
Abstract
There is a current interest in Co-based superalloys hardened by a L12-γ′ phase because Co has a higher melting point than Ni and is more resistant against sulfidation attack. However, the Co-Al-W system many of those γ′ hardened Co-based superalloys are based on, has a number of drawbacks. The γ′ phase Co3(Al,W) is not stable at high temperature, the density of the alloys is very high and the oxidation resistance is insufficient. Due to this, there is an ongoing interest to develop γ′-hardened Co-based superalloys based on other systems. Here, first principles calculations are presented to investigate the properties of the γ′ L12-(Co0.5,Ni0.5)3(Al0.5,Ti0.5) phase and related L12 structures. (Co0.5,Ni0.5)3(Al0.5,Ti0.5) exhibits a lower energy of formation than Co3Ti and Co3(Al0.5,W0.5). Nevertheless, Ni3(Al0.5,Ti0.5) has an even lower energy of formation which is further lowered if Ti is enriched on the second sublattice. This finding is supported by analyzing the electronic densities of states. Ni3(Al0.5,Ti0.5) and especially Ni3(Al0.25,Ti0.75) exhibit Fermi levels close to the gap between binding and antibinding states, which is an indicator for stability. In addition to the stability of the γ′-phase in dependence on Ni and Ti content, also the elastic properties were calculated. Ni3(Al0.25,Ti0.75) is less elastic anisotropic and has higher Young’s and shear modulus compared to Ni3(Al0.5,Ti0.5) and (Co0.5,Ni0.5)3(Al0.5,Ti0.5).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The discovery of the ternary compound Co3(Al,W) with L12 crystal structure (γ′ phase) in Co-based alloys and especially the fact that a fine structure of cuboidal precipitates of that type could be embedded coherently in a Co-matrix[1,2] triggered research in this class of alloys. In Ni-based superalloys, the hardening by such a coherent precipitate phase causes extraordinarily high creep and high temperature deformation resistance. The Co-based system exhibits a number of advantages making the development of L12-phase hardened Co-based superalloys an attractive prospect. Co has a slightly higher melting point than Ni[3] and at least pure Co is more resistant against sulfidation attack due to the higher melting point of CoS than NiS[4,5] and the higher melting point of the eutectic composition in the Co-S system.[6] It was also reported that the defect-free casting of large single crystal parts is easier using such Co-based alloys.[7] In addition, Co has a lower stacking fault energy than Ni[8] which should be beneficial for creep resistance because it hampers dislocation climb and associated recovery processes.
Nevertheless, the L12-hardened Co-based superalloys of the Co-Al-W system as initially proposed by Sato et al.[2] have also a number of disadvantages. The two most important are the insufficient long-term stability of the L12-phase[9,10,11] and the high density due to high alloying content of heavy W.[12] The latter is especially detrimental for fast moving parts as, for example, turbine blades. Therefore, there is a current search to replace W by other alloying elements[13,14,15,16,17,18,19] to generate lower density L12-phases with better stability while retaining or even increasing the solvus temperature and volume fraction of this L12-phase compared to the Co-Al-W-based system.
Alloy development trends into this direction are the addition of Ni to increase phase stability and solvus temperature[20] or the replacement of W by Nb, Mo, Ti or V.[13,14,15,16,17,18,19] Nearly from the beginning of the current alloy development of those L12-hardened Co-based superalloys, simulation and modeling methods played an important role. They are used to better understand the system,[21,22,23,24,25,26,27] predict attractive alloy compositions[28,29,30,31,32,33] or answer questions, which are experimentally not directly accessible. For example, the questions if the L12-Co3(Al,W) phase is metastable at 0 K and which effects could stabilize it at higher temperatures and if those are sufficient to stabilize it compared to competing phases were early addressed by density functional theory (DFT) calculations.[21,34] Since then numerous theoretical as well as experimental works were published which shed light on different properties of the L12-Co3(Al,W) phase as well as other Co-containing L12-phases.[9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34] For Ni-based superalloys being the technological more important system hardened by the L12-Ni3Al phase, DFT was also widely used to predict the properties of this compound. In Ni-base superalloys in industrial use, the L12-phase in reality is not Ni3Al but more akin to a pseudo binary Ni3(Al1−x,Xx) with X being mainly Ti and Ta. DFT was used to predict lattice constants, formation energies, elastic properties and planar fault energies of such pseudo binary compounds (e.g.,[35,36,37,38,39]).
Some of the authors of the present study investigated the possibility to develop a L12-hardened Co-base superalloy based on the Co–Ni-Al-Ti-Cr system.[13,40] The L12-phase formed in this system showed some intriguing peculiarities. Foremost, while the overall alloy composition is Co-rich Ni is the majority element in the L12-phase.[35] To further elucidate the properties of the L12-phase in this alloy system DFT calculations are employed and the most important results of these are presented in this paper. This was helped by the experimental finding that Cr is mainly enriched in the Co solid solution matrix in this alloy system.[35] Thus, DFT calculations of the L12-phase could be restricted to the quaternary system Co–Ni-Al-Ti. For this system, the effect of different contents of those elements on the stability of the phase as well as on other properties, for example elastic constants, were determined and are presented in this work.
