (d)). The generation of O2 under limited H2 is due to O2 release during LiCoO2 decomposition where CoO is the main cobalt product at H2 < 0.5 mol. Above 0.5 mol H2, O2 generation is limited to the small amount of O2 released from CoO to Co and would be consumed to produce water vapor. Water vapor formation is predicted to be directly proportional to H2 amount in reaction up to 1.5 mol. Above 1.5 mol H2 results in unutilized H2 after full conversion to Co (Figure 4(d)).

(a) Remaining LiCoO2. Gas generated from LiCoO2 hydrogen reduction under 0 to 2 mol H2 per 1 mol of LiCoO2 (b) O2(g) (c), (d) H2O(g)
Experimental Results
To test the thermodynamic assessments, as well as to understand the reduction behavior and mechanism, high-temperature experiments were carried out. LiCoO2 powder was characterized by Hot-stage XRD to investigate its evolution upon heating under inert atmosphere. Furthermore, pellets of LiCoO2 were reduced isothermally at temperatures 600 °C to 1000 °C for 30 to 180 minutes. Based on the pellet appearance and phases formed, the reduction mechanisms were classified into three categories: Category 1 (at 600 °C to 700 °C), Category 2 (at 800 °C to 900 °C), and Category 3 (at 950 °C to 1000 °C).
High-temperature XRD experiments
Figure 8 shows the results of phase changes during LiCoO2 heating for 230 minutes at 900 °C. Results showed the decomposition of the LiCoO2 to CoO (and Li2O) at just under 900 °C (see dashed line in Figure 8 which indicates the temperature at which additional CoO begins to be observed). However, LiCoO2 remained the dominant phase throughout the experiment at 900 °C. At 1000 °C (Figure 9), the same decomposition products were observed which confirm that the LiCoO2 is unstable. It was also observed that LiCoO2 began to melt over time and the amount of LiCoO2 decreased with a concomitant increase in the amount of CoO and Li2O.

Results of the hot-stage XRD experiment LiCoO2 under inert atmosphere, showing accumulated XRD data in the range 25 °C to 900 °C with a holding time of 230 min at the peak temperature

Results of the hot-stage XRD experiment LiCoO2 under inert atmosphere, showing accumulated XRD data in the range 25 °C to 1000 °C with a holding time of 300 min at the peak temperature
Category 1 (600 °C to 700 °C)
The appearances of the reduced pellets were observed using an optical microscope and the results are presented in Tables IV and V. The results of the XRD are shown in Figure 10(a) and (b), summarized in Table VI. From the low-magnification observations, several distinctive phases were present in the reduced pellets. On the top surface of the pellet, white phase (determined as Li2O by XRD spectrum, Figure 10(c)) formed progressively over time. On the outer circumference, some horizontal cracks were observed to form followed by minor formation of Li2O (white phase). The bottom surface of the pellet was primarily metallic Co.

XRD analysis of the reduced samples at (a) 600 °C, (b) 700 °C after 30 to 180 min (c) white phase
The transformation began with the conversion of LiCoO2 to non-stoichiometric LiCoO2 after 30 minutes. Along with this conversion, a small amount of Li2O was formed. Afterwards, Co and CoO were formed during 60 to 90 minutes and Li2O formation increased. At later stage up to 180 minutes, LiOH formed alongside Li2O while Co and CoO were still present with more dominance of Co compared to CoO. At 600 °C, the Co had peaks corresponding to both cubic and hexagonal form as soon as Co emerged after 60 minutes. At 700 °C, Co peaks corresponded to only the cubic phase up to 90 minutes then the Co transformed to hexagonal. Co3O4 formed later, after 150 and 180 minutes, at 700 °C and 600 °C, respectively.
At 30 minutes, the conversion from LiCoO2 to non-stoichiometric LiCoO2 can be explained by its thermal decomposition. The conversion started with O2 release alongside Li2O separation from LiCoO2 (Eqs. [7], [7a] and [7b]). The formation of Li2O was mainly on the top surface of the pellet where this was favorable for O2 release. Some cracks were observed on the outer circumference of pellet which was possibly due to the heating-induced stress relief from pelletizing process.
Table VII shows the change amounts of Li and O amounts during the transformation of LiCoO2 to non-stochiometric Li(1-i)Co(1-j)O. During the changes from LiCoO2 to Li(1-i)Co(1-j)O, there is a release of Li and O (denoted by the negative sign). From the O/Li mass ratio, the released elements have more O (1.6 O/Li ratio) compared to Li2O (1.2 O/Li). In other words, during this transformation, O2 was released along with Li2O. After 60 minutes, the cracks became more apparent on the side of the pellet as well as at the bottom surface. Li2O was still observed along the cracks as well as at the bottom. Grayish phase was observed predominantly across the pellet, identified as CoO, explained by overall reaction (8) from subsequence reactions (8a) and (8b). Co micro-grain formation was not obvious by macroscopic observation, but was clarified by microstructure observation, as explained in the see next sections.
A significant increase of Co was observed after 90 minutes, especially in the mid cross section where the major cracks were located. Heating beyond 90 minutes seemed to cause a thickening of the Co mass while the CoO amount was decreased by the reduction (reaction (9)). The cracks at the bottom surface also grew over time which would be beneficial for further reduction from CoO to Co. New phases of Co3O4 and LiOH were found after 120 minutes. The possible reactions for the generation of these phases are described in reactions (10) and (11).
The thermodynamic calculations on LiCoO2 decomposition show that the phases of cobalt oxide can vary between CoO and Co3O4, depending on pO2, as shown in Figure 11. At the same temperature, less reducing atmosphere (higher pO2) will result in the formation of Co3O4 instead of CoO. This might explain the late appearance of Co3O4 (only after 150 to 180 minutes). With the formation of H2O, the local effective pO2 could be higher, thus, favoring Co3O4 formation on the middle-upper part of the pellet.

