
Abstract
Background/Aims
Acute hyperglycemia is known to worsen ischemia/reperfusion (I/R) injury following myocardial infarction and stroke. We investigated whether acute hyperglycemia worsens injury and amplifies the inflammatory response evoked by hepatic I/R.
Methods
Rats were pretreated with an intraperitoneal injection of 25% glucose or 0.9% sodium chloride (10 ml/kg BW). Subsequently, rats underwent partial (70%) hepatic ischemia for 45 min. After 4 h of reperfusion, hepatic injury, oxidative stress, inflammation, and heat shock protein expression were assessed.
Results
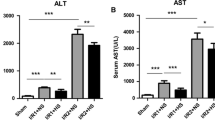
Liver injury was increased in the hyperglycemic group with alanine aminotransferase (ALT) and aspartate aminotransferease (AST) serum concentrations of 7,832 ± 3,374 and 10,677 ± 4,110 U/L compared to 3,245 ± 2,009 and 5,386 ± 3,393 U/L (p < 0.05 vs. control). Hyperglycemic I/R was associated with increased liver nitrotyrosine concentrations and increased neutrophil infiltration. I/R upregulated the protective heat shock proteins HSP32 and HSP70 in control animals, but this protective mechanism was inhibited by hyperglycemia: HSP32 expression decreased from 1.97 ± 0.89 (control) to 0.46 ± 0.13 (hyperglycemia), HSP70 expression decreased from 18.99 ± 11.55 (control) to 3.22 ± 0.56 (hyperglycemia), (expression normalized to sham, both p < 0.05 vs. control I/R).
Conclusions
Acute hyperglycemia worsens hepatic I/R injury by amplifying oxidative stress and the inflammatory response to I/R. The increase in injury is associated with a downregulation of the protective heat shock proteins HSP32 and HSP70.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute hyperglycemia is frequently seen in hospitalized patients and induced by stressors such as acute illness and surgical trauma. Such transient increases in blood glucose concentrations may put patients at risk for adverse outcomes. Hyperglycemia independent of preexisting diabetes mellitus is an established risk factor for increased mortality and morbidity after cardiac surgery.1 Patients without a history of diabetes who were hyperglycemic at admission to the hospital had higher mortality and lower functional outcomes than normoglycemic and even hyperglycemic diabetic patients.2 Van den Berghe et al.3 showed that intensive insulin therapy (IIT) reduces in-hospital mortality in surgical intensive care unit patients by 34% with subsequent investigations confirming that maintaining normoglycemia rather than glycemia-independent effects of insulin is responsible for the beneficial effects of IIT.4,5 These findings emphasize the potential hazards of poor glucose control on patient outcome.
The detrimental effects of hyperglycemia do not require chronic exposure or preexisting diabetes. Animal models of acute hyperglycemia confirm the deleterious effects of even short episodes of hyperglycemia on cerebral6 and renal ischemia/reperfusion (I/R) injury.7 Proposed mechanisms for the detrimental effects of acute hyperglycemia are increased oxidative stress, an enhanced inflammatory response with cytokine activation8,9 and impaired blood flow with reperfusion.10
Diabetic mice have been shown to be more susceptible to liver ischemia,11,12 but so far, the effects of acute hyperglycemia on liver I/R injury have not been addressed. We therefore used a rat model of acute hyperglycemia to investigate its effects on hepatic I/R injury.
Material and Methods
Animal Model
All animal experiments were carried out with approval by the local committee on animal research. Animal care was in agreement with the National Institutes of Health guidelines for ethical research (NIH publication no. 80-123, revised 1985). Inbred male Lewis rats (Harlan, Indianapolis, IN, USA) were used for this study. Animals’ weights on arrival at our facility were 250–300 g. Animals had access to standard laboratory diet and were maintained on a light–dark cycle. They were fasted 12 h prior to the start of the experiments. Prior to the study, animals spent several days in the animal care facility for acclimatization.
