
Abstract
We have successfully ported an arbitrary high-order discontinuous Galerkin method for solving the three-dimensional isotropic elastic wave equation on unstructured tetrahedral meshes to multiple Graphic Processing Units (GPUs) using the Compute Unified Device Architecture (CUDA) of NVIDIA and Message Passing Interface (MPI) and obtained a speedup factor of about 28.3 for the single-precision version of our codes and a speedup factor of about 14.9 for the double-precision version. The GPU used in the comparisons is NVIDIA Tesla C2070 Fermi, and the CPU used is Intel Xeon W5660. To effectively overlap inter-process communication with computation, we separate the elements on each subdomain into inner and outer elements and complete the computation on outer elements and fill the MPI buffer first. While the MPI messages travel across the network, the GPU performs computation on inner elements, and all other calculations that do not use information of outer elements from neighboring subdomains. A significant portion of the speedup also comes from a customized matrix–matrix multiplication kernel, which is used extensively throughout our program. Preliminary performance analysis on our parallel GPU codes shows favorable strong and weak scalabilities.
Similar content being viewed by others

1 Introduction
Computer simulations of seismic wavefields have been playing an important role in seismology in the past few decades. However, the accurate and computationally efficient numerical solution of the three-dimensional elastic seismic wave equation is still a very challenging task, especially when the material properties are complex, and the modeling geometry, such as surface topography and subsurface fault structures, is irregular. In the past, several numerical schemes have been developed to solve the elastic seismic wave equation. The finite-difference (FD) method was introduced to simulate SH and P-SV waves on regular, staggered-grid, two-dimensional meshes in Madariaga (1976) and Virieux (1984, 1986). The FD method was later extended to three spatial dimensions and to account for anisotropic, visco-elastic material properties (e.g., Mora 1989; Igel et al. 1995; Tessmer 1995; Graves 1996; Moczo et al. 2002). The spatial accuracy of the FD method is mainly controlled by the number of grid points required to accurately sample the wavelength. The pseudo-spectral (PS) method with Chebychev or Legendre polynomials (e.g., Carcione 1994; Tessmer and Kosloff 1994; Igel 1999) partially overcomes some limitations of the FD method and allows for highly accurate computations of spatial derivatives. However, due to the global character of its derivative operators, it is relatively cumbersome to account for irregular modeling geometries, and efficient and scalable parallelization on distributed-memory computer clusters is not as straightforward as in the FD method. Another possibility is to consider the weak (i.e., variational) form of the seismic wave equation. The finite-element (FE) method (e.g., Lysmer and Drake 1972; Bao et al. 1998) and the spectral-element (SE) method (e.g., Komatitsch and Vilotte 1998; Komatitsch and Tromp 1999, 2002) are based on the weak form. An important advantage of such methods is that the free-surface boundary condition is naturally accounted for even when the surface topography is highly irregular. And in the SE method, high-order polynomials (e.g., Lagrange polynomials defined on Gauss–Lobatto–Legendre points) are used for approximation, which provides a significant improvement in spatial accuracy and computational efficiency.
The arbitrary high-order discontinuous Galerkin (ADER-DG) method on unstructured meshes was introduced to solve two-dimensional isotropic elastic seismic wave equation in Käser and Dumbser (2006). It was later extended to three-dimensional isotropic elastic case in Dumbser and Käser (2006) and to account for viscoelastic attenuation (Käser et al. 2007), anisotropy (la Puente De et al. 2007) and poroelasticity (la Puente et al. 2008). The p-adaptivity (i.e., the polynomial degrees of the spatial basis functions can vary from element to element) and locally varying time steps were addressed in Dumbser et al. (2007). Unlike conventional numerical schemes, which usually adopt a relatively low-order time-stepping method such as the Newmark scheme (Hughes 1987) and the 4th-order Runge–Kutta scheme (e.g., Igel 1999), the ADER-DG method achieves high-order accuracy in both space and time by using the arbitrary high-order derivatives (ADER), which was originally introduced in Toro (1999) in the finite-volume framework. The ADER scheme performs high-order explicit time integration in a single step without any intermediate stages. In three dimensions, the ADER-DG scheme achieves high-order accuracy on unstructured tetrahedral meshes, which allows for automated mesh generation even when the modeling geometry is highly complex. Furthermore, a majority of the operators in the ADER-DG method are applied in an element-local way, with weak element-to-element coupling based on numerical flux functions, which results in strong locality in memory access patterns. And the high-order nature of this method lets it require fewer data points, therefore, fewer memory fetches, in exchange for higher arithmetic intensity. These characteristics of the ADER-DG method make it well suited to run on massively parallel graphic processing units (GPUs).
In the past four decades, the development in the computing chip industry has been roughly following the Moore’s law. Many of the performance improvements were due to increased clock speeds and sophisticated instruction scheduling in a single core. As the transistor density keeps increasing, the industry is now facing a number of engineering difficulties with using a large number of transistors efficiently in individual cores (e.g., power consumption, power dissipation). The effect is that clock speeds are staying relatively constant, and core architecture is expected to become simpler, if changes much at all. As a consequence, when we consider future platforms for high-performance scientific computing, there are some inevitable trends, for instance, the increase in the number of cores in general-purpose CPUs and the adoption of many-core accelerators (e.g., Field Programmable Gate Array, Graphic Processing Unit, Cell Broadband Engine) due to their smaller footprints and lower power consumptions than general-purpose CPUs. The users who want to once again experience substantial performance improvements as before need to learn how to exploit multiple/many cores. The GPU is becoming an attractive co-processor for general-purpose scientific computing due to its high arithmetic computation power, large memory bandwidth, and relatively lower costs and power consumptions per floating-point operation (FLOP), when compared with a typical CPU. A typical GPU (e.g., NVIDIA GeForce 9800) can reach a peak processing rate of 700 GFLOPS (1 GFLOPS = 109 floating-point-operations per second) and a peak memory bandwidth of 70 Gigabyte/s. Unlike in a conventional CPU, in a GPU, many more transistors are dedicated for data processing rather than data caching or flow control, which makes GPUs particularly well suited to address problems that can be expressed as data-parallel computations. Recent efforts by GPU vendors, in particular, NVIDIA’s CUDA (Compute Unified Device Architecture) programming model, the OpenCL (Open Computing Language) framework, and the OpenACC compiler directives and Application Programming Interfaces (APIs), have significantly increased the programmability of commodity GPUs. Using these tools, a programmer can directly issue and manage data-parallel computations on GPUs using high-level instructions without the need to map them into a set of graphic-processing instructions.