Phase stability diagram of LiCoO2/O2 at different temperatures and oxygen partial pressures, at 1 atm total pressure
To analyze the microstructure evolution of the phases over temperature and time, high-magnification SEM secondary electron characterization was conducted. As mentioned previously, in the early stage of reduction at 600 °C, LiCoO2 started to decompose into Li2O and CoO. A volumetric change in early stages of the reduction would likely to cause a compressive stress in the produced Li2O. The appearance of Li2O is shown in Figure 12 after 15 minutes reduction at 600 °C. The plate-like phases formed and grew along the crevices of the initial LiCoO2. The plate-like Li2O grew at weaker spots on the LiCoO2 broken surface due to the decomposition. These spots became localized stress relief locations. There were stress gradients arose due to the different compressive and tensile stresses between the plate-like root and the surrounding microstructure. Similar phenomenon was observed in tin whiskers formation, reported by Hektor et al.[24] that stress gradients were responsible for the growth of tin whiskers. This difference in stress caused a diffusion of nucleating atoms towards the whisker root, ultimately resulting in the growth of the whisker. The evolution of plate-like Li2O over time is presented in the electronic supplementary material (refer to the Figures S-1 to S-2).

The decomposition of LiCoO2 leads to the segregation of Li-rich thin plane phase which demonstrates the early stage of plate-like Li2O formation, photograph taken from reduction at 600 °C after 15 min: (a) the segregation happens non-uniformly while the remaining LiCoO2 grains are still coalescing, (b) the massive segregation causes defect on the former LiCoO2 surface
In the early reduction stages at 600 °C, the concentration of oxygen atoms in the CoO phase decreased and fine nuclei of Co formed. The cobalt nuclei underwent shape transformation when the reduction time was prolonged (Figure 13) but still had the same cubic structure. At 60 minutes, the grain showed a few diagonal cracks through the symmetry center of the cubic (Figure 13(a)). After 120 minutes, the cracks were reduced and shallower and the surface had a smoother appearance (Figure 13(b)). At 180 minutes, the cracks were pushed towards the edges of the cubes and small holes appeared in these locations (Figure 13(c)). There was an increase in the grain size between 60 and 120 minutes during which the size was approximately doubled. At 180 minutes, there appeared to be grain recrystallization where new grains formed with size similar to the grain size at 60 min.