The rats were divided into hyperglycemic and control group. In the hyperglycemic group (HG, n = 8), 2.5 g/kg glucose (25% solution) was injected intraperitoneally following the assessment of the baseline glucose serum concentration. The control group (CON, n = 8) received 10 ml/kg 0.9% saline instead. Thirty minutes later, rats were anesthetized with isoflurane. Following liver exposure through a midline incision and collection of blood samples, hepatic ischemia was induced. Applying a 70% liver ischemia model, the liver was mobilized, and vascular structures to the left and median lobe were identified and clamped for 45 min using a bulldog clamp. The unoccluded right and caudate lobe allow outflow from the splanchnic circulation, thus avoiding venous congestion. For the duration of hepatic ischemia, the abdominal cavity was closed with clamps. Rectal temperature was continuously assessed using an electronic thermometer (RSP TM-200D, Respiratory Support Products Inc., Santa Ana, CA, USA using a Mallinckrodt probe, cat no. 502-0401, Mallinckrodt Inc., St. Louis, MO, USA) and held constant at 37°C using a heating lamp.
Following reperfusion, the animals received 5 ml of normal saline intraperitoneally, and the incision was closed in two layers. Animals were killed following a 4 h observation period. Blood and tissue were harvested. All tissue was immediately frozen in liquid nitrogen and stored at −80°C until further processing.
Sham experiments (Sham, n = 5) served as reference for subsequent analysis. Sham experiments were identical to control I/R experiments except that hepatic vessels were not clamped. Hyperglycemic sham experiments (HG Sham, n = 4) were added to the protocol to identify the effects of hyperglycemia alone.
Biochemical Markers of Liver Injury
Serum levels of aspartate aminotransferease (AST) and alanine aminotransferase (ALT) were determined at baseline and following 4 h of reperfusion. The analysis was done in the General Laboratory, San Francisco General Hospital, University of California, San Francisco.
Histology
Liver samples were fixed in 10% buffered formalin and processed for routine histology. Five-micron paraffin-embedded tissue sections were stained with hematoxylin and eosin and examined using standard light microscopy by a pathologist (R.R.) who was blinded to the experimental condition of the animals. Sections were scored from 0–4 for sinusoidal congestion, vacuolization of hepatocyte cytoplasm, and parenchymal necrosis as described by Suzuki et al. (Table 1).13
Intrahepatic Neutrophil Accumulation Assessment
Activity of myeloperoxidase (MPO), an enzyme stored in the azurophilic granules of neutrophils, was used to measure tissue neutrophil sequestration. We used a spectrophotometric method to assay tissue MPO activity. Frozen livers were thawed and extracted for MPO following homogenization and sonication. The assay is based on the oxidation of 3,3′,5,5′-tetramethyl benzydine by MPO in the presence of H2O2. Units of MPO activity were calculated using a standard curve derived from a MPO standard sample (Calbiochem, EMD Bioscience, La Jolla, CA, USA). MPO data are expressed as microunits per milligram of tissue per minute.
Protein Isolation and Western Blots
All steps for protein isolation were conducted at 4°C. Snap-frozen liver sections were homogenized in Tissue Protein Extraction Reagent (Pierce Biotechnology, Rockford, IL) containing 1 mm EDTA and 1:100 Protease Cocktail Inhibitor (Sigma, St. Louis, MO) and were centrifuged at 10,000×g for 5 min. The supernatant was aliquoted, snap-frozen, and stored at −80°C. Protein concentrations of liver homogenates were measured by the Pierce bicinchoninic acid protein assay with bovine serum albumin as the standard. Fifty micrograms of liver homogenates was separated on a Novex-NuPAGE 10% Bis-Tris sodium dodecyl sulfate polyacrylamide gel electrophoresis gel (Invitrogen) and transferred to nitrocellulose membrane using the XCell SureLock system (Invitrogen). A mouse anti-heat shock protein 70 (HSP70) monoclonal antibody (SC-24) and a goat anti-actin antibody (SC-1616) from Santa Cruz Biotechnology (Santa Cruz, CA) were used. In addition, a mouse anti-heat shock protein 32 (HSP32) monoclonal antibody from Stressgen (Ann Arbor, MI), a mouse anti-nitrotyrosine antibody from Abcam Inc. (Cambridge, MA), and a rabbit anti-cleaved caspase-3 monoclonal antibody (CST 9661) from Cell Signaling Technology (Danvers, MA) were used for the Western blots. The membranes were incubated with a 1:100 or 1:1,000 dilution of the primary antibody followed by a 1:10,000-fold dilution of a secondary anti-mouse or anti-goat immunoglobulin G from Santa Cruz Biotechnology. Immunoreactive proteins were developed using SuperSignal West Dura (Pierce Biotechnology) and visualized on the FluorChem 5500 Imaging system from Alpha Innotech (San Leandro, CA). Band intensities were quantified via spot densitometry.
