
Abstract
Recycling of Li-ion battery cathode materials using carbon from the anode materials via carbothermic reduction would provide a reduction process option that could be carried out without introducing any external reactants. From this basis, this study investigated and examined the kinetics of carbothermic reduction of LiCoO2 at 700 °C to 1100 °C under inert atmosphere up to 240 minutes reaction time using an isothermal mass change analysis combined with detailed microstructure evolution observation. The overall reduction mechanism appeared to involve diffusion of oxygen in LiCoO2 during its thermal decomposition in the first stage, followed by the nucleation of cobalt in the second stage. The activation energy of the diffusion and nucleation stages were calculated to be 121 and 95 kJ/mol, respectively. The microstructure analyses showed a complex evolution of phases. At 700 °C to 900 °C, Li2CO3 and Co phases were observed as the product of the reductions; while at 1000 °C to 1100 °C, Li2O and Co phases were observed. The information and data obtained are useful when comparing different recycling methods and optimizing the carbothermic reduction parameters for recycling cathode materials from spent Li-ion batteries.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recycling of spent Li-ion batteries is inevitable to conserve raw materials and to avoid leaching and emission of harmful elements in scrap batteries from landfills into the environment. The main components of Li-ion batteries are typically cathode, anode, a current collector, electrolyte, separator, and other components for safety structure.[1] Amongst the elements in Li-ion batteries, cobalt, nickel, and lithium are currently the focus in battery recycling due to their scarcity, importance, and high economic value. These metals are mainly located on the cathode and anode parts of the battery. The anode and cathode are made of active materials held together by carbon black and pasted on a current collector. Lithium metal oxides (LiCoO2, LiMn2O4, LiNixMnyCo1-x-yO2, LiNiCoAlO2) or phosphates (LiFePO4) based materials are usually used for the cathode, while graphite or petroleum coke is commonly used as the active material for the anode.[2]
Currently, cobalt has the highest market price of all the metals in the typical Li-ion battery and provides the key economic driver for battery recycling. In addition, around 70 pct of the global cobalt mine production in 2020 was supplied by the Democratic Republic of the Congo, a politically vulnerable country with human right concerns related to mining.[3] Such a dominant share held by one unstable country possesses a high supply risk.[4] Lithium was added to the critical raw material list for the first time by the European Commission in 2020.[5] The global demand for lithium has been forecast to increase from 305,000 tons of lithium carbonate equivalent in 2020 to 486,000 tonnes in 2021.[6] Growing interest in offtake agreements is indicating that securing the metal supply is a critical issue, while the COVID-19 pandemic has caused significant disruption of mine and brine operations.[6] Recycling and recovering lithium from all possible Li-containing resources is critical for securing its availability in the future, and as Li-ion batteries contain 5 to 7 wt pct Li, that is much higher concentration than the lithium content in natural resources,[7] batteries must be seen as valuable Li-resources.
There are a number of metallurgical routes for recycling of spent Li-ion batteries.[8,9] The pyrometallurgical route of recycling has some benefits compared to other metallurgical routes and includes simplicity and high productivity. In the pyrometallurgical route, a high temperature is applied to break the chemical bonds of cathode materials so that the valuable metals such as cobalt and lithium can be recovered as metals or other usable products. Metal oxides in spent batteries can be reduced by carbon through a carbothermic reduction route. Considering that Li-ion batteries contains graphite for the anode material, this reductant can be used without a need of any additional external reactants.
Understanding the kinetics of the carbothermic reduction of metal oxides in spent Li-ion batteries is essential for mechanism and ensuring that the proposed recycling method can be optimised and operated on an industrial scale. In general, kinetic studies on the carbothermic reduction between cathode and anode materials aim to understand the underlying mechanism(s) of the reactions, identify order of the mechanism(s), observe the possibility of any intermediate processes involved, and identify the rate limiting step and key parameters.
Only a few studies have reported the kinetics of the carbothermic reduction of LiCoO2, covering results on the reduction mechanisms and the activation energy with different variables, including temperature, atmospheric gas, and the molar ratio of reactant (LiCoO2) and reductant. Kwon and Sohn[10] carried out an iso-conversional kinetic study of carbothermic reduction of LiCoO2 under inert (argon) atmosphere by measuring the mass change and analysing the gas composition. They added Co3O4 as the starting material along with the LiCoO2. The study proposed that during heating from 25 °C to 1500 °C, carbothermic reduction of LiCoO2 occurred in five stages. It was reported that Co3O4 reduction to CoO occurred in the temperature range of 500 °C to 610 °C. In the second stage, LiCoO2 started to reduce to form Li2CO3 and CoO at higher temperatures of 610 °C to 690 °C. In the third stage, Li2CO3 decomposed to Li2O and CO2 while LiCoO2 reduction was still progressing. In the fourth stage, at 882 °C to 1128 °C, CoO was reduced to metallic cobalt. At higher temperature (the final stage), an excessive amount of CO was produced as a consequence of the Boudouard reaction. It was concluded that the overall mechanism followed a uniform internal reduction model with a single activation energy of 165.8 kJ/mol. Another iso-conversion kinetic study was reported by Li et al.,[11] where they investigated carbothermic reduction of LiCoO2 employing a thermogravimetric technique and gas spectrometry analysis under inert (argon) atmosphere at 25 °C to 1500 °C. They reported that the reduction progressed in three stages: i.e. LiCoO2 decomposition to simpler oxides of Li2O and CoO, then Li2CO3 formation from Li2O and CO2(g) and in the third stage CoO reduction to metallic cobalt and CO(g). They reported activation energies of 389.61 and 405.67 kJ/mol from Kissinger–Akahira–Sunose and Flynn–Wall–Ozawa models, respectively.
Pindar and Dhawan[12] conducted carbothermic reduction experiments of mixed cathode materials of LiCoO2, LiMn2O4 and LiNi0.5Mn1.5O4 at 600 °C to 1000 °C under argon atmosphere using TGA-DTA analysis and also found LiCoO2 decomposition similar to Li et al. However, the activation energy was reported to be much lower, ranging from 204.4 to 234.2 kJ/mol obtained with the Kissinger, Kissinger–Akahira–Sunose, Flynn–Wall–Ozawa, and Friedman models. They also conducted isothermal experiments at 700 °C to 900 °C for 30, 45, and 60 minutes to investigate the kinetics using the fraction of metal extracted (by water leaching) as the degree of reaction parameter. They applied the model-fitting method and proposed that the metal reduction followed a diffusion-controlled reduction mechanism by the Ginstling–Brounstein model. They reported activation energies of Li and Co diffusion to be 71.0 and 52.1 kJ/mol, respectively. However, they did not report the detailed microstructure and phase evolution in relation to the diffusion mechanism.
Zhi-Kang et al.[13] studied the kinetics of carbothermic reduction of LiCoO2 by TGA-DTA at 400 °C to 1000 °C and 10 °C to 20 °C/min heating rate for 6 h under inert atmosphere (nitrogen). They investigated the effect of pelletizing pressure (45, 50, 60 MPa), reduction time (2 to 10 h), and mass ratio of 1/1, 1/1.5, and 1/2 of LiCoO2/C (equal to 0.12/1, 0.08/1, and 0.06/1 molar ratio). Activation energy was found to be 280.68 kJ/mol by Kissinger method. The study also attempted to investigate the reaction mechanism using data derived from the TGA-DTA curve at 15 °C/min heating rate using Coats-Redfem formula and concluded that the data fitted the Jander model of three-dimensional diffusion mechanism. However, the model-fitting covered only one mechanism, namely LiCoO2 thermal decomposition, and no value of activation energy was concluded from this method.
Xiao et al.[14] studied the kinetics of carbothermic reduction of another cathode material (LiMn2O4) at 30 °C to 1000 °C and 10-25 °C/min heating rate under inert atmosphere (argon). Based on the iso-conversional method (Friedman), the activation energy was found to be 163.74 kJ/mol. Further, using Sestak-Berggren model-fitting, they concluded that three stages occurred during the reduction processes: namely: (I) nucleation reaction control (0.1 < α < 0.32), (II) diffusion reaction control (0.32 < α < 0.72), and (III) nucleation reaction control (0.72 < α < 0.90). However, they did not report any mechanisms corresponding to the three stages.
There is no clear, unanimous agreement on the reduction mechanism and whether multiple steps occur simultaneously. Most previous studies used an iso-conversion kinetic analysis approach to investigate the carbothermic reduction of LiCoO2 over a wide temperature range. This approach is not appropriate for a system with multiple reactions occurring at the same time and that exhibits more than one rate controlling mechanism over different temperature ranges. No detailed phase and microstructure evolution analyses have been presented, except from the work of the current authors.[15] A more systematic kinetic study is needed to reveal the rate limiting step and detailed micro-mechanism of the reduction process.
The current study systematically investigated the carbothermic reduction of LiCoO2 through isothermal kinetic analysis combined with detailed microstructure and phase analysis to identify the different stages of reduction, the rate limiting steps and the overall mechanism. This paper is part of a larger study on the investigation of carbothermic reduction of LiCoO2, which focuses on the kinetics study. A separate work has been presented on the detailed investigation of the macro- and micro-mechanisms supported by thermodynamics analyses and microstructure evolution.[15]
Materials and Method
Analytical grade LiCoO2 and graphite powders from Sigma Aldrich® with purity of 99.8 and 99.99 pct, respectively, were used. The typical morphology of the particles of LiCoO2 and graphite are shown in Figures 1(a) and (b), respectively. A mixture of LiCoO2 and graphite with molar ratio LiCoO2: C = 1: 4.83 was utilised for the experiments to simulate the typical ratio found in spent Li-ion batteries.[16] Based on our previous thermodynamic calculations, 0.8 mol C per mol of LiCoO2 is adequate to reduce cobalt stoichiometrically.[15] Hence, the amount of C used in the current study represents an excess quantity. The powders were weighed and mixed in a 146 mm × 203 mm long ball-mill using 100 alumina balls of 8 mm diameter. The powders and balls were placed into the milling jar and rotated for 3 h with rotation speed of 100 rpm. The mixed powders were then pelletised using a cold press with a load of 10 tons for 5 minutes at room temperature. The final pellets had a dimension of approximately 13 mm in diameter and 12 mm in thickness. The morphology of pressed mixture pellet is presented in Figure 1(c).