2 Methodology
DFT calculations were done with version 5.4.4 of the Vienna Ab Initio Simulation Package (VASP)[41,42,43] using projector augmented waves within the Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation (GGA) as exchange function.[44] For Al, the electrons in the 3s and 3p states and for W the electrons in the 5d and 6s states are considered as valence electrons while for Ti, Ni and Co the electrons in the 3p, 3d and 4s states are treated as valence electrons. To take into account the magnetic nature of Co and Ni all calculations were performed spin-polarized with an initial paramagnetic configuration attributing magnetic moments of a magnitude of ± 1.7 μB to each Co and ± 0.5 μB to each Ni atom as starting configuration. A 5 × 5 × 5 k-point mesh[45] and a cut off energy of 380 eV were used for the majority of the calculations based on a convergence test. Further increasing the k-point density or cut off energy yielded differences of less than 0.2 meV/atom. To achieve at least the same k-point density the k-point mesh for the pure element reference states (hcp-Co, fcc-Ni, fcc-Al, hcp-Ti, bcc-W) to calculate formation energies was 15 × 15 × 15, which was also used for the calculations of electronic densities of states. The determination of the elastic tensor was done automatically by VASP and to ensure convergence of the stress tensor the cut off energy was increased to 500 eV for those calculations. All equilibrium volumes and energies were determined by calculating energies for different volumes while allowing to relax the ionic positions using the conjugate gradient scheme to a precision of below 0.1 meV/Å and subsequently fitting the Murnaghan equation of states to the generated energy-volume data.
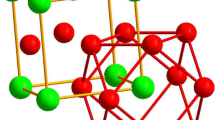
The investigated L12 structures could contain up to four atomic species two of each being present on one of the both sublattices. To depict this so-called special quasi-random structures (SQS) of 32 atoms (i.e., 2 × 2 × 2 L12 unit cells) were constructed using the mcsqs routine of the ATAT software package.[46] SQS for the following compositions were constructed: (A0.5,B0.5)3(C0.5,D0.5), (A0.25B0.75)3(C0.5,D0.5) and (A0.5,B0.5)3(C0.25,D0.75). If only one atomic species was present in the L12 structure on one sublattice the A and B or C and D positions, respectively, were both filled with this one atomic species. For the construction of the SQS, the pair interactions up to the 2nd shell and triple interactions up to the next shell were considered. The quality of the SQS supercells was validated by exchanging Co and Ni between the A and B positions and Al and Ti between the C and D positions as well as extending pair and triple interactions to the 3rd neighbor shell for the 32 atom SQS supercells. In addition, a comparison to 64 atoms SQS supercells, considering the same pair and triple interactions was made. The resulting differences of formation energy are less than 2.5 meV/atom, which is significantly below all energy differences discussed later. An example of the 32 atom (A0.5,B0.5)3(C0.5,D0.5) SQS supercell is shown in Figure 1.

32-atom SQS supercell of (A0.5,B0.5)3(C0.5,D0.5) stoichiometry—C and D positions in red and blue and A and B positions in green and purple (Color figure online)
3 Results and Discussion
3.1 Energies of Formation and Mixing
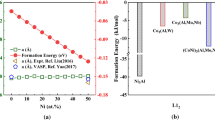
In the multicomponent system Co-Ni-Al-Ti, there are a number of combinations to build L12 phases from those four elements. Looking at the binary systems, Ni3Al and Co3Ti are known to be stable L12 phases. For the Co-Ni-Al-Ti system, L12 phases are of interest where Ti and Al mix on the one and Co and Ni on the other sublattice of L12. Therefore, phases of the types Ni3(Al0.5,Ti0.5) and (Co0.5,Ni0.5)3(Al0.5,Ti0.5) were also considered and in addition the Co3(Al0.5,W0.5) L12 phase as reported by Sato et al.[2] The latter is well suited to validate the SQS approach used in the present work, because a number of articles report different properties of this phase determined by DFT calculations. It is also of interest, whether L12 phases based on the Co-Ni-Al-Ti system are more stable than the Co3(Al0.5,W0.5) phase. In the Co-Ni-Al-Ti system as well as in the Co-Al-W system, there are a number of other competing phases not having a L12 structure. These are CoTi with B2 structure and Ni3Ti with D024 structure in the C-Ni-Al-Ti system, CoAl with B2 structure in both and Co3W with D019 structure in the Co-Al-W system. For all these phases, the energies of formation as a measure of their stability were calculated being defined as the energy of the compound minus the weighted sum of the energies of the constituting elements in their respective stable ground state (i.e., hcp-Co, fcc-Ni, fcc-Al, hcp-Ti and bcc-W). The results are shown in Figure 2. All the phases shown in Figure 2 are stable indicated by their negative energies of formation. This is also true for Ni3Al, Co3Ti, Ni3(Al0.5,Ti0.5) and (Co0.5,Ni0.5)3(Al0.5,Ti0.5), which are possible L12 phases in the Co-Ni-Al-Ti system. For (Co0.5,Ni0.5)3(Al0.5,Ti0.5) also the energies of formation determined with two 64-atom SQS are shown in Figure 2 as a diamond (2 × 2 × 4 orthorhombic supercell) and circle (2 × 2 × 4 triclinic supercell). The results do not significantly differ between the 32- and the two 64-atom SQS supercells.