Statistical Analysis
All data are presented as mean ± SD. Comparison between study groups was performed using analysis of variance with post hoc Dunnett correction, with normoglycemic sham animals serving as controls. Comparison of the two ischemic groups alone was done using a two-tailed unpaired t test. p values < 0.05 were considered as being statistically significant.
Results
Intraperitoneal injection and surgery alone resulted in an increase in serum glucose concentrations from 89 ± 19 baseline to 182 ± 32 mg/dL in the saline pre-treated group. In the glucose-pretreated group, three animals were not considered for subsequent analysis due to an only moderate increase in serum glucose concentrations (<250 mg/dL, 30 min after treatment). In the five remaining animals, serum glucose concentrations before ischemia increased from 107 ± 32 to 360 ± 32 mg/dL. At the end of the 4-h reperfusion period, serum glucose concentrations remained higher in the glucose pretreated group (Fig. 1).

Intraperitoneal injection of saline and surgery alone increased serum glucose concentrations in control animals. Intraperitoneal injection of 2.5 g glucose/kg resulted in significantly higher serum glucose concentrations. After 4 h of reperfusion, glucose concentrations were still significantly higher in hyperglycemic animals. *p < 0.05 vs. control.
Serum Marker of Liver Injury
Serum transaminase concentrations following I/R were higher in the glucose-pretreated animals: 7,832 ± 3,374 vs. 3,245 ± 2,009 U/L (ALT, p < 0.05) and 10,677 ± 4,119 vs. 5,385 ± 3,393 U/L (AST, p < 0.05). Transaminase concentrations after 4 h of reperfusion were correlated with glucose concentrations before ischemia of all animals that entered the study (Fig. 2).

Correlation of serum glucose concentrations before the start of ischemia and serum concentrations of ALT (a) and AST (b) after 4 h of reperfusion. Liver injury, as assessed by transaminase concentrations and glucose concentrations, was correlated with correlation coefficients of r = 0.70 (ALT) and r = 0.68 (AST).
Histology
Both experimental groups showed liver damage including vacuolization and at least minimal congestion and single-cell necrosis (Fig. 3). Damage was graded using the Suzuki score. There was no statistical difference between hyperglycemic animals and control animals in Suzuki scores (6.0 ± 2.2 vs. 6.1 ± 1.8) or necrosis scores (2.0 ± 0.8 vs. 2.0 ± 0.8). Whether individual cell death after 4 h of reperfusion was attributable to necrosis or apoptosis could not be determined by histology. Using nuclear features to distinguish between types of cell death is not considered reliable,14 and both experimental groups demonstrated zonal as well as spotty areas of dead hepatocytes (Fig. 3).

Sham-treated animals show no significant congestion at low power (a ×100). Vacuolization and cellular necrosis are not evident in periportal hepatocytes (b ×200) or in centrizonal areas. Hepatic architecture is unremarkable. In contrast, control animals following 45 min of ischemia and 4 h of reperfusion show diffuse, moderate sinusoidal congestion, with numerous sinusoidal channels distended by red blood cells in several areas of the liver section (c ×200). Two dying hepatocytes (likely evolving into Councilman bodies) are visible in the center of the field (c). Mild parenchymal vacuolization is visible in some hepatocytes, with several hepatocytes in this field showing irregular nuclear contours, chromatin condensation, pyknosis, and nuclear dust, histologic evidence of cell damage, and evolving cell death (d ×400). Moderate sinusoidal congestion also is seen in hyperglycemic animals treated with high levels of dextrose before ischemia (e ×200). This particular animal showed both zones of necrosis and patchy single-cell necrosis. Cell damage is seen in several adjacent hepatocytes containing vacuolated cytoplasm, pyknotic nuclei, and nuclear dust. These dying hepatocytes also show paler cytoplasm than their undamaged counterparts nearby (f ×400).