(a) LiCoO2 (b) graphite (c) mixture of LiCoO2 and graphite, pressed as a pellet
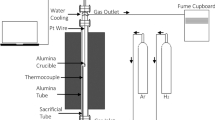
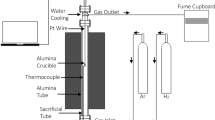
The experiments were performed in a water-cooled vertical tube furnace integrated with an analytical balance (FX-300i with 1 mg accuracy) for mass change recording. A pellet sample was placed inside an 18 mm diameter cylindrical alumina crucible, which was suspended from the digital balance using a platinum wire, as shown in Figure 2. A K-type thermocouple was used for temperature calibration and measurement. The temperature and mass were recorded using data acquisition before and during the experiments.

A schematic of the experimental setup
The experiment was commenced by loading the sample to the furnace from the top water-cooled zone. Once the furnace was sealed and while the sample was still in the cold zone, argon was passed into the furnace tube at a flow rate of 0.5 L/min. This flow rate of gas was applied to eliminate possible gas phase mass transfer control. After the gas atmosphere was stabilised, the sample was lowered to the hot zone of the furnace (and this represent time zero of the reaction) and heated isothermally at 700, 800, 900, 1000, and 1100 °C for maximum 240 minutes while the mass change was recorded every minute by the digital balance. The mass recording was repeated three times for all experiments to ensure the repeatability and calculate the errors. The approach used in the current study was an isothermal kinetic analysis for solid-state reactions where a set of conversion values (α) versus time were produced at each temperature and fit into existing general kinetic models.[17]
X-ray Powder Diffraction (XRD) was used to identify the phases present using Cu-Kα radiation and a 2θ 5-140 deg at a step size of 0.02 deg (~ 2 hours per scan) at 40 kV and 40 mA. Phase identification was performed using DIFFRAC.EVA V4 software equipped with the International Centre of Diffraction Data PDF2 + database. The sample was prepared by crushing the pellets using mortar and pestle to form a homogenous powder. During data collection, divergence slit was set at 1° to limit the total irradiation area of the sample.
Scanning Electron Microscope (SEM) analyses (Zeiss SUPRA-40) were conducted in conjunction with energy Dispersive X-ray spectroscopy (EDX) (INCA Suite 5.05) to obtain the elemental mapping and semi-quantitative chemical compositions. These EDX results (ratio of different elements) were used to deduce the possible phases during reaction. For the sample preparation, the pellets were coated with a 10 nm layer of gold and pasted on a layer of carbon tape. Secondary Electrons (SE) and Angle Selective Backscatter (AsB) images were used for the study of the microstructure. The SE images were used to study the topography and morphology of the phases in the sample and to obtain high resolution SEM imaging. The AsB images were used to observe elemental distribution in the sample. The SEM and EDX images analyses were carried out using an accelerating voltage of 20 kV, a working distance of 12 to 17 mm, an emission current of 276 pA, and a vacuum of 2.06 × 10–6 mbar.
Results and Discussion
Kinetics Data and Assessment of a Suitable Kinetic Model
Figure 3 shows the mass loss as a function of reaction time during the isothermal heating at 700 °C to 1100 °C. It can be seen that the mass loss increases with reaction time at each temperature, and that total mass loss at 240 minutes increases with increasing reaction temperature. The mass loss trend was similar at all temperatures; i.e. the greatest mass loss occurred at the early stage of reduction, after which the mass gradually decreased. However, the transition between the early and later stage was unique to each temperature.

Recorded mass change as a function of reaction time at each temperature (700 °C to 1100 °C)
The degree of reaction (α) was defined as the ratio of mass loss at particular time t to the maximum mass loss when the reaction was complete:
where mt is the mass of pellet at time t, m0 is the initial mass, and m∞ is the mass when the reaction is complete, i.e. the theoretical mass loss when oxygen removal is at its maximum such as at equilibrium. The theoretical total mass loss (at equilibrium) was determined by equilibrium reaction calculations using the FactSage™ thermochemical package. The databases of FactPS (database for pure substances, sourced from SGTE and JANAF) and FToxide (database for solid and liquid oxide compounds and solutions) were used in the modelling and the results are presented in Table I. The recorded mass loss (in Figure 3) and theoretical mass loss (in Table I) were then used to calculate α using Eq. [1]. The calculated degrees of reactions are presented in Figure 4.

The degree of reaction (α) as a function of reaction time at each temperature (700 °C to 1100 °C)
Generally, the trends of α were similar. The increment of α was significant in the early period then became slower in the later period. It can also be seen from Figure 4 that higher temperature caused an increase in α at the same time period. After 240 min, the final α at 700 °C to 800 °C was below 0.6 (0.19 and 0.54, respectively) while the values from the reduction at 1000 and 1100 °C were close to 1 (0.92 and 0.94, respectively).
A distinctive trend of α was observed at 900 °C where the mass change did not significantly decelerate in the second period (compared to at 700 °C and 800 °C). Based on the thermodynamic assessment from (Table I), the reduction product of lithium phase at 900 °C was predicted to be Li2O, as opposed to Li2CO3 at lower temperatures (700 °C and 800 °C). The formation of Li2O would cause a greater mass loss compared to Li2CO3. The proposed explanation on the cause of this distinctive trend at 900 °C will be discussed in the next section and correlated to the XRD analysis. Overall, it can be concluded that temperature plays an important role in the reduction progress.
Considering the kinetics data (in Figures 3 and 4), the XRD and microstructure analyses (both presented in the next Sections), it is clear that there were different major reduction steps and sequence of reactions occurring during reaction at each temperature. Hence, it is very unlikely that a single kinetic model can explain the kinetics at different time at each reaction temperature. Therefore, the kinetics analyses were carried out by considering two stages of reaction period: early and later stage. The onset of the transition from early to later stage is different at each reaction temperature and was defined by considering the α and the phases observed from characterisation results. The onsets were determined to be at 16, 23, 25, 19, 10 minutes for reaction temperature of 700 °C, 800 °C, 900 °C, 1000 °C and 1100 °C, respectively. In the reactions at 900 °C to 1100 °C, there appeared to be the third/final stage (after the nucleation) in which the kinetics are slow.
Different kinetic models were tested on the kinetics data using linear regression analysis to find a suitable model. The models tested were nucleation models (2D and 3D Avrami–Erofeyev); interface zero order, contracting area, and contracting volume; and diffusion-controlled models (1D, 2D, 3D, Wagner, Holt-Cutler-Wadsworth, Jander, Ginstling-Brounshtein, and Valensi-Carter). In the early stage, the best fitted model was found to be the diffusion-controlled following the Ginstling-Brounshtein (GB) model.[18] In the later stage, the kinetics data were found to be well-fitted with the nucleation-controlled following Avrami–Erofeyev/A3 model.[19] For the GB model, the overall reaction is governed by Eq. [2] while nucleation model is described by Eq. [3], both in integral form:
The plot of the left-hand-side of Eqs. [2] and [3] with time for the early stage and later stage reaction period are presented in Figures 5 and 6, respectively. The coefficient of determination (R2) and k values for both models at two stages of reaction are presented in Tables II and III, along with the onset transition times.

Early stage kinetics date fitted with a diffusion-controlled (GB) model

Later stage kinetics data fitted with a nucleation-controlled model
Phases and Microstructure Observations for Samples Reacted at 700 °C to 900 °C
To understand the reaction mechanism (correlated to the kinetics), the phases formed in each corresponding stage at particular time were determined and analysed using XRD, SEM and EDX. Considering the predicted reaction products at equilibrium are different at different temperatures, the discussion is presented for temperature range 700 °C to 900 °C and 1000 °C to 1100 °C. Figures 7 through 9 show the XRD spectra of the samples reacted at 700 °C to 900 °C after 15 minutes for the early stage and 180 and 240 minutes for the later stage.