With the rapid development of the GPU programming tools, various numerical algorithms have been successfully ported to GPUs, and GPU-CPU hybrid computing platforms and substantial speedups, compared with pure-CPU implementations, have been achieved for applications in different disciplines. The discontinuous Galerkin (DG) method for solving the 3D Maxwell’s equations, which are linear, hyperbolic systems of conservation laws similar to the seismic wave equation, has been successfully mapped to GPU using NVIDIA’s CUDA framework and achieved more than an order of magnitude speed-up compared with the same implementation on a single current-generation CPU (Klöckner et al. 2009). The GPU implementation was done on a single NVIDIA GTX 280, which costs around $400. A significant portion of the speed-up came from the high-order nature of the DG method. In the area of acoustic/elastic seismic wave propagation simulations, the finite-difference and the spectral-element methods have been successfully implemented on CPU-GPU hybrid clusters using the CUDA programming model (e.g., Abdelkhalek et al. 2009; Komatitsch et al. 2009; Komatitsch et al. 2010; Michéa and Komatitsch 2010; Okamoto et al. 2010; Wang et al. 2010). The speedup obtained varies from around 20× to around 60× depending on several factors, e.g., whether a particular calculation is amenable to GPU acceleration, how well the reference CPU code is optimized, the particular CPU and GPU architectures used in the comparisons, and the specific compilers, as well as the compiler options, used for generating the binary codes.
In Mu et al. (2013), we successfully ported the ADER-DG method for solving three-dimensional elastic seismic wave equation to a single NVIDIA Tesla C2075 GPU using CUDA and obtained a speedup factor of about 24 when compared with the serial CPU code running on one Intel Xeon W5880 core. In this article, we explore the potential of accelerating the ADER-DG method using multiple NVIDIA Fermi GPUs with CUDA and the Message-Passing Interface (MPI). Our reference CPU code is a community code named “SeisSol.” The “SeisSol” code was written in Fortran 90 and parallelized using the Message-Passing Interface (MPI). It implements the ADER-DG method for solving the three-dimensional seismic wave equation in different types of material properties (e.g., elastic, visco-elastic, anisotropic, poroelastic). It has been optimized for different types of CPU architectures and applied extensively in seismic wave propagation simulations related to earthquake ground-motion prediction, volcano seismology, seismic exploration, and dynamic earthquake rupture simulations. For a complete list of the references of its applications, please refer to the “SeisSol Working Group” website or la Puente et al. (2009). The lessons learned in our implementation and optimization experiments may also shed some light on how to port this type of algorithms to GPUs more effectively using other types of GPU programming tools such as OpenCL and OpenACC.
2 CUDA programming model
For readers who are not familiar with CUDA or GPU programming, we give a very brief introduction about the programming model in this section. The CUDA software stack is composed of several layers, including a hardware driver, an application programming interface (API), and its runtime environment. There are also two high-level, extensively optimized CUDA mathematical libraries, the fast Fourier transform library (CUFFT) and the basic linear algebra subprograms (CUBLAS), which are distributed together with the software stack. The CUDA API comprises an extension to the C programming language for a minimum learning curve. The complete CUDA programming toolkit is distributed free of charge and is regularly maintained and updated by NVIDIA.
A CUDA program is essentially a C program with multiple subroutines (i.e., functions). Some of the subroutines may run on the “host” (i.e., the CPU), and others may run on the “device” (the GPU). The subroutines that run on the device are called CUDA “kernels.” A CUDA kernel is typically executed on a very large number of threads to exploit data parallelism, which is essentially a type of single-instruction-multiple-data (SIMD) calculation. Unlike on CPUs where thread generation and scheduling usually take thousands of clock cycles, GPU threads are extremely “light-weight” and cost very few cycles to generate and manage. The very large amounts of threads are organized into many “thread blocks.” The threads within a block are executed in groups of 16, called a “half-warp,” by the “multiprocessors” (a type of vector processor), each of which executes in parallel with the others. A multiprocessor can have a number of “stream processors,” which are sometimes called “cores.” A high-end Fermi GPU has 16 multiprocessors, and each multiprocessor has two groups of 16 stream processors, which amounts to 512 processing cores.
The memory on a GPU is organized in a hierarchical structure. Each thread has access to its own register, which is very fast, but the amount is very limited. The threads within the same block have access to a small pool of low-latency “shared memory.” The total amount of registers and shared memory available on a GPU restricts the maximum number of active warps on a multiprocessor (i.e., the “occupancy”), depending upon the amount of registers and shared memory used by each warp. To maximize occupancy, one should minimize the usage of registers and shared memory in the kernel. The most abundant memory type on a GPU is the “global memory”; however, accesses to the global memory have much higher latencies. To hide the latency, one needs to launch a large number of thread blocks so that the thread scheduler can effectively overlap the global memory transactions for some blocks with the arithmetic calculations on other blocks. To reduce the total number of global memory transactions, each access needs to be “coalesced” (i.e., consecutive threads accessing consecutive memory addresses), otherwise the access will be “serialized” (i.e., separated into multiple transactions), which may heavily impact the performance of the code.