The evolution of cobalt-rich grain at 600 °C (a) 60 min: cubical shape with diagonal crack; (b) 120 min: smoother surface with less crack appearance; (c) 180 min: the expulsion of crack toward the edges of the cubical shape, showed by the appearance of small holes; (d) FCC Co-based on the Wulff construction, adapted from Ref. [25]
Cobalt exhibits more than one allotrope: HCP under 422 °C and FCC above 422 °C until the melting point at 1495 °C.[26,27,28] Schuett et al.[28] investigated the growth of cobalt single crystals using the flame fusion method under controlled hydrogen–oxygen atmosphere. Cobalt crystallization were observed in the FCC structure temperature range where crystals with triangle shape were observed when the cobalt was annealed just below its melting point.[28] The FCC-structured cobalt adopts an octahedron-like shape with high symmetry. It has four types of facets which are (111) for 8 planes, (100) for 6 planes, (311) for 8 planes, and (110) for 8 planes, shown in Figure 13(d).[25] As identified by XRD, both hexagonal and cubic structures for Co were found in this study, but mainly cubic phase is shown in Figure 13.
The growth of Co was more profound when the temperature was increased to 700 °C and resulted in an extensive dense metallic mass. The mass was observed in the middle cross section of pellet where cracks were predominantly located, as shown in Figures 14(a) and (b). This is consistent with macroscopic observations in Tables IV and V, and metallic cobalt was more predominantly seen in the middle to bottom part of the pellet rather than in the middle upward part. The dense cobalt mass was surrounded by smaller cobalt grains, as shown in Figure 14(c). It appeared that this dense mass was a result of sintering and densification of the smaller cobalt grains.

Middle cross section of pellet reduced at 700 °C after 150 min (a) upper part, (b) lower part, (c) SEM secondary electron image of upper part
Considering the XRD results, the macro-observation of the pellets and the SEM analyses, the generic macro-mechanism of the reduction in the Category 1 is summarized in Figure 15. The phenomenon of the variation of metallic cobalt formation across the pellet is explained by Figure 15 (at reduction time 60 to 90 minutes) where extensive cracks assist the H2 flow into the pellet and form high amounts of H2O(g). When the H2O vapor moves upwards, the middle to top area would be in the condition of lacking H2 and creating a low pO2 atmosphere; hence, limited reduction was observed in the upper half of the pellet.

Schematic of macroscopic evolution during reduction at 600 °C to 700 °C after 0 to 180 min
Category 2 (800 °C to 900 °C)
The XRD analyses results of the reduced pellets at 800 and 900 °C are shown in Figure 16 and summarized in Table VIII. The phases recovered from the reductions in this temperature range were identical for all heating times. The heating period influenced the degree of conversion of CoO to Co, as well as the amount of Li2O. Compared to Category 1, and significant metallic Co was formed earlier, after 30 minutes. However, unreactive LiCoO2 remained even after 180 minutes, and there was no transformation to the non-stoichiometric form.

XRD analysis of the reduced samples at (a) 800 °C, (b) 900 °C after 30 to 180 min
From the macroscopic observation, as shown in Tables IX and X, formation of Co was rapid, resulting in a thick mass of Co, mainly at the bottom and side of the pellet. The intensity of cracks was greater than at lower temperatures which led to earlier Co formation due to easy access of H2 to the interior. The only lithium phase observed was Li2O which was present uniformly on all the surfaces, but mainly on the top surface where no cracks were found. In general, the cracks on the side of the pellet eased all the reactions because they provided passage for H2 to the interior and H2O release (Eqs. [12] and [13]).
The LiCoO2 decomposition on the top surface of the pellet resulted in the formation and growth of Li2O across the top (Eq. [12]). Once the surface was covered by Li2O, further CoO reduction to Co was hindered as H2 could not easily interact with CoO as it had to pass through the Li2O layer. The direct reduction reaction could result in Co formation without intermediate CoO (Eq. [14]) on the side and the bottom of the pellet. Indeed, the majority of Co was found on the side and bottom surfaces where the formation was supported by the significant cracks that allow the transport of H2 and water vapour.
The microscopic observations of the phases formed on the top surface of the pellet at 800 °C are shown in Figure 17. The overall phases, as captured at lower magnification (Figure 17(a)), show a clear segregation of agglomerated Li2O phase (white-colored phase) on the top surface of the pellet. Figure 17(b) with a higher magnification of the top surface shows a similar structure of plate-like Li2O as found at lower temperatures. The base surface of the pellet contained CoO and the remaining LiCoO2. A higher magnification of the base surface is shown in Figure 17(c) and presents a similar appearance of CoO as at lower temperatures. However, on some areas, the segregation between Li2O and CoO was not well distinguished, representing the early period of LiCoO2 decomposition, as shown in Figure 17(d). More images on the structure of plate-like Li2O on the top surface at 900 °C are shown in the electronic supplementary material: Figure S-3.