Energies of formation for different phases in the systems Co–Ni-Al-Ti and Co-Al-W (diamond and circle indicate the values determined using 64-atom SQS supercells)
(Co0.5,Ni0.5)3(Al0.5,Ti0.5) and Ni3(Al0.5,Ti0.5) have significantly lower energies of formation than Co3(Al0.5,W0.5) indicating that they are more stable types of L12 phases compared to Co3(Al0.5,W0.5). However, Ni3(Al0.5,Ti0.5) exhibits the lowest energy of formation of all L12 phases shown in Figure 2 being lower than the one of (Co0.5,Ni0.5)3(Al0.5,Ti0.5) thus indicating that a mixing of Co and Ni on the first sublattice is energetically not favorable while a mixing of Al and Ti on the second is. The D024-Ni3Ti phase has only a slightly lower energy of formation than Ni3(Al0.5,Ti0.5) and L12-Ni3Ti. However, CoAl with B2 structure is the most stable compound of all investigated phases. B2-CoTi is energetically less favored. Therefore, if one would try to increase the γ′ volume fraction in the system Co-Ni-Al-Ti by adding γ′ builders Al and Ti, the γ/ γ′ phase stability is probably restricted by the nucleation of B2-CoAl if too much Al is added. The trends and values of formation energies shown in Figure 2 correspond with respective trends and values reported in literature. Chandran and Sondhi determined a formation energy of 0.435 eV/atom for L12-Ni3Al,[35] which is nearly equal to that reported by Breidi et al.[47] and very close to the value shown in Figure 2 while Wu and Li report slightly lower values.[36] Nevertheless, also the value of 0.4558 eV/atom given by Wu and Li is still higher than the formation energies for L12-Ni3Ti and L12-Ni3(Al0.5,Ti0.5) found in the present work. An addition of Ti may stabilize L12-Ni3(Al1−x,Tix) as found here is also in agreement with literature. Wu and Li report a decrease in formation energy for Ti addition,[36] which is a trend also resembled by the cohesive energies if Ti is added shown by Vamsi and Karthikeyan.[37] One is tempted to speculate that an increase in APB energy if Ti is added as reported by Chandran and Sondhi[35] may also indicate a stabilizing effect of Ti additions to the L12-Ni3Al phase because a higher APB energy means that disordering is energetically less favorable. How the substitution of Ni by Co on the first sublattice influences the phase stability is less frequently reported in literature. The higher (i.e., less negative) formation energy of L12-Co3Ti compared to L12-Ni3Ti as well as the decrease in the energy of superintrinsic stacking faults in L12-(Ni1−x,Cox)Al (indicating a tendency to transform to the D019 structure) both reported by Breidi et al.[38,46] support the destabilizing effect of Co replacing Ni as found in the present work. The formation energies calculated for Co3(Al0.5,W0.5) are in good agreement with results of Saal et al.[27] and Jiang.[21] Also, for Co3(Al0.5,W0.5) and Co3Ti a good agreement with results from Koßmann et al.[24] and Jin et al.[25] is found. The energy of formation of D019-Co3W calculated by Jin et al. and Saal et al. (judged from the convex hull plotted in the latter work) agrees with the value reported here while Jiang[21] found a slightly lower energy of formation for D019-Co3W. In the present work as well as in the literature references, the formation energy of D019-Co3W is always higher (less negative) than the one of L12-Co3(Al0.5,W0.5)[21,27] indicating that L12-Co3(Al0.5,W0.5) is more stable. Nevertheless, it was reported that the D019-Co3W phase forms at expense of the L12-Co3(Al0.5,W0.5) phase in Co-Al-W alloys.[11] From comparing energies of formation, this is not understandable and indicates that formation energies alone are no good measures for the stability of a phase in a system because they do not consider the combination of competing phases in a reasonable way: While a transformation of Co3(Al0.5,W0.5) to Co3W leads to an energy loss, it also sets Al free. This can now form B2-CoAl with its much lower energy of formation and remaining Co, Al and W can be incorporated in a Co solid solution. Thus, even if one of the transformation products is less energetically stable, a phase will dissolve if the combination of all transformation products leads to an energy gain for the system. While a complete thermodynamic treatment based on first principles is far beyond the scope of the present paper by considering the so-called mixing energies, it can be better judged how stable a specific L12-structure is than by comparing formation energies alone. The energy of mixing is the difference between the energy of the compound in question and a weighted sum of the energies of competing compounds, which combined result in the same chemical composition as the compound in question. i.e., the mixing energy of a ternary Co3(Al0.5,Ti0.5) compound relative to the binary compounds Co3Al and Co3Ti would be calculated as:
with \(E_{{{\text{Co}}_{3} \left( {{\text{Al}}_{0.5} ,{\text{Ti}}_{0.5} } \right)}}\) being the energy of the Co3(Al0.5,Ti0.5) compound and \(E_{{Co_{3} Al}}\) l and \(E_{{Co_{3} Ti}}\) being the energies of the two competing binary compounds. If this mixing energy is negative, mixing of elements Al and Ti on the second sublattice is energetically favorable and Co3(Al0.5,Ti0.5) will not transform into a mixture of Co3Al and Co3Ti.