In addition to data-parallelism, GPUs are also capable of task parallelism which is implemented as “streams” in CUDA. Different tasks can be placed in different streams, and the tasks will proceed in parallel despite the fact that they may have nothing in common. Currently task parallelism on GPUs is not yet as flexible as on CPUs. Current-generation NVIDIA GPUs now support simultaneous kernel executions and memory copies either to or from the device.
3 Overview of the ADER-DG method
The ADER-DG method for solving the seismic wave equation is both flexible and robust. It allows unstructured meshes and easy control of accuracy without compromising simulation stability. Like the SE method, the solution inside each element is approximated using a set of orthogonal basis functions, which leads to diagonal mass matrices. These types of basic functions exist for a wide range of element types. Unlike the SE or typical FE schemes, the solution is allowed to be discontinuous across element boundaries. The discontinuities are treated using well-established ideas of numerical flux functions from the high-order finite-volume framework. The spatial approximation accuracy can be easily adjusted by changing the order of the polynomial basis functions within each element (i.e., p-adaptivity). The ADER time-stepping scheme is composed of three major ingredients, a Taylor expansion of the degree-of-freedoms (DOFs, i.e., the coefficients of the polynomial basis functions in each element) in time, the solution of the Derivative Riemann Problem (DRP) (Toro and Titarev 2002) that approximates the space derivatives at the element boundaries and the Cauchy-Kovalewski procedure for replacing the temporal derivatives in the Taylor series with spatial derivatives. We summarize major equations of the ADER-DG method for solving the three-dimensional isotropic elastic wave equation on unstructured tetrahedral meshes in the following. Please refer to Dumbser and Käser (2006) for details of the numerical scheme.
The three-dimensional elastic wave equation for an isotropic medium can be expressed using a first-order velocity-stress formulation and written in a compact form as
where is a 9-vector consisting of the six independent components of the symmetric stress tensor and the velocity vector , and , and are space-dependent 9 × 9 sparse matrices with the nonzero elements given by the space-dependent Lamé parameters and the buoyancy (i.e., the inverse of the density). Summation for all repeated indices is implied in all equations. The seismic source and the free-surface and absorbing boundary conditions can be considered separately as shown in Käser and Dumbser (2006) and Dumbser and Käser (2006).
Inside each tetrahedral element , the numerical solution can be expressed as a linear combination of space-dependent and time-independent polynomial basis functions of degree N with support on ,
where are time-dependent DOFs, and , , are coordinates in the reference element . Explicit expressions for the orthogonal basis functions on a reference tetrahedral element are given in Cockburn et al. (2000) and the appendix A of Käser et al. (2006). Bring Eq. (2) into Eq. (1), multiplying both sides with a test function , integrate over an element , and then apply integration by parts, we obtain,
The numerical flux between the element and one of its neighboring elements, , j = 1, 2, 3, 4, can be computed from an exact Riemann solver,
where is the rotation matrix that transforms the vector from the global Cartesian coordinate to a local normal coordinate that is aligned with the boundary face between the element and its neighbor element . Bring Eq. (4) into Eq. (3) and convert all the integrals from the global xyz-system to the -system in the reference element through a coordinate transformation, we obtain the semi-discrete discontinuous Galerkin formulation,
where is the determinant of the Jacobian matrix of the coordinate transformation being equal to 6 times the volume of the tetrahedron, is the area of face j between the element and its neighbor element , , , and are linear combinations of , , and with the coefficients given by the Jacobian of the coordinate transformation, , , and are the mass, stiffness, and flux matrices are given by
The mass, stiffness, and flux matrices are all computed on the reference element which means that they can be evaluated analytically beforehand using a computer algebra system (e.g., Maple, Mathematica) and stored on disk.
If we project Eq. (1) onto the DG spatial basis functions, the temporal derivative of the DOF can be expressed as
and the m-th temporal derivative can be determined recursively as
The Taylor expansion of the DOF at time is,
which can be integrated from to ,
where , and can be computed recursively using Eq. (8).
Considering Eq. (9), the fully discretized system can then be obtained by integrating the semi-discrete system, Eq. (5), from to
Equation (10), together with Eqs. (8) and (9), provides the mathematical foundation for our GPU implementation and optimization.
4 Implementation and optimization on multiple GPUs
Prior to running our wave-equation solver, a tetrahedral mesh for the entire modeling domain was generated on a CPU using the commercial mesh generation software “GAMBIT.” The mesh generation process is fully automated, and the generated tetrahedral mesh conforms to all discontinuities built into the modeling geometry, including irregular surface topography and subsurface fault structures. The entire mesh was then split into subdomains, one per GPU, using the open-source software “METIS” which is a serial CPU program for partitioning finite-element meshes in a way that minimizes inter-processor communication cost while maintaining load balancing.
4.1 Pre-processing
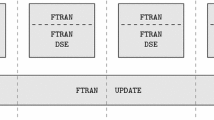
In Fig. 1, we listed the major steps in the reference parallel CPU code, “SeisSol” la Puente De et al. (2009), and those in our parallel CPU-GPU hybrid implementation. In our parallel CPU-GPU hybrid implementation, we assume that each MPI process has access to only one device, and each device is controlled by only one MPI process. At the start of the calculation, a sequence of pre-processing steps is executed on the CPUs. The pre-processing sequence includes:

The flowcharts of the major steps in the reference parallel CPU codes (left) and those in our CPU-GPU hybrid implementation (right). The whole calculation can be separated into three sections: pre-processing, time-stepping, and post-processing. The pre-processing section reads and calculates all the data that the time-stepping section will use. The time-stepping section updates the DOFs of each tetrahedral element according to Eqs. (8)–(10) and has been ported to the GPU. The post-processing section is in charge of writing out the DOFs and/or the seismograms at the pre-specified locations
-
(1)
reading and processing a control file;
-
(2)
reading and processing geometric information, which include the tetrahedral mesh, the boundary conditions, the material properties (i.e., density and Lamé parameters) for each element, and the mesh partitioning information generated by METIS;
-
(3)
for the elements in each subdomain, creating a list of all the elements that are in contact with elements in other subdomains, which we call the “outer” elements, and those that are not, which we call the “inner” elements;
-
(4)
reading and processing the DG matrices, which include the mass, stiffness, and flux matrices, which were pre-computed and stored on the disk; and
-
(5)
reading and processing the files describing the seismic source and seismic receivers.