XRD analysis of the reduced samples under argon at 700 °C after (a) 15 min (b) 240 min

XRD analysis of the reduced samples under argon at 800 °C after (a) 15 min (b) 180 min (c) 240 min

XRD analysis of the reduced samples under argon at 900 °C after (a) 15 min (b) 240 min
Early stage
In all samples (15 min, 700 °C to 900 °C), unreacted LiCoO2 and carbon were detected, and no metallic cobalt was found. Some decomposition products from LiCoO2 were found, Li2O and CoO, along with non-stoichiometric LiCoO2. Li2CO3 was also found indicating that there existed a reaction between lithium and carbon, most likely between Li2O (as the decomposition product from LiCoO2) and carbon (gas–solid reaction via CO2).[20] Although it is thermodynamically feasible to obtain Co3O4 during LiCoO2 decomposition at 700 °C,[15] the phase did not appear during the early stage.
Considering the XRD spectra, there are two possible reactions during the early stage: LiCoO2 decomposition into Li2O and CoO (with some oxygen release), and Li2CO3 formation from the reaction between Li2O and CO2 (CO2 was forming from carbon gasification). These two mechanisms are relevant to early stage reaction at temperature range of 700 °C to 900 °C. Increasing the temperature did not results in different phases, rather it only affected the degree of reaction, where the reaction at 700 °C was quite slow compared 800 °C and 900 °C. The XRD spectra suggest that Li2CO3 was more dominant compared to Li2O, and this may suggest that the Li2CO3 formation was quite fast and reaction with CO2 occurred immediately after Li2O formed from LiCoO2 decomposition. Additionally, the presence of remaining LiCoO2 and non-stoichiometric LiCoO2 (LixCo1-xO) also suggests that the decomposition was still progressing at this stage.
Figures 10(a) and (b) show the SEM images taken from sample reduced at 700 °C for 15 minutes and shows a distinct segregation between light and dark phases. There were two different light phases; type 1—small globular particles connected with each other forming a porous structure (Figure 10(a)) and type 2—a stem-like structure with elongated grain of bigger size growing from the type 1 globular particles (Figure 10(b)). The dark phase, comparatively larger in mass and had two distinctive surfaces: smooth and flaky.

Early stage microstructures at 700 °C, 15 min (a) the appearance of light phase (CoO) and dark phase, mixed of Li2O and remaining C (b) growth of CoO
Higher magnification SEM images of the light phases along with the elemental mapping and EDX point analyses are presented in Figure 11. Semi-quantitative EDX analyses were carried out to deduce the possible phases in the micrographs. The phases were deduced by considering the atomic ratio of Co, O, and C (noting that Li could not be analysed using standard SEM/EDX equipped with Be window used in the current study) and cross checked with the phases identified by XRD. The results of the EDX point analysis are presented in Table IV.

Phase segregation during early stage at 700 °C, 15 min (a) SE image of the appearance of the phases: CoO as the middle light phase, LiCoO2 as the small globular light phase, Li2O as the dark phase, remaining C as the flaky dark phase (b) AsB image of image a, correlated with elemental point analysis shown in Table IV, (c) Co and O elemental mapping (d) C elemental mapping
The small globular light phases were identified as LiCoO2. Compared to the initial morphology of LiCoO2 (Figures 1(a) and (c)), the particle changed into more porous grains during decomposition after 15 minutes of reaction. The change of LiCoO2 may be explained by its transformation to non-stoichiometric LiCoO2 (phase 1* from Figure 7(a)) by the release of oxygen during the decomposition. The bigger structure of the stem-like light phase is rich in Co and deduced as CoO (note: although the concentration of oxygen is quite small compared to that for stoichiometric CoO as shown in Table IV, no Co was detected in XRD).
The dark phase with smooth surface, situated close to the LiCoO2 and CoO, contained oxygen and was deduced to be Li2O with a small amount of carbon present. The other dark phase had a glassy and flaky texture compared to the smooth dark phase of Li2O, and was deduced to be C.
Figure 11(b) shows the AsB image and selected EDX point analysis locations for the phases in Figure 11(a). Figures 11(c) and (d) show the EDX elemental mapping of Co–O and C, respectively. Cobalt was concentrated in the stem-like light phase, while oxygen was concentrated in the smooth dark phase. The remaining carbon was seen to be attached with the Li2O but was still concentrated in some area with flaky structure.
The microstructures observed during the early stage reaction at 900 °C (Figure 12(a)) show some differences compared to samples reduced at 700 °C. The dark phase attached to the light phase contained lower Co percentage (Figures 12(b), (c)): however, from EDX point analysis this phase can still be deduced as LiCoO2 (Table V). At 700 °C, LiCoO2 appeared as a porous grain in globular spheres and still having some clear particle boundaries. However, at 900 °C, LiCoO2 was quite smudged and becoming a matrix surrounding the light phases. This could happen due to the sintering/densification effect at higher temperature which kept LiCoO2 as a dense phase. The light phases had similar Co concentration with Co-rich phase in 700 °C and was also deduced as CoO (note that the XRD did not detect any Co only CoO in the early stage of reaction). Unlike its stem-like and elongated shape at 700 °C, the CoO particle at 900 °C showed more uniform growth in all direction before colliding with the other CoO particles and was more distributed on the LiCoO2 matrix.

Early stage microstructures at 900 °C, 15 min (a) SE image of the appearance of the phases: CoO as the middle light phase confined by LiCoO2 matrix (b) elemental mapping of picture a, Co-red, O-green (c) higher magnification image with elemental point analysis shown in Table V (Color figure online)
Figures 13(a) and (b) show the Li2CO3 appearance at 700 °C and 900 °C, consistent with the XRD analysis. The morphology appeared to follow a spherulitic growth structure on top of denser matrix (Figure 13(b)). This phenomenon could be explained with the fact that Li2CO3 could originally form as a liquid but then form the spherulite shape upon cooling.