In the present case, the mixing energies of different L12 structures with mixing of Co and Ni on the first sublattice and/or Al and Ti on the second sublattice were calculated relative to combinations of Ni3Al, Ni3Ti, Co3Al and Co3Ti. It is noteworthy that the energy of the D024-Ni3Ti phase is close to the one of the L12-Ni3Ti compound evidenced by their similar energies of formation as shown in Figure 2. Thus, calculating the energies of mixing using the equilibrium D024 ground state of Ni3Ti instead of L12 would not change the overall trends. The energies of mixing are shown in Figure 3. In addition to compounds with equiatomic mixing on the second sublattice, also compounds with content ratios of 3 to 1 on the second sublattice are shown. In the different L12-structures, a mixture of Al and Ti on the second sublattice is always favored. For a first sublattice occupied only by Co, an Al-rich composition on the second sublattice and for a first sublattice occupied only by Ni, a Ti-rich composition on the second sublattice yields the lowest energies of mixing. Considering the first sublattice, compounds only containing Ni or Co are energetically favorable compared to a mixture of Co and Ni. From these phases, the energetically most stable one is Ni3(Al0.25,Ti0.75).

Energies of mixing of different L12-structures of Co3(Al1−x,Tix) (red symbols), Ni3(Al1−x,Tix) (blue symbols) and (Co0.5,Ni0.5)3(Al1−x,Tix) (black symbols) type relative to a combination of binary Ni3Al, Ni3Ti, Co3Al and Co3Ti (Color figure online)
Vamsi and Karthikeyan reported energies of mixing for L12-Ni3(Alx−1,Tix) compounds.[37,39] Qualitatively their results resemble the stabilizing effect of Ti in the compound. However, in their first publication[37] while the curve for energy of mixing shows a slight asymmetry to the Ti-rich side still a Ti content of 0.5 on the second sublattice is predicted to be the most stable. In their newer publication the minimum of the energy of mixing curve is at a content of 0.75 Ti corresponding to the results shown here while the value of about 0,02 eV/atom found by them is slightly higher. The discrepancy in the Ti content yielding the lowest energy of mixing may stem from the fact that a regular solution model was used in one reference[37] while a full DFT treatment using a supercell approach was used in the other reference.[39] Only the approach using supercells could resemble the deviation of Ti mixing from a regular solution behavior.
As already mentioned, the above authors performed experimental studies on an alloy with the nominal composition Co-30Ni-15Cr-5Al-5Ti (all at. pct).[35] The alloy exhibited a two phase microstructure consisting of a Co solid solution matrix (γ phase) and two size fractions of L12-type precipitates (primary and secondary γ′). The chemical compositions of those phases were measured with atom probe and the results are listed in Table I. It is noteworthy that Cr is mainly enriched in the γ matrix and thus it seems justified to model the γ′-L12 phase as only containing Co, Ni, Al and Ti for the DFT calculations. Taking a closer look especially at the data for the primary γ′ it is found that some of the findings of the experimental measurement are in agreement with the DFT calculations while others are not. The experimentally found mixture of Al and Ti with a preference for an increased content of Ti on the second sublattice can be explained directly by the energy of the different L12 compounds (even if the tendency to enrich Ti on the second sublattice is more pronounced in the calculations). However, the mixing of Ni and Co on the first sublattice found in the experiment is not predicted. There are different possible explanations for this difference. One is that the role of the γ matrix as a competing phase was not considered in the DFT calculations. If it is energetically unfavorable for the system consisting of both γ and γ′ phase to enrich all Co in the γ matrix, it could be that some Co is pushed back into the γ′ phase to optimize the energy balance of the whole alloy. In addition, the specimen measured by atom probe was heat treated at 900 °C for 200 hours. Thus, it is necessary to also discuss thermal contributions to the energy of the phases while the DFT calculations only include the electronic contribution to the energy at 0 K. While as already stated a complete first principle-based thermodynamic description of the phases is far beyond the scope of the present paper, a quick estimation about the effect of configurational entropy, which should favor mixing in a phase, is possible. The configurational entropy can be calculated using the following formula:
Here k is the Boltzmann constant, i the sublattice, fi the fraction of the sublattice i and cj the concentration of element j on the respective sublattice. If applying this formula on the compounds Ni3(Al0.25,Ti0.75) and (Ni0.75,Co0.25)3(Al0.25,Ti0.75), the former being the most stable one predicted by DFT and the latter being similar to the composition as found by atom probe, it yields a contribution to free energy of − 0.01421 eV/atom for Ni3(Al0.25,Ti0.75) and − 0.05684 eV/atom for (Ni0.75,Co0.25)3(Al0.25,Ti0.75) at 900 °C. Thus, the contribution of configurational entropy can explain the mixing of Co and Ni on the first sublattice at 900 °C as the gain in free energy due to mixing based on the calculated configurational entropy is in the range of differences in mixing energies as shown in Figure 3.