Our CUDA program adopts the typical CUDA programming model. After the pre-processing sequence is carried out on the host CPUs, the arrays needed by the CUDA kernels are then copied to the global memory of the devices using “cudaMemcpy.” The hosts then call a sequence of CUDA kernels in every time step. The results of the simulation (e.g., the synthetic seismograms) are stored on the devices during the time loop and copied back to the hosts after all time steps are completed.
In our implementation, the calculation for each tetrahedral element is carried out by one thread block. Within each thread block, the number of threads depends upon the dimension of the element’s DOFs. The DOFs, , are allocated and initialized in the global memory of the device using “cudaMalloc” and “cudaMemset.” For a 5th-order scheme which is sufficiently accurate for most of our applications, the number of DOFs per component per element is 35. Considering the nine components of the governing PDE (i.e., six stress components and three velocity components), the DOFs of each element consist of a 9 × 35 matrix which is represented in memory as a one-dimensional array of length 315 organized in the column-major ordering. To obtain better memory alignment, we padded five zeros behind the DOFs of each element so that the length of the one-dimensional DOF array of each element is increased to 320, which is 10 times the number of threads in a warp. For a subdomain of “nElem” elements, the length of the one-dimensional DOF array for the whole subdomain is, therefore, “nElem × 320.” The amount of memory that is wasted on purpose is less than 1.6 %; however, the better memory alignment improved the performance of some simple operations such as summation operations and scalar-product operations by around 6.3 %.
4.2 Matrix–matrix multiplications
The implementation and optimization details for steps (1)–(5) are documented in Mu et al. (2013). In this section, we give a very brief summary. In those steps, most of the total wall-time is spent on matrix–matrix multiplications. We use step (2) which computes the volume contribution, as an example. A flowchart of the major calculations in step (2) is shown in Fig. 2a. Considering the first term on the right-hand-side of Eq. (10), the calculations in step (2) involve mathematical operations in the form of , where the time-integrated DOF , denoted as “dgwork” in Fig. 2a, is computed in step (1) and has the same dimension and memory layout as the DOF array. The multiplication between , denoted as “Kxi” in Fig. 2a, and the time-integrated DOF is different from the normal matrix–matrix product in linear algebra. This multiplication involves three steps: first, transpose the time-integrated DOF matrix, second, multiply with the stiffness matrix following the usual matrix–matrix product rule, third, transpose the matrix obtained in the previous step. We call this multiplication the “left-multiplication.” A code segment for the baseline CUDA implementation of the left-multiplication is shown in Fig. 2b. This left-multiplication operation is used extensively through the calculations in steps (1)–(4) and deserves more optimization effort. First, we can reduce the number of floating-point operations by exploiting the fact that some of the matrices in this operation are sparse; second, the order of the floating-point operations can be rearranged in a way such that the accesses to “dgwork” in the global memory are as coalesced as possible. The DOF, its temporal derivatives, and the time-integrated DOF are generally dense. However, the stiffness matrices and the Jacobians have fill-ratios ranging from 8.8 % to 29.6 %. To take advantage of this sparsity, one possibility is to adopt an existing sparse linear algebra library such as “CUSP” (Bell and Garland 2009) or cuSPARSE. However, the result we obtained using “CUSP” was not very satisfactory. The best performance gain, which was obtained using the “HYB” matrix format, was about 36.2 % compared with the baseline implementation shown in Fig. 2b. This is largely due to the very irregular distribution of the non-zeros in our matrices, which caused a large number of uncoalesced accesses to the time-integrated DOF arrays, and the amount of arithmetic calculations was not large enough to hide the memory access latencies due to the low fill-ratio in the stiffness matrix. Considering the fact that the locations of the non-zero elements in the stiffness matrices can be determined beforehand and are fixed throughout the program, the results of the left-multiplications can be evaluated analytically beforehand and expressed in terms of the non-zero elements in those matrices using a computer algebra system. The expressions of the left-multiplication results, which are linear combinations of the time-integrated DOF with coefficients given by the non-zero elements of the stiffness matrices, can be hardwired into the CUDA kernels. This implementation eliminates all redundant calculations involving zero elements and by carefully arranging the order of the calculations in accordance with the thread layout, we can also minimize the number of uncoalesced memory accesses to the time-integrated DOF array. A code segment of the optimized left-multiplication is shown in Fig. 2c, which is about 4 times faster than the baseline implementation shown in Fig. 2b. This approach can also be applied to normal matrix–matrix product and is used throughout steps (1)–(4) in our optimized CUDA codes. A drawback of this approach is that the resulting kernel source code is quite long, and some manual editing is required to ensure coalesced memory accesses. However, modern computer algebra systems (e.g., Mathematica, Maple) usually have automated procedures for translating long mathematical expressions into the C language which is usually error-proof and can be directly incorporated into the CUDA kernels with minimal effort.