Apoptosis
Cleaved caspase-3 expression was higher in control animals (2.12 ± 0.47) vs. hyperglycemic animals (1.49 ± 0.42) when compared to sham animals (1.00 ± 0.10), indicating more apoptotic cells in livers from control animals Fig. (4)

Cleaved caspase-3 expression was highest in control animals, suggesting preferential apoptotic cell death in animals that were not pretreated with glucose. Data are presented as mean ± SD. *p < 0.05 vs. sham.
Oxidative Stress
Nitrotyrosine concentrations after 4 h of reperfusion were higher in hyperglycemic animals (1.63 ± 0.54-fold when compared to control animals (1.00 ± 0.30), p < 0.05) indicating increased oxidative stress resulting in nitration of tyrosine residues of proteins by peroxynitrite.
Inflammation
MPO activity in the liver after 4 h of reperfusion was higher in glucose-pretreated animals (5,383 ± 2,512 vs. 2,219 ± 2,086 mU/mg protein−1 min−1, p < 0.05), indicating increased neutrophil migration into the hepatic tissue of hyperglycemic animals (Fig. 5).

Myeloperoxidase activity was increased in liver homogenates of hyperglycemic animals when compared to control animals, indicating increased neutrophil accumulation after 4 h of reperfusion. Data are presented as mean ± SD. *p < 0.05 vs. sham, # p < 0.05 vs. control I/R.
Heat Shock Protein activation
I/R increased HSP32 expression in control but suppressed HSP32 expression in hyperglycemic animals (1.97 ± 0.89-fold vs. 0.46 ± 0.13-fold when normalized to sham animals, p < 0.05; Fig. 6a). Hyperglycemia alone without I/R (hyperglycemic sham) did not affect HSP32 expression (0.92 ± 0.20 vs. 1.00 ± 0.16 when compared to control sham). I/R increased HSP70 expression in control animals more than in hyperglycemic animals (19.99 ± 11.55-fold vs. 3.22 ± 0.56-fold when normalized to sham, p < 0.05; Fig. 6b). Again, hyperglycemia alone without I/R did not affect HSP70 expression (0.94 ± 0.08 vs. 1.00 ± 0.15) when compared to control sham.

Heat shock protein expression as assessed by Western blot for HSP32 (hemeoxygenase-1) (a) and HSP70 (b). I/R resulted in a distinct activation of both HSP32 and HSP70 expression in control animals. However, hyperglycemia ameliorated the activation of HSP70 by I/R and suppressed HSP32 expression. Densitometric values were normalized to actin and are expressed as ratios of sham ± SD. *p < 0.05 vs. sham, # p < 0.05 vs. control I/R.
Discussion
Acute hyperglycemia during hepatic ischemia amplified the inflammatory response and resulted in elevated transaminase concentrations following I/R. The elevated serum glucose concentration at the start of ischemia seemed to be responsible for the increase in injury, as there was a strong correlation between serum glucose concentrations before ischemia and transaminase concentration after 4 h of reperfusion. We used a transient model of hyperglycemia starting only shortly before ischemia. Serum glucose concentrations were still higher in hyperglycemic animals at the end of the 4 h reperfusion period, albeit the graph (Fig. 1) clearly demonstrates a declining trend.