3.2 Electronic Densities of States
To gain a deeper insight into the stability and bonding conditions of the L12 phases in the Co–Ni-Al-Ti system total and partial electronic densities of states (tDOS and pDOS) were calculated for (Co0.5,Ni0.5)3(Al0.5,Ti0.5), Ni3(Al0.5,Ti0.5) and Ni3(Al0.25,Ti0.75). These three compounds were chosen because Ni3(Al0.25,Ti0.75) is the most stable one predicted by the DFT calculations while Ni3(Al0.5,Ti0.5) and (Co0.5,Ni0.5)3(Al0.5,Ti0.5) should indicate the effects of mixing on only one and on both sublattices. The tDOS are shown in Figure 4. All compounds show a clear separation between binding states below the Fermi level (EF indicated as dashed line in Figure 4) and anti-binding states above the Fermi level separated by a pseudo gap. This indicates that the compounds all show a covalent contribution to the character of bonding.[48] However, the compounds differ if we take a closer look at the density of states directly at the Fermi level and the distance between the Fermi level and the pseudo gap. In general, a phase is considered to be the more stable, the lower the density of states is at the Fermi level and the closer the Fermi level is located to the pseudo gap.[49,50,51] Accordingly the Ni3(Al0.25,Ti0.75) compound should be the most stable followed by Ni3(Al0.5,Ti0.5) and (Co0.5,Ni0.5)3(Al0.5,Ti0.5) being the least stable of the three. This is also mirrored in the order of formation energies which is − 0.4849, − 0.4730, and − 0.3200 eV/atom, respectively. The trend that Co addition on the first sublattice causes a higher density of states at the Fermi level and a larger separation between pseudo gap and Fermi level is also present if only a fraction of 0.25 (i.e., about the Co content found in experiments) of the atomic positions of the first sublattice are occupied by Co (results shown in supplementary material). The more clearly pronounced pseudo gap located closer to the Fermi level EF in both Co-free L12-phases stems from the more pronounced hybridization of binding states in those two compounds compared to (Co0.5,Ni0.5)3(Al0.5,Ti0.5). The origin of this more pronounced hybridization of binding states can be deduced by taking a closer look at the pDOS.

Total electronic density of states (tDOS) for (from top to bottom) (Co0.5,Ni0.5)3(Al0.5,Ti0.5), Ni3(Al0.5,Ti0.5) and Ni3(Al0.25,Ti0.75)
From the pDOS shown in Figure 5, it is clearly visible that the overlap between Al p-orbitals and especially Ti d-orbitals with the Ni and Co d-orbitals in (Co0.5,Ni0.5)3(Al0.5,Ti0.5) is not very localized. However, in Ni3(Al0.5,Ti0.5) and most pronounced in Ni3(Al0.25,Ti0.75) the Ti d-orbitals and Ni d-orbitals show a strong localized overlap below the Fermi level. This results in the strong hybridization of states below the Fermi level in both Co-free L12 phases explaining their higher stability compared to (Co0.5,Ni0.5)3(Al0.5,Ti0.5).

Partial electronic density of states (pDOS) for (from left to right) (Co0.5,Ni0.5)3(Al0.5,Ti0.5), Ni3(Al0.5,Ti0.5) and Ni3(Al0.25,Ti0.75), Co in purple, Ni in green, Al in red and Ti in blue (Color figure online)
3.3 Elastic Properties
In Table II, different elastic constants for the three compounds (Co0.5,Ni0.5)3(Al0.5,Ti0.5), Ni3(Al0.5,Ti0.5) and Ni3(Al0.25,Ti0.75) are listed. It is important to note that the tensor of elastic constants calculated using a SQS supercell does not resemble the symmetry expected for the cubic L12-structure (i.e., C11=C22=C33, C12=C13 = C23 and C44=C55=C66) because this cubic symmetry is broken in the SQS supercell. The elastic tensor reported here was symmetrized using the procedure as proposed in.[52] The polycrystalline elastic constants bulk modulus B, shear modulus G and Young’s modulus Y are calculated according to Hill’s method.[53] All three compounds fulfill the elastic stability criterion for cubic crystals with C11 + 2C12 > 0, C11–|C12|> 0 and C44 > 0.[54] The so-called Pugh ratio B/G is often considered as an indicator of ductile or brittle behavior of a phase. Possessing a B/G above 1.754 a phase is considered as ductile.[55] Another indicator for ductility and the character of bonding is the so-called Cauchy pressure C12–C44. A positive Cauchy pressure is interpreted as a more metallic bonding character and it was proposed by Pettifor[56] to be also a sign for ductile behavior. Therefore, all three compounds can be considered as ductile owing to the more metallic like character of their bonding. It is noteworthy that the Cauchy pressure and the Pugh ratio between the three compounds show the same trend. Ni3(Al0.5,Ti0.5) with the highest Pugh ratio has also the highest Cauchy pressure and Ni3(Al0.25,Ti0.75) the lowest of both values.
If Co and Ni are mixed on the first sublattice C11 and C12 tend to decrease while C44 increases judging from the comparison of values for (Co0.5,Ni0.5)3(Al0.5,Ti0.5) and Ni3(Al0.5,Ti0.5) in Table I. Also a decrease in the bulk modulus and an increase of shear and Young’s modulus is found. It is noteworthy that an addition of 0.25 Co on the first sublattice still exhibits C11 close to Ni3(Al0.5,Ti0.5) while C12 is closer to (Co0.5,Ni0.5)3(Al0.5,Ti0.5) and C44 in between. Also the shear and Young’s modulus of a (Co0.25,Ni0.75)3(Al0.5,Ti0.5) compound are close to the values of (Co0.5,Ni0.5)3(Al0.5,Ti0.5) but the bulk modulus shows a more linear dependence from Co content on the first sublattice (see data in supplementary material).