a The flowchart of the calculations in step (2), the volume contributions. “dudt” is the volume contribution, “Kxi,” “Keta,” “Kzeta” correspond to the stiffness matrices, , , , in the text. “JacobianDet” is the determinant of the Jacobian . “nElem” is the number of tetrahedral elements in the subdomain, “nDegFr” is the number of DOFs per component per tetrahedral element, “nVar” is the number of components in the governing equation. “AStar,” “BStar,” “CStar” correspond to A*, B*, C* in the text. Code segments for the calculations in the dark-gray box are listed in b and c. b. Baseline implementation of the CUDA kernel for the “left-multiplication” between the time-integrated DOF and the stiffness matrix . “Kxi_dense” corresponds to the dense matrix representation of , “dgwork” corresponds to the time-integrated DOF, and the result of the multiplication is stored in “temp_DOF.” c A segment of the optimized CUDA kernel for the “left-multiplication” between the time-integrated DOF and the stiffness matrix . “Kxi_sparse” corresponds to the sparse matrix representation of . Meanings of other symbols are identical to those in Fig. b
Our own matrix–matrix multiplication scheme greatly contributes our CUDA codes. Before we settled for the final version of our single-GPU code, we also complete two other versions of CUDA implementations, one is the baseline implementation, which adapts conventional CUDA dense matrix–matrix multiplication, and the other is the median implementation which uses sparse matrix–matrix multiplication scheme. As a result, compared with the CPU based ADER-DG code, the baseline implementation obtains a speedup factor of 9.7×, and the median implementation improved the speedup factor from 9.7× to 13.2×. This improvement mainly gains from getting rid of all zeros elements’ computation; however, due to the very irregular distribution of non-zeros locality, this median implementation still almost 2 times slower than our final implementation, which enjoys a speedup factor of 24.3×.
4.3 Overlapping communication with computation
Considering Eqs. (8)–(10), the calculations on the devices within each time step can be organized into five major steps:
-
(1)
calculating the time-integrated DOFs, i.e., the term using the DOFs at the current time step through the Cauchy-Kovalewski procedure, i.e., Eqs. (8) and (9),
-
(2)
calculating the volume contributions, i.e., the first term on the right-hand-side of Eq. (10), using the time-integrated DOFs obtained in step (1),
-
(3)
calculating the first numerical flux term, i.e., the second term on the right-hand-side of Eq. (10), using the time-integrated DOFs obtained in step (1),
-
(4)
calculating the second numerical flux term, i.e., the third term on the right-hand-side of Eq. (10), using the time-integrated DOFs of the four neighboring elements obtained in step (1),
-
(5)
updating the DOFs to the next time step using the DOFs at the current time step , the volume contributions obtained in step (2) and the numerical flux terms obtained in steps (3) and (4), as well as any contributions from the seismic source, by using Eq. (10) which also involves inverting the mass matrix , which is diagonal.
All the calculations in steps (1), (2), (3), and (5) can be performed in an element-local way and require no inter-element information exchange, which is ideal for SIMD-type processors such as GPUs. The calculations in step (4) need to use the time-integrated DOFs from all neighboring elements which in our distributed-memory, parallel implementation requires passing time-integrated DOFs of the outer elements of each subdomain across different MPI processes. Most of this communication overhead can be hidden through overlapping computation with communication.
In our implementation, we calculate the time-integrated DOFs for all the outer elements of a subdomain first. The calculation of the time-integrated DOF requires access to the DOF array in the global memory. The DOFs of the outer elements are usually scattered throughout the entire DOF array of the subdomain. To avoid non-coalesced memory accesses, which could impact performance by up to 54 %, the entire DOF array is split into two sub-arrays, one for DOFs of all the outer elements and the other for the DOFs of all inner elements. Once we complete the calculations of the time-integrated DOFs of the outer elements, the device starts to compute the time-integrated DOFs of the inner elements of the subdomain right away. At the same time, the time-integrated DOFs of the outer elements are assembled into a separate array which is then copied into the host memory asynchronously to fill the MPI buffer using a separate CUDA stream, and then the host initiates a non-blocking MPI data transfer and returns. While the messages are being transferred, the device completes the calculations of the time-integrated DOFs of the inner elements, combines them with the time-integrated DOFs of the outer elements into a single time-integrated DOF array and proceeds to calculations of the volume contributions in step (2) and the first numerical flux term in step (3). On the host, synchronization over all the MPI processes is performed; once the host receives the array containing the time-integrated DOFs of the outer elements on the neighboring subdomains, it is copied to the device asynchronously using a separate CUDA stream. After completing step (3), the device synchronizes all streams to make sure that the required time-integrated DOFs from all neighboring subdomains have arrived and proceed to calculate the second numerical flux term in step (4) and then update the DOFs as in step (5). The overhead for splitting the entire DOF array into two sub-arrays for inner and outer elements and for combining the time-integrated DOFs of the outer and inner elements into a single array amounts to less than 0.1 % of the total computing time on the device. The entire process is illustrated in Fig. 1.
There are many factors can influent the speedup factor contribution by overlapping communication with computation; the overlapping benefit almost differs from computer to computer. Based on our completed experiments, the best scenario which all the GPUs located on the same node, the communication time takes 11.7 % of total computation time, while when each GPU locate on the different node, the ratio could rise up to 25.1 %. However, since we apply the multiple streams and the overlapping techniques, the overhead caused by communication could be eliminated.
To ensure effective communication-computation overlap, the ratio of the number of the outer to inner elements must be sufficiently small. An upper bound of this ratio can be estimated based on both the processing capability of the devices and the speed of the host-device and host–host inter-connections. On the NVIDIA Fermi M2070 GPUs that we experimented with, we achieved nearly zero communication overheads when this ratio is below 2 %. We note that if the same approach is implemented using a classic CPU cluster, this ratio can be much larger, since the calculations for the inner elements and steps (2) and (3) are over an order of magnitude slower on a CPU core.
5 Performance analysis
In this study, the speedup factor is defined as the ratio between the wall-time spent on running a simulation on a number of CPU cores with the wall-time spent on running the same simulation on the same or less number of GPUs. The CPU used as the reference is the Intel Xeon W5660 (2.80 GHz/12 MB L2 cache) processor, and the GPU is the NVIDIA Tesla C2070 (1.15 GHz/6 GB 384bit GDDR5) processor. Specifications of our CPU and GPU processors can be found on Intel and NVIDIA’s websites.