(a) The formation of white agglomerates floating on the pellet surface, captured at 800 °C after 180 min, (b) plate-like Li2O forming agglomerate, (c) the morphology of the base surface, (d) the distinction between plate-like Li2O and CoO
A dense metallic Co mass was found in the middle cross section of a pellet at 180 minutes as presented in Figures 18(a) and (b). Figure 18(c) shows an area that was not fully covered by dense Co which appeared to have micro-grains of Co. This might indicate that initially there were Co micro-grains that underwent coalescence and densification as part of sintering process. At the earlier time of 120 minutes, shown in Figures 19(a) and (b) for 800 °C and 900 °C, respectively, the Co mass still showed the outline of Co grains that constructed the mass. To understand the development of Co mass vertically across the pellet, the corresponding cross-sectional area is shown in the electronic supplementary material in Figure S-4. From the SEM back-scattered image, a full coverage of the Co mass was found on the bottom surface where cracks were evident in the area (Tables VIII and IX). On the upper part where the denser structure was, the development of the Co mass was not as progressive compared to the continuous mass in the bottom.

Middle cross section of a pellet reduced at 800 °C after 180 min (a) macroscopic observation, (left) the upper half of the pellet; (right) the bottom part of the pellet, (b) SEM secondary electron image of upper part, (c) higher magnification of b

The appearance of the development of Co mass, captured at middle part of pellet (a) 800 °C, (b) 900 °C after 120 min
The overall macro-mechanism of the pellet reduction in this temperature category is summarized in Figure 20.

Schematic of macroscopic evolution during reduction at 800 °C to 900 °C during heating times between 0 and 180 min
Category 3 (950 °C to 1000 °C)
The XRD results of the reduced pellets at 950 °C and 1000 °C are shown in Figures 21(a) and (b) and also summarized in Table XI. A unique characteristic of this temperature range was the formation of Li6CoO4 and generation of a molten phase in all heating periods. XRD spectra for the molten phase are shown in Figure 21(c), which was taken from samples heated for 60 minutes at 950 °C and 1000 °C. The molten phase was non-homogenous, containing the compounds Li6CoO4, LiOH.H2O, and CoO. Nevertheless, the CoO spectra appeared to have come from the CoO underneath/nearby due to the highly mixed nature of these phases. Between Li6CoO4 and LiOH.H2O, the molten structure was likely to be LiOH.H2O considering its melting point of 462 °C and its decomposition temperature of 924 °C.[29] Thermodynamically, LiOH.H2O is supposed to decompose in this temperature range, but slower kinetic is suspected to cause a delay in the decomposition.

XRD analysis of the reduced samples at (a) 950 °C, (b) 1000 °C after 30 to 180 min, (c) molten phase
From macroscopic observations, shown in Tables XII and XIII, a significant shrinkage and densification of the pellet happened as early as 30 minutes at both 950 °C and 1000 °C. The evidence of molten phase was found all over the outer surface, resulting in the bottom part of the pellet following the shape of the crucible. Some parts of the pellet not covered by the molten phase showed a presence of metallic Co. This phenomenon specifically happened at 950 °C, while most of the pellets at 1000 °C were covered by the molten phase. Additionally, there was a consistent appearance of a cavity at the bottom of the pellet in all heating periods. The inside wall of the cavity was smooth indicating that a gas phase might have caused this structure. The trapped gas was most likely H2O vapor resulting from the reaction between H2 and O2.
Large amounts of remaining LiCoO2 and non-stoichiometric LiCoO2 were still observed, even after 180 minutes. Although Co was recovered, the progress was not as fast as those reduced at lower temperatures. The molten phase covering the surface might have limited LiCoO2 exposure to H2 which would explain the non-progressive reaction on the remaining LiCoO2. The trapped H2O (causing the cavity) inside the pellet would also contribute to the less reducing condition.
Unlike LiOH from the reduction at 600 °C to 700 °C, the hydroxide formed at 950 °C to 1000°C was already generated as early as 30 minutes. From the thermodynamic assessment (Figure 2, Eq. [17]), direct formation of LiOH from LiCoO2 decomposition is favorable and has more negative ∆rG° with increase of temperature. The main reactions ruling the mechanism in this temperature range derived from Eq. [17] are
The top surface of a pellet reduced at 950 °C for 30 minutes is shown in Figure 22(a) which shows solidified molten phase (shiny light gray color) and the solid phase appeared in dark gray. Another surface of the side of the pellet showed similar characteristic at 120 minutes (Figure 22(b)). At higher magnification, the presence of plate-like Li2O can be seen (Figure 22(c)). The observation of the pellet reduced at 1000 °C is included in the electronic supplementary material; Figures S-5 to S-6. The schematic macro-mechanism of the reduction in this temperature category is presented in Figure 23.