Early stage microstructures after 15 min: SE image of Li2CO3 (a) 700 °C (b) 900 °C
Later stage
After 240 min, at 700 °C, a small amount of LiCoO2 still remained along with the excess carbon while at higher temperatures (800 °C to 900 °C) LiCoO2 was fully decomposed. Lithium was recovered mainly as Li2CO3 at 700 °C to 900 °C and cobalt was recovered as metallic cobalt except at 700 °C where both Co and Co3O4 were formed as the intermediate reduction product.
A similar trend of co-existence of Li2O and Li2CO3 at both early and later stage was observed at 800 and 900 °C. However, the crystal structure of Li2O appeared to be different at these two temperatures. The Li2O crystal structure at 800 °C was cubic and changed to rhombohedral at 900°C. As for the lithium phases at 700 °C, Li2O was observed only after 15 minutes and not after 240 minutes. Mosqueda et al.[20] investigated the behaviour of Li2O under CO2 atmosphere using TGA analysis up to 1000 °C. During the CO2 incorporation with Li2O, the formation of Li2CO3 was revealed to be a surface reaction, generating a shell of Li2CO3 which covered the unreacted Li2O.[20] This would impede the absorption of CO2 which likely explained the Li2O existence in the early stage of reaction. At 900 °C, it is expected from thermodynamic assessment that lithium would be recovered as Li2O instead of Li2CO3. Although the presence of Li2O was detected up to 900 °C, the fraction of Li2CO3 was more dominant. This indicates that at 900 °C, the reaction between Li2O and CO2 forming Li2CO3 was still occurring.
After 240 min, Li2CO3 would have transformed to its liquid phase, experimentally observed between 700 and 731 °C.[21,22,23,24,25] The Li2CO3 would apparently decompose into Li2O above 727 °C[24,26,27] which is accompanied by CO2 release. Hence, the order of Li2CO3 melting and decomposing might happen interchangeably. If the decomposition happened before the melting, Li2CO3 transformation to liquid would be affected by the degree of the decomposition as the melting point was approached.[28] Additionally, the excess carbon could trigger the decomposition on which Li2CO3 could react with carbon to form Li2O and CO.[26,29]
Although the coexistence of Li2O and Li2CO3 was observed at both 800 and 900 °C, the degree of reaction at 900 °C (as previously mentioned by the unique α trend) was larger compared to 800 °C. This phenomenon might be explained by the possibility of an acceleration of Li2CO3 decomposition at 900 °C. At 850 °C to 950 °C, molten Li2CO3 was observed to have significant increase on mass loss.[30] It was also observed by Timoshevskii et al.[31] that during Li2CO3 decomposition, the CO2 pressure abruptly increased by increasing temperature, indicating a larger degree of decomposition.
Contrary to the XRD analysis results at the early stage after 15 min, which showed the presence of CoO, Co3O4 was observed after 240 minutes at 700 °C and was strongly correlated with the oxidation of CoO.[32] Moreover, both CoO and Co3O4 can coexist at 700 °C.[33,34,35,36,37] This also indicates that at 700 °C, the reduction of CoO to metallic cobalt was slower than at higher temperatures and allowed sufficient time for CoO to oxidize to Co3O4.
The SEM-SE-micrographs of the top surface of the sample reduced at 900 °C after 240 minutes is shown in Figure 14(a). The final products observed were cobalt and Li2CO3 along with the remaining carbon mixed with Li2O. Figure 14(b) shows a magnified Li2CO3 particle with the location of EDX point analyses and the corresponding C and O mapping in Figures 14(c) and (d), respectively. Table VI shows the corresponding EDX analyses and the associated deduced phases. A similar spherulitic structure of Li2CO3 was also found in the sample reduced at 800 °C after 180 minutes (Figures 15(a) and (b)).

(a) Later stage microstructures at 900 °C after 240 min (b) structure of Li2CO3 and its elemental point analysis shown in Table VI (c) C elemental mapping of b (d) O elemental mapping of b

Later stage microstructures showing: (a) appearance of Li2CO3 at 800 °C at 180 min, lower magnification; (b) Li2CO3 at 800 °C at 180 min, higher magnification; (c) Co nucleation from CoO at 800 °C at 180 min, lower magnification; (d) Co nucleation from CoO at 800 °C at 180 min, higher magnification; (e) Co growth on Li2O + C matrix at 700 °C at 240 min; (f) Co growth on Li2O + C matrix at 900 °C at 240 min
Another reaction occurring during the later stage was cobalt nucleation from CoO as shown in Figures 15(c) and (d) which were taken from samples at 800 °C after 180 minutes reaction to show the appearance while the nucleation was still progressing. After 240 min, it can be seen that the cobalt nuclei have grown on the surface. At 700 °C (Figure 15(e)), the presence of metallic cobalt is shown by the light nuclei which appeared and grew at the surface of dark phases composed of Li2O and remaining carbon. At 900 °C (Figure 15(f)), some part of the cobalt particles have undergone Ostwald ripening phenomenon forming larger grains.
Phases and Microstructure Observations for Samples Reacted at 1000 °C to 1100 °C
Figures 16 and 17 show the results of the XRD analyses of samples reduced at 1000 °C to 1100 °C reacted up to 10 minutes for the early stage and 180 and 240 minutes for the later stage.