Theoretical predictions how the elastic constants of a L12-Ni3Al compound should change if Ti is added are reported by Wu and Li[36] and Vamsi and Karthikeyan.[39] The results in Reference 36 partly correspond with the values reported here also showing a slight increase in C11 but C12 stays nearly on the same level while C44 increases instead of being nearly unaffected as found here. It has to be noted that the values reported in[36] stem from a 32 atom supercell with only one Al atom replaced by Ti, i.e., a strongly diluted solution, which may explain the discrepancies. The results of Vamsi and Karthikeyan are of more interest because values for Ni3(Al0.5,Ti0.5) and Ni3(Al0.25,Ti0.75) are provided.[39] Thus, a direct comparison with the results presented here is possible. Judging from the graphs in[39] C11 increases from about 250 GPa to slightly more than 270 GPa if the Ti content on the second sublattice is raised from 0.5 to 0.75. This is exactly the change, which is also found in the present paper. Also C12 is lowered from slightly above 150 GPa for Ni3(Al0.5,Ti0.5) with increasing Ti content to 0.75 on the second sublattice but only by about 5 GPa and not to the extent found here. Thus C44 is nearly unaffected by changes of the Ti content and exhibits values slightly below 130 GPa, which is a common result of[39] and the present work. Thus, it can be concluded that the results reported here are in good agreement with literature data in[39] and the discrepancies if comparing with[36] are most probably due to the fact that the change in elastic constants follows no linear trend with Ti addition and thus values from strongly diluted compounds cannot be extrapolated.
It is intriguing to speculate how the elastic constants could influence the strength and the microstructure. The shear modulus is a parameter occurring in the equation for the line energy of dislocations[57] and thus also in different equations for hardening contributions against dislocation movement.[58,59] Therefore, the higher shear modulus of Ni3(Al0.25,Ti0.75) compared to Ni3(Al0.5,Ti0.5) and (Co0.5,Ni0.5)3(Al0.5,Ti0.5) should be favorable with respect to mechanical strength of the γ′ precipitates. However in superalloys, dislocation movement is usually initiated in the softer γ-matrix and only subsequently dislocations cut into the γ′-precipitates.[60] Due to this, a larger Young’s and shear modulus may also be advantageous due to the larger coherency stresses they induce to hinder dislocations to cut into the γ′ precipitates.[61] However, in this consideration of course also the elastic properties of the matrix and the lattice mismatch between the phases γ and γ′ play a role. It could be that one of those parameters or a combination of them counter balance the beneficial effect of a higher Young’s and shear modulus of γ′. In addition, while a phase having higher Young’s and shear modulus at 0 K may retain this advantage also at elevated temperatures there is a certain possibility that the order is changed at higher temperature due to thermal effects.
The shape of γ′ precipitates is influenced by a number of factors including elastic constants of γ and γ′, the mismatch between the two phases, the γ′ precipitate size and the crystallographic anisotropy of the elastic tensor of the γ and γ′ phase.[62] The fact that frequently cubic γ′ precipitates form in superalloys instead of globular ones, while the latter would exhibit a more favorable ratio between surface and volume, is caused by the elastic anisotropy.[55] For a given lattice mismatch, the resulting coherency stresses depend on the elastic constant parallel to the precipitate matrix interface. Thus, a γ′ morphology with interfaces parallel to directions of low elastic moduli results. These are the < 001 > directions and the resulting shape is cubic.[55] Comparing the anisotropy of the elastic properties of the three compounds (Co0.5,Ni0.5)3(Al0.5,Ti0.5), Ni3(Al0.5,Ti0.5) and Ni3(Al0.25,Ti0.75) they exhibit different levels of elastic anisotropy as visible by the anisotropy factors AU and A in Table II: Different elastic properties of the compounds (Co0.5,Ni0.5)3(Al0.5,Ti0.5), Ni3(Al0.5,Ti0.5) and Ni3(Al0.25,Ti0.75)—all values except for Pugh ratio B/G and anisotropy factors AU/A in GPa (Table II). These are defined as \(A^{U} = 5\left( {G_{V} /G_{R} } \right) + \left( {B_{V} + B_{R} } \right) - 6\) with \(G_{V}\), \(G_{R}\), \(B_{V}\) and \(B_{R}\) being the shear and bulk moduli in the Voigt and Reuss averaging[63] and \(A = \left( {2C_{44} } \right)/\left( {C_{11} - C_{12} } \right)\).[25,57] AU as proposed by Zener is only applicable for cubic crystals. The higher the value the more pronounced the anisotropy indicating that (Co0.5,Ni0.5)3(Al0.5,Ti0.5) has the highest with Ni3(Al0.5,Ti0.5) nearly on the same level while the one of Ni3(Al0.25,Ti0.75) is significantly lower. Also an addition of 0.25 Co on the first sublattice exhibits a low anisotropy as can be seen from data provided in supplementary. This is also evident from the 3D representations of the Young’s modulus as shown in Figure 6. If the γ′ phase exhibits a higher elastic anisotropy this should promote a more cubic γ′ precipitate morphology.

3D Young’s modulus bodies of (from left to right) (Co0.5,Ni0.5)3(Al0.5,Ti0.5), Ni3(Al0.5,Ti0.5) and Ni3(Al0.25,Ti0.75)
4 Conclusions
Different properties of L12-phases in the Co-Ni-Al-Ti system were calculated using DFT and SQS supercells to depict the partially disordered structures. The calculation results were compared with experimental findings on a similar system and the main results are as follows:
-
A Ni3(Al0.25,Ti0.75) L12-phase showed the highest stability. While it is energetically favorable to have only Ni occupying the first sublattice, mixing of Al and Ti on the second sublattice with an enrichment of Ti is promoted.
-
With respect to mixing of Al and Ti, the enrichment of Ti as predicted by the DFT calculations is in good agreement with experimental results.
-
A small content of Co on the first sublattice found in the experiment and in contradiction with DFT calculations can be explained by an effect of configurational entropy.