(a) Secondary electron image of pellet’s top surface, taken at 950 °C reduction after 30 min, showing the segregation of molten and solid phase, (b) molten phase appearance at outer circumference at 950 °C reduction after 120 min, (c) higher magnification of -b, showing the appearance of plate-like Li2O

Schematic of macroscopic evolution during reduction at 950 °C to 1000 °C, over heating time between 0 and 180 min
Conclusions
A systematic study of the reduction of LiCoO2 cathode material using hydrogen at 600 °C to 1000 °C has been carried out. The following conclusions were drawn from the study:
-
1.
Based on the thermodynamic assessments, it was predicted that the effect of H2 is quite strong in terms of reduction of LiCoO2. Metallic Co is generated at 400 °C and it stayed stable until 1200 °C. Along with metallic Co, CoO was formed as the decomposition product. As for lithium, LiOH was stable up to 1035 °C, then gradually transformed to Li2O.
-
2.
By assessing the effect of H2 amount on the reduction, it was predicted that the cobalt products were strongly dependent on the molar amount of H2. On the other hand, the formation of lithium phases was more dependent on the temperature rather than H2 molar amount. The effect of H2 was significant only when the amount was below 0.5 mol. The amount of H2 above 1.5 mol was predicted to increase unutilized H2.
-
3.
Experimental results showed that the mechanism of H2 reduction of LiCoO2 could be classified into three categories, namely Category 1 (600 °C to 700 °C), Category 2 (800 °C to 900 °C), and Category 3 (950 °C to 1000 °C). Cobalt was generally recovered as metallic Co and CoO in all temperature categories, while re-oxidized Co3O4 was found at 600 °C to 700 °C after prolonged reduction time. The metallic Co crystal growth was found to be most progressive at 800 °C to 900 °C. The molten phase generated at 950 °C to 1000 °C provided a barrier on the surface of the sample which seemed to significantly hinder the reduction of CoO to metallic Co.
-
4.
Lithium was generally present as Li2O, with the exemption of LiOH generation at 600 °C to 700 °C, and LiOH·H2O at 950 °C to 1000 °C. The reduction at 600 °C to 700 °C showed an intermediate transformation of LiCoO2 to its non-stoichiometric form before the formation of Li2O. Most of the Li2O was found on the top surface of pellets where the initial release of O2 was most favorable.
-
5.
The thermodynamic study combined with experimental work revealed possible mechanisms of H2 reduction of LiCoO2 in the different temperature categories. The results of this investigation can provide guidance on the development of processes for recycling of LiCoO2. It was found that temperature range 800 °C to 900 °C offered more benefit as Co could be generated as a larger mass with more progressive reduction. The mechanism at 600 °C to 700 °C showed more gradual evolution through non-stoichiometric LiCoO2 and transformed to CoO before finally being reduced to Co. The highest temperature (950 °C to 1000 °C) had the disadvantage of molten phase generation which hindered further reduction of CoO to metallic Co.
This study has provided the suitable conditions for the formation of reduced products of Co, Li2O, and LiOH. Further processing can be carried out to separate these phases. There are numbers of ways to separate the products, i.e., cobalt separation by magnetic force, and Li-containing phase separation by water leaching and acid leaching.[14,17] It is reported that the recovery rate after separation by magnetic force and water leaching could achieve 96.8wt pct Li and 99.4wt pct Co.[17]
References
IEA: Global EV Outlook 2022, IEA, Paris, 2022.
M. Chen, X. Ma, B. Chen, R. Arsenault, P. Karlson, N. Simon, and Y. Wang: Joule, 2019, vol. 3, pp. 2622–46.