Hepatic I/R has been reported to result in hepatocyte death by two different pathways, necrosis and apoptosis. Whether apoptotic or necrotic cell death predominates following liver I/R has been the subject of debate. Based on terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, it was suggested that sinusoidal endothelial cells and then subsequently hepatocytes undergo apoptosis but rarely necrosis following 60 min of liver ischemia.15 However, a later study applying a very similar ischemia model found only few apoptotic cells and predominantly necrosis following 60 min of ischemia when combining TUNEL with morphological criteria.16 A subsequent review emphasized that apoptosis and necrosis share features and mechanisms that can make discrimination between both forms of cell death very challenging. In particular, the TUNEL assay is not suited to differentiate between necrosis and apoptosis, since DNA fragmentation was reported in apoptosis as well as necrosis.17
In the present study, histological assessment of the liver samples after 4 h of reperfusion could not reliably distinguish between apoptotic and necrotic cell death. A longer reperfusion could potentially facilitate histological analysis, but 4 h of reperfusion was chosen to enable the detection of inflammatory mediators. As a result, the histological scores were basically identical in both experimental groups, in spite of serologic evidence for increased necrotic cell death in hyperglycemic animals. The higher transaminase concentrations measured in the hyperglycemic group after 4 h of reperfusion reflect increased cellular breakdown due to necrosis with release of intracellular enzymes. Apoptosis maintains the barrier function of the cell membrane and would contribute only to a minor extent to elevated transaminase concentrations. Caspase-3 activation is considered the most reliable method for the detection of apoptosis.14 We assessed caspase-3 activation to quantify the amount of apoptotic cell death and found higher apoptosis scores in the control group. The lower caspase-3 activation in the hyperglycemic group may be further evidence that necrosis, not apoptosis, is the preferential form of cell death in hyperglycemic conditions.
Hyperglycemia per se is known to increase oxidative stress and to cause a proinflammatory state.8 Furthermore, hyperglycemia has been shown to amplify the inflammatory response caused by stressors such as LPS administration.18 Our results support the hypothesis that the mechanisms responsible for increased ischemic injury by hyperglycemia are the amplification of oxidative stress and of the inflammatory response normally seen with I/R. The increased concentration of nitrotyrosine containing protein is an established marker for severe oxidative stress. Reactive oxygen species, such as the superoxide radical, react with NO to form the more potent peroxynitrite species, which then subsequently nitrate tyrosine residues of proteins, leading to inactivation of key cellular proteins, DNA damage, and eventually cell death.19 While Kupffer cell-induced oxidative stress is considered the first step in I/R injury,20 it is followed by a profound inflammatory response that is largely responsible for the extent of I/R injury. This inflammatory response culminates in the hepatic accumulation of neutrophils, which directly damage hepatocytes by releasing oxidants and proteases. The MPO assay confirmed an increased neutrophil infiltration in the liver tissue of hyperglycemic animals. This neutrophil migration and infiltration is initiated by the production and release of cytokines such as tumor necrosis factor alpha and interleukin-6. Earlier studies demonstrated that hyperglycemia enhances cytokine production in response to stress.21
A surprising finding of the present investigation was the downregulation of HSP32 and HSP70 in hyperglycemic animals undergoing I/R. Hepatic I/R normally results in upregulation of HSPs, and the observed effects in livers from hyperglycemic animals differ distinctly from the situation in kidneys7 and the brain,22 where hyperglycemic I/R injury is associated with an increased activation of HSPs. We could demonstrate in sham experiments that hyperglycemia alone was not responsible for the downregulation of HSPs (data not shown) but that the combination of hyperglycemia and I/R is required to block or even suppress HSP activation. Since HSPs are one of the most potent protective mechanisms against I/R injury, it can be assumed that their suppression in hyperglycemic I/R contributes to the increased injury during acute hyperglycemia. Inhibition of HSP activation in response to ischemia has so far not been described in other organs and may represent a liver-specific (mal-)adaptation to hyperglycemia: It has been described before that diabetes does inhibit hepatic HSP70 activation by heat stress,23 although subsequent studies did not confirm this finding.24,25
The mechanism responsible for the downregulation of HSPs remains to be defined. The expression of the heat shock genes encoding the different HSPs is regulated by heat shock transcription factors (HSFs), which are normally bound to HSPs within the cytosol. When cells are exposed to stress, HSFs are phosphorylated and form trimers that enter the nucleus and bind the heat shock elements located within the promoter of heat shock genes, thus initiating increased expression of HSPs.26 It has been hypothesized that, in diabetes, the activation of HSF is inhibited in insulin-sensitive tissue.27 In type 2 diabetic primates, livers had reduced HSP70 and HSP90 tissue concentrations that were related to 50% lower levels of the transcription factor heat shock factor 1.28 But again, these results are challenged by a study that showed similar heat shock factor 1 content in livers from control and streptozotocin treated rats following heat stress.24 Further interventional studies with activation of HSPs are planned to show whether the suppression of HSP activation is responsible for the worsened injury during hyperglycemia and whether activation of HSPs is capable of reversing such detrimental effects.