-
The stability of the L12-phase Ni3(Al0.25,Ti0.75) is also reflected in the electronic densities of states showing a low density at the Fermi level, which is located close to the pseudo gap. This stems from the hybridization of the Ti-d and Ni-d orbitals. While Ni3(Al0.5,Ti0.5) shows a similar shape of the density of states, the density of states at the Fermi level is higher for (Co0.5,Ni0.5)3(Al0.5,Ti0.5) and the Fermi level is farer away from the pseudo gap explaining the lower stability of this phase.
-
The Ni3(Al0.25,Ti0.75) L12-phase exhibits the highest Young’s and shear modulus compared to Ni3(Al0.5,Ti0.5) and (Co0.5,Ni0.5)3(Al0.5,Ti0.5) but the lowest elastic anisotropy.
References
Ch. S. Lee: Precipitation-Hardening Characteristics of Ternary Cobalt-Aluminium-X Alloys, Dissertation, The University of Arizona, 1971.
J. Sato, T. Omori, K. Oikawa, I. Ohnuma, R. Kainuma, and K. Ishida: Science, 2006, vol. 312, pp. 90–91.
M. Ooshima, K. Tanaka, N.L. Okamoto, K. Kishida, and H. Inui: J. Alloys Compds., 2010, vol. 508, pp. 71–78.
J.R. Craig, D.J. Vaughan, and J.B. Higgins: Mater. Res. Bull., 1979, vol. 14, pp. 149–54.
H. Okamoto: J. Phase Equilib. Diffus., 2009, vol. 30, p. 123.
Q.X. Fan, S.M. Jiang, H.J. Yu, J. Gong, and C. Sun: Appl Surf Sci, 2014, vol. 311, pp. 214–23.
M. Tsunekane, A. Suzuki, and T.M. Pollock: Intermetallics, 2011, vol. 19, pp. 636–43.
P. Liu, R. Zhang, Y. Yuan, Ch. Cui, F. Liang, X. Liu, Y. Gu, Y. Zhou, and X. Sun: J. Mater. Sci. Technol., 2021, vol. 77, pp. 66–81.
S. Kobayashi, Y. Tsukamoto, T. Takasugi, H. Chinen, T. Omori, K. Ishida, and St. Zaefferer: Intermetallics, 2009, vol. 17, pp. 1085–89.
E.A. Lass, M.E. Williams, C.E. Campbell, K.W. Moon, and U.R. Kattner: J. Phase Equilib. Diffus., 2014, vol. 35, pp. 711–23.
Y. Li, F. Pyczak, M. Oehring, L. Wang, J. Paul, U. Lorenz, and Z. Yao: J. Alloy Compd., 2017, vol. 729, pp. 266–76.
H.-Y. Yan, V.A. Vorontsov, and D. Dye: Intermetallics, 2014, vol. 48, pp. 44–53.
J.P. Minshull, S. Neumeier, M.G. Tucker, and H.J. Stone: Adv. Mater. Res., 2011, vol. 278, pp. 399–404.
S.K. Makineni, B. Nithin, and K. Chattopadhyay: Scr. Mater., 2015, vol. 98, pp. 36–39.
A. Bhowmik, St. Neumeier, S. Rhode, and H.J. Stone: Intermetallics, 2015, vol. 59, pp. 95–101.
F. Xue, H.J. Zhou, Q.Y. Shi, X.H. Chen, H. Chang, M.L. Wang, and Q. Feng: Scr. Mater., 2015, vol. 97, pp. 37–40.
Ch.. Zenk, I. Povstugar, R. Li, F. Rinaldi, St.. Neumeier, D. Raabe, and M. Göken: Acta Mater., 2017, vol. 135, pp. 244–51.
P. Pandey, S. Mukhopadhyay, C. Srivastava, S.K. Makineni, and K. Chattopadhyay: Mater. Sci. Eng. A, 2020, vol. 790, p. 139578.
F.R. Long, S.I. Baik, D.W. Chung, F. Xue, E.A. Lass, D.N. Seidman, and D.C. Dunand: Acta Mater., 2020, vol. 196, pp. 396–408.
K. Shinagawa, T. Omori, J. Sato, K. Oikawa, I. Ohnuma, R. Kainuma, and K. Ishida: Mater. Trans., 2008, vol. 49, pp. 1474–79.
C. Jiang: Scripta Mater., 2008, vol. 59, pp. 1075–78.
M. Chen and Ch.-Y. Wang: J. Appl. Phys., 2010, vol. 107, p. 093705.
A. Mottura, A. Janotti, and T.M. Pollock: Intermetallics, 2012, vol. 28, pp. 138–43.
J. Koßmann, T. Hammerschmidt, S. Maisel, S. Müller, and R. Drautz: Intermetallics, 2015, vol. 64, pp. 44–50.
Q. Yao, S.L. Shang, K. Wang, F. Liu, Y. Wang, Q. Wang, T. Lu, and Z.-K. Liu: J. Mater. Res., 2017, vol. 32, pp. 2100–08.
W. Li and C. Wang: Chin. Phys. B, 2020, vol. 29, p. 026102.
P. Pandey, S.K. Makineni, A. Samanta, A. Sharma, S.M. Das, B. Nithin, C. Srivastava, A.K. Singh, D. Raabe, B. Gault, and K. Chattopadhyay: Acta Mater., 2019, vol. 163, pp. 140–53.