XRD analysis results of the reduced samples under argon at 1000 °C after (a) 10 min (b) 180 min (c) 240 min

XRD analysis results of the reduced samples under argon at 1100 °C after (a) 10 min (b) 240 min
Early stage
Figures 16(a) and 17(a) show the phases present during the early stage, at 10 min, at 1000 °C and 1100 °C, respectively. Similar to the previous temperature range of 700 °C to 900 °C, decomposition products Li2O and CoO were observed in this stage. The crystal structure of the Li2O was rhombohedral which was the same as at 900 °C. The phases observed indicate that higher temperature did not result in different decomposition products in the early stage (compared to 700 °C to 900 °C), except that the rate of the reaction was faster than those reduced at lower temperatures (Figure 4). At 1000 °C to 1100 °C, Li2CO3 was not present in the system anymore, and the intensities of the Li2O peaks slightly increased. At this temperature range, although excess carbon was available, the presence of CO2(g) was thermodynamically not favourable due to the Boudouard reaction and Li2O was unreactive with only CO.[15]
The conversion of Li2CO3 to Li2O at higher temperature might be explained due to its catalytic effect to the Boudouard reaction. An experimental study by Yokoyama et al.[38] concluded that C/CO2 reaction was catalysed by carbonates of Li, Na, K, Cs, and Rb and their rate were found to be equivalent to the amount of oxygen absorbed on the solid surface during the reaction. It was quite possible in the system studied that the Li2CO3 produced acted as catalyst to increase the conversion of CO2 to CO in the Boudouard reaction. Hence facilitated its decomposition to Li2O.At higher temperatures (1000 °C to 1100 °C), selected image taken from samples reduced at 1100 °C after 10 minutes is shown in Figure 18(a) to represent the early stage. The distinction between the Co-rich phase (deduced as CoO) and O-rich phase (deduced as Li2O), was not significant compared to the phases in the sample reduced at 900 °C (Figure 12). The remaining carbon was present as a flaky dark phase and appeared to be mixed with the Li2O (Table VII, Point 4 and 5). The CoO was growing into much larger size interconnected particles when compared to those found at 900 °C indicating more extensive decomposition at higher temperature.

(a) LiCoO2 decomposition at 1100 °C after 10 min (b) Co elemental mapping (c) O elemental mapping (d) C elemental mapping
Later stage
Figures 16(c) and 17(b) show the XRD analyses of samples reduced after 240 minutes at 1000 and 1100 °C, respectively. The cobalt was fully recovered as metallic cobalt and no cobalt oxides were observed. As for the lithium product, there was no phases observed other than Li2O. At an earlier time of 180 minutes at 1000 °C, Li2O, CoO and Co were observed. Therefore, it can be concluded that the reaction was more dominated by the conversion of CoO to Co, while there was no change in Li2O.
The SEM-SE images of the sample reacted at 1000 °C for 180 minutes are shown in Figures 19(a) and (b) where the Co nucleation from CoO is observed. The microstructure at 1100 °C after 240 minutes is shown in Figure 20(a) and shows the phases Li2O and cobalt which were deduced from the EDX point analyses shown in Table VIII. The cobalt nuclei show slightly different structure (a more distinct shape) compared to those observed in samples reduced at lower temperatures (700 °C to 900 °C, Figures 15(c) and (d)) and possibly due had undergone a recrystallization from the effect of higher temperature. Schuett et al.[39] investigated the growth of a single crystal of cobalt via controlled-atmosphere flame fusion method. They concluded that cobalt recrystallization happened during the FCC phase temperature range (422 °C to 1495 °C), and the cobalt single crystal with a triangular shape was observed when they annealed cobalt just below its melting point. The FCC cobalt adopted an octahedron-like shape with high symmetry which corresponds to the structure of the observed cobalt nuclei in Figure 20(a).

Later stage microstructures at 1000 °C after 180 min showing the Co nucleation from CoO at (a) low magnification, and (b) high magnification

Later stage microstructures at 1100 °C, 240 min (a) the appearance of Co particle (right) and Li2O spheres (left) (b) enlargement image of Li2O (c) O elemental mapping of b (d) C elemental mapping of b
Activation Energy and Mechanism
The values of apparent activation energies for the reduction stages were calculated using the Arrhenius equation given in Eq. [4]:
where A is the reaction constant, Ea is the activation energy (kJ/mol), R is the ideal gas constant (8.3145 J/mol K), k is the rate constant (shown in Tables II and III) and T is temperature (K). Figure 21 presents the plot of \( \ln {k} \) vs \(1/T\), for kinetic data in the early stage (diffusion-controlled step) showing the slope as a negative value of the activation energy. It can be seen that the plot is quite well-fitted with R2=0.976. The calculation showed that the activation energy during diffusion stage was 121 kJ/mol with a cumulative error of ± 14 kJ/mol. The error was evaluated with the Chi-square method.[40] For comparison, some literature values for the diffusion of associated elements are summarized in Table IX.

Activation energy derived from ln k and inverse temperature during the diffusion stage
The present value of activation energy falls in the range of the activation energies of oxygen diffusion through non-stoichiometric LiCoO2 of Furushima et al.[45] This might indicate that during the early stage of the reduction, the dominant mechanism is oxygen diffusion in the LiCoO2 during its decomposition. The period of diffusion-control was different at each reaction temperature, as shown in Table II. Generally, the transition from diffusion-control to the next stage occurred earlier at higher temperatures which implies a higher rate at higher temperature.
The later stage was nucleation-controlled, with the activation energy calculated to be 95 kJ/mol with cumulative error of ± 18 kJ/mol (Figure 22). Although the early diffusion-controlled stage appeared to occur within a short period of time, it is likely that the diffusion was still occurring into the nucleation stage. This is supported by the observation of the unreacted LiCoO2 at 700 °C at 240 min, which indicates that the decomposition of LiCoO2 was still occurring at this time. Since the diffusion stage is correlated to the oxygen diffusion during LiCoO2 decomposition, the subsequent Co nucleation would be contributed by the region in the sample where the LiCoO2 had completely decomposed to CoO.