The speedup factor depends strongly upon how well the reference CPU code is optimized and sometimes also on the specific CPU compiler and compiling flags. The fastest executable on our CPU was obtained using the Intel compiler “ifort” with the flag “–O3.” The wall-time for running the CPU code in double-precision mode is only slightly longer than running the CPU code in single-precision mode by around 5 %. Our GPU codes were compiled using the standard NVIDIA “nvcc” compiler of CUDA version 4.0. Throughout this article, we use the double-precision version of the fastest CPU code as the reference for computing the speedup factors of our single- and double-precision GPU codes.
The accuracy of our single-precision GPU codes is sufficient for most seismological applications. The computed seismograms have no distinguishable differences from the seismograms computed using the double-precision version of the reference CPU code, and the energy of the waveform differences is much less than 1 % of the total energy of the seismogram.
5.1 Single-GPU performance
For our single-GPU performance analysis, we computed the speedup factors for problems with seven different mesh sizes (Fig. 3). The number of tetrahedral elements used in our experiments is 3,799, 6,899, 12,547, 15,764, 21,121, 24,606, and 29,335. The material property is constant throughout the mesh with density 3,000 kg/m3 and Lamé parameters 5.325 × 1010 and 3.675 × 1010 Pascal. We applied the traction-free boundary condition on the top of the mesh and absorbing boundary condition on all other boundaries. The seismic source is an isotropic explosive source buried in the center of the mesh. The wall-time measurements were obtained by running the simulations for 1,000 time steps. The speedup factors were computed for our single-precision GPU code with respect to the CPU code running on one, two, four, and eight cores. For the multi-core runs on the CPUs, the parallel version of the “SeisSol” code is used as the reference. For the seven different mesh sizes, the speedup factor ranges from 23.7 to 25.4 with respect to the serial CPU code running on one core, from 12.2 to 14 with respect to the parallel CPU code running on two cores, from 6.5 to 7.2 with respect to the parallel CPU code running on four CPU cores, and from 3.5 to 3.8 with respect to the parallel CPU code running on eight CPU cores. The speedup factor does not decrease linearly with increasing number of CPU cores. For instance, the speedup factor with respect to eight CPU cores is about 14 % better than what we would have expected considering the speedup factor with respect to one CPU core if we had assumed a linear scaling. For the parallel version of the CPU code, there are overheads incurred by the MPI communication among different cores, while for the single-GPU simulations, such communication overheads do not exist.

Single-GPU speedup factors obtained using 7 different meshes and 4 different CPU core numbers. The total number of tetrahedral elements in the 7 meshes is 3,799, 6,899, 12,547, 15,764, 21,121, 24,606, and 29,335, respectively. The speedup factors were obtained by running the same calculation using our CPU-GPU hybrid code with 1 GPU and using the serial/parallel “SeisSol” CPU code on 1/2/4/8 CPU cores on the same compute node. The black columns represent the speedup of the CPU-GPU hybrid code relative to 1 CPU core, the dark-gray columns represent the speedup relative to 2 CPU cores, the light gray column represents the speedup relative to 4 CPU cores and the lightest gray columns represent the speedup relative to 8 CPU cores
Since most of the calculations in the ADER-DG method are carried out in an element-local way, and there are no inter-process communication overheads on a single GPU, we expect the “strong scaling” on a single GPU, defined as the total wall-time needed to run the application on one GPU when the total number of elements (i.e., the amount of workload) is increased (e.g., Michéa and Komatitsch 2010), to be nearly linear. In Fig. 4, we show the total wall-time for 100 time steps as a function of the total number of tetrahedral elements. As we can see, the strong scaling of our codes on a single GPU is almost linear. The calculations in step (4) (i.e., the second term in the numerical flux) involve time-integrated DOFs of direct neighbors; however, this inter-element dependence keeps its spatially local character, while the number of elements increases and does not affect the scaling.

Strong scalability of our single GPU code with different mesh sizes. The black squares show the average wall-time per 100 time steps, and the number of elements varies from 800 to 57,920
5.2 Multiple-GPU performance
To analyze the performance of the parallel version of our CUDA codes, we use a simplified version of the SEG/EAGE salt model (Käser et al. 2010) as the benchmark. This model is geometrically complex, as shown in Fig. 5a and b. However, the generation of the tetrahedral mesh for such a complex model is highly automatic, once the geometries of the structural interfaces are imported into the meshing software. The material properties of the different zones in this model are summarized in Table 1. We note that a thin layer of water lies on top of the three-dimensional model. The ADER-DG method can accurately handle seismic wave propagation in water simply by setting the shear modulus of the elements in the water region to zero (Käser and Dumbser 2008).

This salt model is discretized into tetrahedral meshes with different number of elements. In Fig. 6, we show the speedup factors obtained for two different mesh sizes, one with 327,886 elements, and the other with 935,870 elements. The simulations were run on 8, 16, 32, and 48 CPU cores using the parallel version of the “SeisSol” code. And the speedup factors were obtained by running the same simulations on the same number of GPUs. On average, the speedup factor for our parallel GPU codes is around 28, which is slightly higher than the speedup factor obtained in the single-GPU-single-CPU comparison. This may due to the fact that in the parallel CPU code, the outer elements of a subdomain are not treated separately from the inner elements, which do not allow the parallel CPU code to overlap the computation on the inner elements with the communication of the time-integrated DOFs of the outer elements.