W.W. Xu, J.J. Han, Z.W. Wang, C.P. Wang, Y.H. Wen, X.J. Liua, and Z.Z. Zhu: Intermetallics, 2013, vol. 32, pp. 303–11.
S.R. Joshi, K.V. Vamsi, and S. Karthikeyan: MATEC Web Conf., 2014, vol. 14, p. 18001.
M. Jin, N. Miao, W. Zhao, J. Zhou, Q. Du, and Z. Sun: Comput. Mater. Sci., 2018, vol. 148, pp. 27–37.
W.Y. Wang, F. Xue, Y. Zhang, S.-L. Shang, Y. Wang, K.A. Darling, L.J. Kecskes, J. Li, X. Hui, Q. Feng, and Z.-K. Liu: Acta Mater., 2018, vol. 145, pp. 30–40.
Z. Fan, X. Wang, Y. Yang, H. Chen, Z. Yang, and C. Zhang: Metall. Mater. Trans. A, 2020, vol. 51A, pp. 2495–2508.
W.-W. Xu, Z.Y. Xiong, X.G. Gong, G.H. Yin, L.J. Chen, C.P. Wang, and X.J. Liu: Materialia, 2021, vol. 18, p. 101171.
J.E. Saal and C. Wolverton: Acta Mater., 2013, vol. 61, pp. 2330–38.
M. Chandran and S.K. Sondhi: Modell. Simul. Mater. Sci. Eng., 2011, vol. 19, p. 025008.
Q. Wu and Sh. Li: Comput. Mater. Sci., 2012, vol. 53, pp. 436–43.
K. V. Vamsi and S. Karthikeyan in: Superalloys 2012: 12th international symposium on superalloys, 2012, eds.: E.C. Huron, R. Reed et al., The Minerals, Metals and Materials Society, pp. 521–30.
A. Breidi, J. Allen, and A. Mottura: Acta Mater., 2018, vol. 145, pp. 97–108.
K.V. Vamsi and S. Karthikeyan: J. Alloys Compds., 2021, vol. 860, p. 158511.
Z.D. Liang, S. Neumeier, Z.Y. Rao, M. Göken, and F. Pyczak: Mater. Sci. Eng. A, 2022, vol. 854, p. 143798.
G. Kresse and J. Furthmüller: Phys. Rev. B, 1996, vol. 54, pp. 11169–86.
G. Kresse and J. Furthmüller: Comput. Mater. Sci., 1996, vol. 6, pp. 15–50.
G. Kresse and D. Joubert: Phys. Rev. B, 1999, vol. 59, p. 1758.
J.P. Perdew, K. Burke, and M. Ernzerhof: Phys. Rev. Lett., 1996, vol. 77, p. 3865.
H.J. Monkhorst and J.D. Pack: Phys. Rev. B, 1976, vol. 13, p. 5188.
A. van de Walle: Calphad, 2009, vol. 33, pp. 266–78.
A. Breidi, J. Allen, and A. Mottura: Phys. Status Solidi B, 2017, vol. 254, p. 1600839.
C. Wolverton and A. Zunger: Phys. Rev. B, 1995, vol. 52, pp. 8813–28.
J.-H. Xu, T. Oguchi, and A.J. Freeman: Phys. Rev. B, 1987, vol. 36, pp. 4186–89.
W. Lin, J.-H. Xu, and A.J. Freeman: Phys. Rev. B, 1992, vol. 45, pp. 10863–71.
J.-H. Xu, W. Lin, and A.J. Freeman: Phys. Rev. B, 1993, vol. 48, pp. 4276–86.
J. von Pezold, D. Alexey, M. Friak, and J. Neugebauer: Phys. Rev. B, 2010, vol. 81, p. 094203.
G. Grimvall: Thermophysical Properties of Materials, North-Holland, Amsterdam, 1999.
M. Born and K. Huang: Dynamical Theory of Crystal Lattices, Clarendon Press, Oxford, 1954.
H. Niu, X.Q. Chen, P. Liu, W. Xing, X. Cheng, D. Li, and Y. Li: Sci. Rep., 2012, vol. 2, p. 718.
D.G. Pettifor: Mater. Sci. Technol., 1992, vol. 8, pp. 345–49.
J. Friedel: Dislocations, in Elastic Theory of Dislocations. J. Friedel, ed., Pergamon Press, Oxford, 1964, pp. 17–48.
J. Friedel: Dislocations, in Nature of the Interaction with Impurities. J. Friedel, ed., Oxford, Pergamon Press, 1964, pp. 351–67.
J. Friedel: Dislocations, in Hardness of a Crystal Containing Uniformly Distibuted Impurities or Precipitates. J. Friedel, ed., Pergamon Press, Oxford, 1964, pp. 368–84.
B. Reppich: Acta Metall., 1982, vol. 30, pp. 87–94.
A.J. Ardell: Metall. Mater. Trans. A, 1985, vol. 16A, pp. 2131–65.
M. Doi, T. Miyazaki, and T. Wakatsuki: Mater. Sci. Eng., 1984, vol. 67, pp. 247–53.
S.I. Ranganathan and M. Ostoja-Starzewski: Phys. Rev. Lett., 2008, vol. 101, p. 055504.
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pyczak, F., Liang, Z., Neumeier, S. et al. Stability and Physical Properties of the L12-γ′ Phase in the CoNiAlTi-System. Metall Mater Trans A 54, 1661–1670 (2023). https://doi.org/10.1007/s11661-022-06949-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-022-06949-y