K. Keen: Battery scrap projection for recycling plummets—Circular Energy Storage exec. (S&P Global Market Intelligence, 2022), https://www.spglobal.com/marketintelligence/en/news-insights/latest-news-headlines/battery-scrap-projection-for-recycling-plummets-8211-circular-energy-storage-exec-71007327. Accessed 30 November 2022
S. Zhao, W. He, and G. Li: Recycling Technology and Principle of Spent Lithium-Ion Battery, Springer, Cham, 2019.
J. Wang and Z. Guo: Hydrometallurgically Recycling Spent Lithium-Ion Batteries, Springer, Cham, 2019.
W. Lv, Z. Wang, H. Cao, Y. Sun, Y. Zhang, and Z. Sun: ACS Sustain. Chem. Eng., 2018, vol. 6, pp. 1504–21.
S. Luidold and H. Antrekowitsch: JOM, 2007, vol. 59, pp. 20–26.
A. Rukini, M.A. Rhamdhani, G.A. Brooks, and A. Van den Bulck: J. Sustain. Metall., 2022, vol. 8, pp. 1–24.
O. Bulavchenko, S. Cherepanova, V. Malakhov, L. Dovlitova, A. Ishchenko, and S. Tsybulya: Kinet. Catal., 2009, vol. 50, pp. 192–98.
N.G. Gallegos and J.M.P. Lopez: Mater. Chem. Phys., 1988, vol. 19, pp. 431–46.
F. Glaser: J. Inorg. Chem., 1903, vol. 36, pp. 1–35.
A. Kuznetsov and N. Kulish: Otdelenie Tekhnichesikikh Nauk Metallutgiya I Toplivo, 1959, vol. 4, pp. 52–58.
H.Y. Lin and Y.W. Chen: Mater. Chem. Phys., 2004, vol. 85, pp. 171–75.
G.S. Bhandari and N. Dhawan: Energy Fuels, 2022, vol. 36, pp. 15188–98.
H. Pinegar, R. Marthi, P. Yang, and Y.R. Smith: ACS Sustain. Chem. Eng., 2021, vol. 9, pp. 7447–53.
G.S. Bhandari and N. Dhawan: J. Sustain. Metall., 2022, vol. 8, pp. 1704–18.
Z. Huang, F. Liu, B. Makuza, D. Yu, X. Guo, and Q. Tian: ACS Sustain. Chem. Eng., 2022, vol. 10, pp. 756–65.
Z. Huang, D. Yu, B. Makuza, Q. Tian, X. Guo, and K. Zhang: Front. Chem., 2022, vol. 10.
M.W. Chase: JANAF Thermochemical Tables, 3rd ed. American Chemical Society, Washington, 1986.
A.T. Dinsdale: Calphad, 1991, vol. 15, pp. 317–425.
I.-H. Jung, S. Decterov, A. Pelton, H. Kim, and Y.-B. Kang: Acta Mater., 2004, vol. 52, pp. 507–19.
Y.C. Zhang, H. Tagawa, S. Asakura, J. Mizusaki, and H. Narita: J. Electrochem. Soc., 1997, vol. 144, pp. 4345–50.
O. Jankovský, J. Kovařík, J. Leitner, K. Růžička, and D. Sedmidubský: Acta Mater., 2016, vol. 634, pp. 26–30.
J. Hektor, J.-S. Micha, S.A. Hall, S. Iyengar, and M. Ristinmaa: Acta Mater., 2019, vol. 168, pp. 210–21.
J.-X. Liu, H.-Y. Su, D. Sun, B.-Y. Zhang, and W.X. Li: J. Am. Chem. Soc., 2013, vol. 135(44), pp. 16284–7.
N.V. Kazantseva, N.N. Stepanova, and M.B. Rigmant: Superalloys: Analysis and Control of Failure Process, 1st ed. CRC Press, Boca Raton, 2018, p. 133.
M. Knapek, P. Minárik, P. Dobroň, J. Šmilauerová, M.M. Celis, E. Hug, and F. Chmelík: Materials, 2020, vol. 13, p. 5775.
F.M. Schuett, D. Esau, K.L. Varvaris, S. Gelman, J. Björk, J. Rosen, G. Jerkiewicz, and T. Jacob: Angew. Chem. Int. Ed., 2020, vol. 59, pp. 13246–52.
ILO and WHO, Lithium hydroxide monohydrate. (ILO, 2021), https://www.ilo.org/dyn/icsc/showcard.display?p_version=2&p_card_id=0914. Accessed 1 September 2022.
Acknowledgments
The authors thank and acknowledge the financial support from Swinburne University’s Automotive Engineering Graduate Program (AEGP) by the Australian Government through the Department of Industry, Science, and Resources, and the Commonwealth Scientific and Industrial Research Organisation (CSIRO). The authors would like to also thank Dr. Nathan Webster and Dr. Shu Huang from CSIRO for the assistance on High-Temperature XRD measurements and analysis.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nuraeni, B.A., Avarmaa, K., Prentice, L.H. et al. Hydrogen Reduction of LiCoO2 Cathode Material: Thermodynamic Analysis, Microstructure, and Mechanisms. Metall Mater Trans B 54, 2011–2036 (2023). https://doi.org/10.1007/s11663-023-02813-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-023-02813-5