Activation energy derived from ln k and inverse temperature during the nucleation stage
Previously reported data on activation energy for nucleation are summarised in Table X showing that, under the same experimental conditions, generally the activation energy for CoO nucleation from Co3O4 is higher than Co nucleation from CoO. Only the study from Gallegos and Lopez[46] reported an activation energy for CoO nucleation slightly lower than for Co. It should be noted that these data were from reduction under hydrogen as opposed to carbothermic reduction.
The activation energy of CoO nucleation from Co3O4 by Lin and Chen[47] is close to our Ea value, although their experiments were performed under H2 and at lower temperatures. However, based on the analysis in the present study, the nucleation stage was shown to occur via Co nucleation/reduction from CoO rather than CoO from Co3O4. The XRD and SEM–EDX analysis showed that CoO was the main cobalt-containing product after LiCoO2 decomposition meanwhile Co3O4 was likely generated from CoO oxidation. According to this study, and supported by the previous work,[15] the reduction of CoO to metallic Co would be the slowest (i.e. the limiting step), and therefore would control the overall kinetics in the nucleation stage. The Ea value determined in the present study also falls into the same range with the activation energy of Co nucleation from CoO presented by Tomić-Tucaković et al.[48]
At 900 °C, 1000 °C and 1100 °C, there appeared to be the third stage after the nucleation (as shown in Figure 6). In this stage, significant deceleration was observed, and the overall kinetics were slow. The rate was found to decrease with increasing of temperature. From the microstructural evaluation, major grain growth and densification of Co were observed. It appeared that this dense Co provide a barrier for further reduction of CoO.
A general carbothermic reduction mechanism is proposed based on the findings in the current study and is summarized in Figure 23 and Table XI. In the early stage of the reduction, the thermal decomposition of LiCoO2 into cobalt oxide (CoO) and Li2O occurred, while in the later stage reduction of CoO to metallic cobalt occurred via nucleation process. At 700 °C to 900 °C, Li2O is proposed to react with CO2 producing Li2CO3. At 1000 °C to 1100 °C, CO was more likely to present compared to CO2, thus preventing further reaction with Li2O.