Conclusions
Acute hyperglycemia worsened liver injury as assessed by increased transaminase concentrations following hepatic I/R in rats. The effects of hyperglycemia on liver injury were associated with increased hepatic oxidative stress, an increased inflammatory response, and a suppression of HSP activation. These results, in spite of their descriptive nature, emphasize the need to better understand the role of hyperglycemia in organ injury, especially in clinical scenarios associated with a risk for ischemia. Glucose concentrations in this study were overall high, suggesting that glucose control may not need to be very aggressive to have beneficial effects. Preventing severe hyperglycemia alone may reduce I/R injury, thus avoiding the inherent risk of an IIT to cause undesired hypoglycemia.
References
Doenst T, Wijeysundera D, Karkouti K, Zechner C, Maganti M, Rao V, Borger MA. Hyperglycemia during cardiopulmonary bypass is an independent risk factor for mortality in patients undergoing cardiac surgery. J Thorac Cardiovasc Surg 2005;130:1144.
Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002;87:978–982.
van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in the critically ill patients. N Engl J Med 2001;345:1359–1367.
Van den Berghe G, Wouters PJ, Bouillon R, Weekers F, Verwaest C, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P. Outcome benefit of intensive insulin therapy in the critically ill: Insulin dose versus glycemic control. Crit Care Med 2003;31:359–366.
Ellger B, Debaveye Y, Vanhorebeek I, Langouche L, Giulietti A, Van Etten E, Herijgers P, Mathieu C, Van den Berghe G. Survival benefits of intensive insulin therapy in critical illness: impact of maintaining normoglycemia versus glycemia-independent actions of insulin. Diabetes 2006;55:1096–1105.
Martin A, Rojas S, Chamorro A, Falcon C, Bargallo N, Planas AM. Why does acute hyperglycemia worsen the outcome of transient focal cerebral ischemia? Role of corticosteroids, inflammation, and protein O-glycosylation. Stroke 2006;37:1288–1295.
Hirose R, Xu F, Dang K, Liu T, Behrends M, Brakeman PR, Wiener-Kronish J, Niemann CU. Transient hyperglycemia affects the extent of ischemia–reperfusion-induced renal injury in rats. Anesthesiology 2008;108:402–414.
Ling PR, Mueller C, Smith RJ, Bistrian BR. Hyperglycemia induced by glucose infusion causes hepatic oxidative stress and systemic inflammation, but not STAT3 or MAP kinase activation in liver in rats. Metabolism 2003;52:868–874.
Iwasaki Y, Kambayashi M, Asai M, Yoshida M, Nigawara T, Hashimoto K. High glucose alone, as well as in combination with proinflammatory cytokines, stimulates nuclear factor kappa-B-mediated transcription in hepatocytes in vitro. J Diabetes Complications 2007;21:56–62.
Giugliano D, Marfella R, Coppola L, Verrazzo G, Acampora R, Giunta R, Nappo F, Lucarelli C, D’Onofrio F. Vascular effects of acute hyperglycemia in humans are reversed by L-arginine. Evidence for reduced availability of nitric oxide during hyperglycemia. Circulation 1997;95:1783–1790.