Speedup factors of our parallel GPU codes obtained using two different mesh sizes. The number of tetrahedral elements used in our experiments are 327,866, 935,870. The speed factors were computed for our single-precision multiple-GPUs code with respect to the CPU code running on 16/32/48/64 cores runs on different nodes
To investigate the strong scalability (i.e., the decrease in wall-time with increasing GPU number, while holding the total workload that is the number of elements and time steps, constant), we discretized the salt model using a mesh with about 1.92 million elements and ran the simulation for 100 times steps. The number of GPUs used in the simulations ranges from 32 to 64. As seen on Fig. 7, the strong scaling of our parallel GPU codes is close to the ideal case with some fluctuations. Our codes start to slightly underperform the ideal case when the number of GPUs used in the simulation is larger than 48. As analyzed in Sect. 4.2, to effectively overlap computation with communication, the ratio between the number of outer elements and the number of inner elements of a subdomain cannot exceed a certain threshold, which is determined by the processing capability of the GPU and the speed of the inter-connections. In our case, when the number of GPUs used in the simulation starts to exceed 48, this ratio becomes larger than 2 %, which we believe is the threshold for our hardware configuration. The performance of our parallel GPU codes depends upon a number of factors, such as load balancing, but we think the extra communication overhead that was not effectively hidden by the computation was the dominant factor for causing our codes to underperform the ideal case. In Fig. 8, we show the results of our weak scaling test (i.e., the workload per GPU is kept about constant while increasing the number of GPUs). By definition, the total number of elements in the weak scaling test increases approximately linearly with the number of GPUs. If the communication cost is effectively overlapped by computation, the weak scaling test should be approximately flat. In our tests, the average number of elements per GPU was kept around 53,000 with about 6 % fluctuation across different simulations. The ratio between the number of outer and inner elements was kept around 1 %. The weak scaling is approximately flat (Fig. 8), with some fluctuation mostly caused by the variation in the number elements per GPU used in each simulation.

Strong scalability of our multiple-GPUs codes with 1.92 million elements, the black line shows the average wall-time per 100 time steps for this size-fixed problem performed by 32–64 GPUs

Weak scalability of our multiple-GPUs code performed by 2–80 GPUs, the black line shows the average wall-time per 100 time steps for these size-varied problems. The average number of elements per GPU is around 53,000 with about 6 % fluctuation
5.3 Application examples
We have applied our multiple-GPUs code on two well-defined models, one is the SEG-EAGE Salt, and the other is marmousi2. All the results with these two models have been compared with CPU code SeisSol for validation.
For the marmousi2 model, we extrude its 2D original profile and make it a 3D model with dimension of 3,500 m in depth, 17,000 m in length, and 7,000 m in width (Fig. 9a). There are 379,039 tetrahedral elements, and each element has its own material property, also 337 receivers locate at 5 m beneath the surface along the A–A′ (yellow) line, and the horizontal interval is 50 m; the explosive source located at 10.0 m(depth), 7,500.0 m(length), 3,500.0 m(width). In this case, we used 16 M2070 Fermi GPUs located on eight different nodes, and each node has 2 GPUs. Our CUDA code uses 16 GPUs spend 4,812.64(s) to calculate 5 s seismogram, (Fig. 9b), while the SeisSol runs on 16 CPU cores need 135,278.50 s, the speedup factor of 28.11×.

a A perspective view of the 3D Marmousi2 model with dimension of 3,500 m in depth, 17,000 m in length, and 7,000 m in width. There are 379,039 tetrahedral elements, and each element has its own material property. There are 337 receivers locate at 5 m beneath the surface along the A–A′ (yellow) line, and the horizontal interval is 50 m; the explosive source located at 10.0 m(depth), 7,500.0 m(length), 3,500.0 m(width). (b). The plot of the marmousi2 model shot gather, computed by our multiple-GPUs code. Our CUDA code uses 16 GPUs spend 4,812.64(s) calculate 5 s seismogram, while the SeisSol runs on 16 CPU cores need 135,278.50(s), it’s a speedup of 28.11×. (Color figure online)
For the SEG/EAGE salt model, we remove the very detailed original structure and only keep the main features, such as the salt body and those main faults (Fig. 10a). This simplified SEG/EAGE salt model with dimension of 4,200 m in depth, 13,500 m in length, and 13,500 m in width. The total tetrahedral elements number is 447,624; each element has its own material property. There are 192 receivers locate at 5 m beneath the surface along A–A′ (yellow) line, and the horizontal interval is 50 m; the explosive source located at 10.0 m(depth), 7,060.0 m(length), 4,740.0 m(width). In this case, we used the same hardware we used for the marmousi2 model. Our CUDA code uses 16 GPUs spend 7,938.56 s calculate 7 s seismogram, (Fig. 10b) while the SeisSol runs on 16 CPU cores need 224,589.80 s, it’s a speedup of 28.29×.

a A perspective view of the simplified SEG/EAGE salt model with dimension of 4,200 m in depth, 13,500 m in length and 13,500 m in width. There are 447,624 tetrahedral elements and each element has its own material property. There are 192 receivers locate at 5 m beneath the surface along A–A′ line and the horizontal interval is 50 m, the explosive source located at 10.0 m (depth), 7,060.0 m (length), 4,740.0 m (width). b The plot of the SEG/EAGE salt model shot gather, computed by our multiple-GPUs code. Our CUDA code uses 16 GPUs spend 7,938.56(s) calculate 7 s seismogram, while the SeisSol runs on 16 CPU cores need 224,589.80(s), it’s a speedup of 28.29×
6 Summary
In this study, we have successfully ported the ADER-DG method for solving the three-dimensional isotropic elastic seismic wave equation on unstructured tetrahedral meshes to CPU-GPU hybrid clusters using NVIDIA’s CUDA programing model and the message-passing interface (MPI). The serial version of our CUDA codes runs approximately 24.3 times faster than the reference serial CPU codes at single precision and about 12.8 times at double precision. The parallel version of our CUDA codes runs about 28.3 times faster than the reference parallel CPU codes at single precision and about 14.9 times at double precision. The increase in speed can be directly translated into an increase in the size of the problems that can be solved using the ADER-DG method. Some preliminary performance analysis shows that our parallel GPU codes have favorable strong and weak scalability as long as the ratio between the number of outer elements and inner elements of each subdomain is smaller than a certain threshold.