Schematics of proposed carbothermic reduction mechanisms at (a) 700 °C to 900 °C (b) 1000 °C to 1100 °C
This study has some implications for the Li-ion battery recycling, especially in the context of lithium and cobalt recovery. The recovered lithium product could be tailored based on the temperature. Lower temperatures would generate Li2CO3 (700 °C to 900 °C) and higher temperatures would generate Li2O (1000 °C to 1100 °C). However, the presence of excess carbon was not beneficial in regard to Li2CO3 formation, especially at 900 °C, due to the acceleration of Li2CO3 decomposition to Li2O. On the other hand, formation of cobalt was found to be more dependent on the reaction time rather than temperature. Metallic cobalt could be obtained from the reduction at all temperatures. However, at lower temperature (700 °C), an intermediate product of cobalt oxide (Co3O4) was still possible to be present.
Additionally, the kinetic study is helpful in determining the critical times over which the reactions progressed. To recover cobalt in its metallic form, the reaction time needs to exceed the early stage period allowing significant LiCoO2 decomposition to CoO. Overall, temperature affected the rate of kinetics in both diffusion and nucleation stage. There was also a high-possibility that diffusion mechanism could overlap with the nucleation stage which would generate metallic cobalt thus hindering further diffusion of oxygen from LiCoO2. The effect of this would be larger at lower temperatures due to lower rate of LiCoO2 decomposition.
Conclusions
A systematic study of carbothermic reduction of LiCoO2 cathode material has been carried out that included kinetics analyses supported by detailed characterization. The kinetics of isothermal reduction of LiCoO2/C at 700 °C to 1100 °C in inert atmosphere were evaluated by measurement of mass change with reaction time. From the results obtained, the reduction can be classified into two major stages:
-
The early stage of reduction of LiCoO2 which follows a diffusion-controlled mechanism and can be explained using the GB model. The kinetics appears to be controlled by oxygen diffusion in non-stoichiometric LiCoO2 during its decomposition. The activation energy was calculated to be 121 kJ/mol.
-
The later stage appeared to be controlled by Co nucleation which happened during the reduction of CoO to Co with an activation energy of 95 kJ/mol.
In terms of products after reaction, different reduction temperatures would generate different Li and Co phases. In the range 700 °C to 900 °C, Li2CO3 and Co phases were observed; while in the range of 1000 °C to 1100 °C, Li2O and Co phases were observed. The presence of excess carbon was not beneficial in regard to Li2CO3 formation, especially at 900 °C, due to the acceleration of Li2CO3 decomposition to Li2O. Metallic cobalt could be obtained from the reduction at all temperatures studied; however at 700 °C, an intermediate product of Co3O4 was observed.
The kinetic study could help in designing the process parameter (e.g. temperature, reaction time, and carbon amount) to obtain particular recovered products of lithium and cobalt. Despite the fact that higher temperature increased the reaction rates of both the diffusion and nucleation stages, increasing the temperature did not significantly affect the final product of cobalt. On the other hand, the lithium products were strongly dependant on the temperature of the reaction.
References
M. Lowe, S. Tokuoka, T. Trigg, and G. Gereffi: Lithium-ion Batteries for Electric Vehicles: the U.S. Value Chain, Center on Globalization, Governance & Competitiveness, Duke University, 2010.
M.A. Hannan, M.S.H. Lipu, A. Hussain, and A. Mohamed: Renew. Sustain. Energy Rev., 2017, vol. 78, pp. 834–54.
Mineral Commodity Summaries 2021, U.S.G. Survey, Reston, VA, 2021.
K.B. Shedd, E.A. McCullough, and D.I. Bleiwas: Min. Eng., 2017, vol. 69, pp. 37–42.
Study on the EU's List of Critical Raw Materials, 2020.
C. Lewis: Resources and Energy Quarterly September 2021, Department of Industry, Science, Energy and Resources, Commonwealth of Australia, 2021.
S.M. Shin, N. Kim, J. Sohn, D. Yang, and Y. Kim: Hydrometallurgy, 2005, vol. 79, pp. 172–81.
H. Pinegar and Y.R. Smith: J. Sustain. Metall., 2020, vol. 6, pp. 142–60.
H. Pinegar and Y.R. Smith: J. Sustain. Metall., 2019, vol. 5, pp. 402–16.
O.S. Kwon and I. Sohn: Resour., Conserv. Recycl., 2020, vol. 158, pp. 104809.