Rogers H 3rd, Zibari GB, Roberts J, Turnage R, Lefer DJ. Nitric oxide attenuates ischaemia–reperfusion (I/R) injury in the diabetic liver. Clin Transplant 2004;18(Suppl 12):7–11.
Elrod JW, Duranski MR, Langston W, Greer JJ, Tao L, Dugas TR, Kevil CG, Champion HC, Lefer DJ. eNOS gene therapy exacerbates hepatic ischemia–reperfusion injury in diabetes: a role for eNOS uncoupling. Circ Res 2006;99:78–85.
Suzuki S, Nakamura S, Koizumi T, Sakaguchi S, Baba S, Muro H, Fujise Y. The beneficial effect of a prostaglandin I2 analog on ischemic rat liver. Transplantation 1991;52:979–983.
Saraste A. Morphologic criteria and detection of apoptosis. Herz 1999;24:189–195.
Kohli V, Selzner M, Madden JF, Bentley RC, Clavien PA. Endothelial cell and hepatocyte deaths occur by apoptosis after ischemia–reperfusion injury in the rat liver. Transplantation 1999;67:1099–1105.
Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of cell death during warm hepatic ischemia–reperfusion in rats: apoptosis or necrosis? Hepatology 2001;33:397–405.
Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology 2003;125:1246–1257.
Brix-Christensen V, Gjedsted J, Andersen SK, Vestergaard C, Nielsen J, Rix T, Nyboe R, Andersen NT, Larsson A, Schmitz O, Tonnesen E. Inflammatory response during hyperglycemia and hyperinsulinemia in a porcine endotoxemic model: the contribution of essential organs. Acta Anaesthesiol Scand 2005;49:991–998.
Raina AK, Perry G, Nunomura A, Sayre LM, Smith MA. Histochemical and immunocytochemical approaches to the study of oxidative stress. Clin Chem Lab Med 2000;38:93–97.
Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia–reperfusion injury in rat liver. Am J Physiol 1991;260:G355–362.
Ling PR, Smith RJ, Bistrian BR. Hyperglycemia enhances the cytokine production and oxidative responses to a low but not high dose of endotoxin in rats. Crit Care Med 2005;33:1084–1089.
Muranyi M, He QP, Fong KS, Li PA. Induction of heat shock proteins by hyperglycemic cerebral ischemia. Brain Res Mol Brain Res 2005;139:80–87.
Yamagishi N, Nakayama K, Wakatsuki T, Hatayama T. Characteristic changes of stress protein expression in streptozotocin-induced diabetic rats. Life Sci 2001;69:2603–2609.
Najemnikova E, Rodgers CD, Locke M. Altered heat stress response following streptozotocin-induced diabetes. Cell Stress Chaperones 2007;12:342–352.
Swiecki C, Stojadinovic A, Anderson J, Zhao A, Dawson H, Shea-Donohue T. Effect of hyperglycemia and nitric oxide synthase inhibition on heat tolerance and induction of heat shock protein 72 kDa in vivo. Am Surg 2003;69:587–592.
Santoro MG. Heat shock factors and the control of the stress response. Biochem Pharmacol 2000;59:55–63.
Hooper PL. Insulin signaling, GSK-3, heat shock proteins and the natural history of type 2 diabetes mellitus: a hypothesis. Metab Syndr Relat Disord 2007;5:220–230.
Kavanagh K, Zhang L, Wagner JD. Tissue-specific regulation and expression of heat shock proteins in type 2 diabetic monkeys. Cell Stress Chaperones 2009;14:291–299.
Acknowledgments
This study was supported by grants from the Research Evaluation and Allocation Committee (REAC) of the University of California San Francisco (M.B.) and the Ministerio de Educacion y Ciencia, Spain (G.M.P.)
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Behrends, M., Martinez-Palli, G., Niemann, C.U. et al. Acute Hyperglycemia Worsens Hepatic Ischemia/Reperfusion Injury in Rats. J Gastrointest Surg 14, 528–535 (2010). https://doi.org/10.1007/s11605-009-1112-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11605-009-1112-3