J. Li, Y. Lai, X. Zhu, Q. Liao, A. Xia, Y. Huang, and X. Zhu: J. Hazard. Mater., 2020, vol. 398, 122955.
S. Pindar and N. Dhawan: Metall. Mater. Trans. B, 2021, vol. 52B, pp. 3078–92.
C. Zhi-Kang, L. Ji-Dong, L. Zhen, W. Xue-Lian, and Y. Ling-Feng: Mater. Express, 2021, vol. 11, pp. 1988–96.
J. Xiao, R. Gao, L. Zhan, and Z. Xu: ACS Sustain. Chem. Eng., 2021, vol. 9, pp. 9418–25.
B.A. Nuraeni, K. Avarmaa, L.H. Prentice, W.J. Rankin, and M.A. Rhamdhani: SM&T, 2022, vol. 34, e00526.
T. Georgi-Maschler, B. Friedrich, R. Weyhe, H. Heegn, and M. Rutz: J. Power Sources, 2012, vol. 207, pp. 173–82.
A. Khawam and D.R. Flanagan: J. Phys. Chem. B, 2006, vol. 110, pp. 17315–28.
A.M. Ginstling and V.I. Brounshtein: J. Appl. Chem. USSR, 1950, vol. 23, pp. 1327–38.
B.V. Erofeyev: Dokl. Akad. Nauk SSSR, 1946, vol. 52, p. 511.
H.A. Mosqueda, C. Vazquez, P. Bosch, and H. Pfeiffer: Chem. Mater., 2006, vol. 18, pp. 2307–10.
L. Shi, T. Qu, D. Liu, Y. Deng, B. Yang, and Y. Dai: Process of Thermal Decomposition of Lithium Carbonate, Springer, Cham, 2020.
A. Bazhenov, V. Miklushevskii, I. Vatulin, E. Kropacheva, and A. Bidylo: Russ. J. Non-Ferr. Met., 2010, vol. 51, pp. 44–48.
P. Pasierb, R. Gajerski, S. Komornicki, and M. Rękas: J. Therm. Anal. Calorim., 2001, vol. 65, pp. 457–66.
P. Pasierb, R. Gajerski, M. Rokita, and M. Rekas: Physica B, 2001, vol. 304, pp. 463–76.
L. Ahamad, S.K. Rakshit, S. Parida, Y. Naik, G. Rao, S. Kulkarni, S. Singh, and S.C. Gadkari: J. Therm. Anal. Calorim., 2012, vol. 112, pp. 17–23.
J.W. Kim and H.G. Lee: Metall. Mater. Trans. B., 2001, vol. 32, pp. 17–24.
Y. Di, N. Feng, W. Dong, J. Peng, and Y. Wang: Nonf. Met. (Extract. Metall.), 2009, vol. 6, pp. 28–30.
H.C. Stober: Lithium Carbonate, Academic Press, 1986.
H. Beyer, S. Meini, N. Tsiouvaras, M. Piana, and H.A. Gasteiger: Phys. Chem. Chem. Phys., 2013, vol. 15, pp. 11025–37.
S. Licht, H. Wu, Z. Zhang, and H. Ayub: Chem. Commun., 2011, vol. 47, pp. 3081–83.
A.N. Timoshevskii, M.G. Ktalkherman, V.A. Emel’kin, B.A. Pozdnyakov, and A.P. Zamyatin: High Temp., 2008, vol. 46, pp. 414–21.
R. Metz, J. Morel, H. Delalu, S. Ananthakumar, and M. Hassanzadeh: Mater. Res. Bull., 2009, vol. 44, pp. 1984–89.
E.A. Gulbransen and K.F. Andrew: J. Electrochem. Soc., 1951, vol. 98, p. 241.
R.E. Carter, F.D. Richardson, and C. Wagner: JOM, 1955, vol. 7, pp. 336–43.
H.S. Hsu and G.J. Yurek: Oxid. Met., 1982, vol. 17, pp. 55–76.
S. Mrowec, T. Walec, and T. Werber: Corros. Sci., 1966, vol. 6, pp. 287–92.
E.M. Fryt: Oxid. Met., 1978, vol. 12, pp. 139–56.
S.-Y. Yokoyama, K. Miyahara, K.-I. Tanaka, and J. Tashiro: J. Jpn. Petrol. Inst., 1983, vol. 26, pp. 455–61.
F.M. Schuett, D. Esau, K.L. Varvaris, S. Gelman, J. Björk, J. Rosen, G. Jerkiewicz, and T. Jacob: Angew. Chem. Int. Ed., 2020, vol. 59, pp. 13246–52.
F. Pyrczak: Making Sense of Statistics: A Conceptual Overview.5th edition, Pyrczak Pub., Glendale, Calif, 2010, pp.
K. Ando, M. Akiyama, and Y. Oishi: J. Nucl. Mater., 1980, vol. 95, pp. 259–64.
K. Ando, Y. Oishi, and T. Yoneda: J. Nucl. Sci. Technol., 1980, vol. 17, pp. 269–74.
Y. Oishi, Y. Kamei, M. Akiyama, and T. Yanagi: J. Nucl. Mater., 1979, vol. 87, pp. 341–44.
A. Swiderska-Mocek and A. Lewandowski: J. Solid State Electrochem., 2017, vol. 21, pp. 1365–72.
Y. Furushima, C. Yanagisawa, T. Nakagawa, Y. Aoki, and N. Muraki: J. Power Sources, 2011, vol. 196, pp. 2260–63.
N.G. Gallegos and J.M.P. Lopez: Mater. Chem. Phys., 1988, vol. 19, pp. 431–46.
H.Y. Lin and Y.W. Chen: Mater. Chem. Phys., 2004, vol. 85, pp. 171–75.
B. Tomić-Tucaković, D. Majstorović, D. Jelić, and S. Mentus: Thermochim. Acta, 2012, vol. 541, pp. 15–24.
Acknowledgments
The authors thank and acknowledge the financial support from Swinburne University’s Automotive Engineering Graduate Program (AEGP) by the Australian Government through the Department of Industry, Science, Energy and Resources (previously the Department of Industry, Innovation, and Science) and the Commonwealth Scientific and Industrial Research Organisation (CSIRO).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nuraeni, B.A., Avarmaa, K., Prentice, L.H. et al. Recovery of Cobalt and Lithium by Carbothermic Reduction of LiCoO2 Cathode Material: A Kinetic Study. Metall Mater Trans B 54, 602–620 (2023). https://doi.org/10.1007/s11663-022-02712-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-022-02712-1