The ADER-DG method has a number of unique characteristics that make it very suitable for acceleration using GPU-type SIMD processors. The majority of the calculations can be carried out in an element-local way with weak inter-element coupling implemented using the numerical flux functions. In particular, as shown in Eq. (10), the only term that involves inter-element information exchange is the second term in the numerical flux, and we have shown that on a distributed-memory parallel system, the communication cost can be effectively overlapped with computation. This locality in the ADER-DG method makes it relatively straightforward to partition the workload among different thread blocks on each GPU and also among different GPUs on the cluster and results in close-to-ideal scalability. The ADER-DG method is also a high-order method, which requires more work per DOF than low-order methods. The increase in arithmetic intensity shifts the bottleneck from the memory bandwidth to the compute bandwidth. The relative abundance of cheap computing power on a GPU makes it favorable for implementing high-order methods.
7 Discussion
Debates still exist in the computer sciences community about how the speedup factor should be defined in a more general and objective way. Some definitions are more favorable to the GPUs, and some definitions are more favorable to the CPUs. But in spite of the debates about the definitions of the speedup factor, a common consensus among both the theoreticians and the practitioners is that GPUs are relatively low-cost, low-power-consumption, powerful co-processors that are suitable for SIMD-type calculations and studies about efficient implementations of numerical algorithms for scientific computing on the GPU architectures are worthwhile. In this sense, the lessons learned through the implementation and optimization process are more important than the exact speedup numbers that we have obtained.
Unlike implementations on a single GPU, to port the codes to multiple GPUs effectively, we need to deal with an extra layer of complexity introduced by inter-process communications. For the ADER-DG method, in which a majority of the calculations are local to each element, we can hide the inter-process communication overhead by overlapping communication with computation. We separate the elements on a subdomain into inner and outer elements. Once the computation on the outer elements is completed, we can fill the MPI buffer using a separate stream on the GPU and issue a non-blocking MPI call on the CPU. While the MPI messages are traveling across the network, the GPU proceeds to perform computations on the inner elements, and all other computations that do not need information from the outer elements. The technologies for multiple-GPU and CPU-GPU inter-connections are rapidly evolving, and GPU-Aware MPI (GAMPI) libraries for CUDA-enabled devices are gradually emerging. It is likely that the process for overlapping communication with computation by taking advantage of the multiple-stream capabilities of GPUs (i.e., task parallelism) will be much simplified and become more efficient in the near future.
A sizeable portion of the speedup we obtained is owed to the use of our customized matrix–matrix multiplication kernels. In our implementation, the results of the multiplications are evaluated analytically in terms of the non-zeros in the matrices whose locations can be determined beforehand and are fixed throughout the program. This approach allows us to condense the calculations by exploiting the sparsity of the matrices and also gives us enough freedom to manually adjust memory access patterns to minimize uncoalesced global memory accesses. This approach is applicable because the matrices involved in the ADER-DG method are element-local and relatively small (e.g., for a 5th-order scheme the size of the stiffness matrices is only 35 by 35), and the structures of these matrices are determined only by the specific forms of the spatial basis functions used to approximate the solution in the reference element. For more general matrix–matrix multiplication problems, some off-the-shelf, pre-tuned GPU linear algebra libraries, such as CUBLAS and cuSPARSE, might be more suitable.
The work described in this article is vender-specific. But we believe that most of the algorithmic analysis and implementation ideas presented here can be reused either identically or with slight modifications to adapt the ADER-DG method to other related architectures. To reinforce this point, we note that the emerging OpenCL industry standard for parallel programming of heterogeneous systems specifies a programming model that is very similar to CUDA. As GPUs are being widely adopted as powerful and energy-efficient co-processors in modern-day computer clusters, the work described here may help to accelerate the adoption of the ADER-DG method for seismic wave propagation simulations on such heterogeneous clusters.
This work provides an accurate yet flexible forward simulation solution other than conventional finite-difference method. With the capability of unstructured mesh and topography, our ADER-DG CUDA code could handle some complex scenario along with a relatively high efficiency and accuracy. This work could be further applied to earthquake related applications, such as full-wave seismic tomography (e.g., Chen et al. 2007; Liu and Tromp 2006; Tromp et al. 2008), accurate earthquake source inversion (e.g., Chen et al. 2005, 2010; Lee et al. 2011), seismic hazard analysis (e.g., Graves et al. 2010), and reliable ground-motion predictions (e.g., Graves et al. 2008; Komatitsch et al. 2004; Olsen 2000).
References
Abdelkhalek R, Calandra H, Coulaud O, Roman J, Latu G (2009) Fast seismic modeling and reverse time migration on a GPU cluster. The 2009 International Conference on High Performance Computing & Simulation, 2009. HPCS’09, pp 36–43
Aminzadeh F, Brac J, Kunz T (1997) 3-D salt and overthrust models. SEG/EAGE 3-D modeling series No. 1, 1997. Society of Exploration Geophysicists and European Association of Exploration Geophysicists
Bao H, Bielak J, Ghattas O, Kallivokas LF, O’Hallaron DR, Shewchuk JR, Xu J (1998) Large-scale simulation of elastic wave propagation in heterogeneous media on parallel computers. Comput Methods Appl Mech Eng 152:85–102
Bell N, Garland M (2009) Efficient sparse matrix-vector multiplication on CUDA. In: Proceedings of ACM/IEEE Conference Supercomputing (SC), Portland, OR, USA
Carcione JM (1994) The wave equation in generalized coordinates. Geophysics 59:1911–1919
Chen P, Jordan T, Zhao L (2005) Finite-moment tensor of the 3 September 2002 Yorba Linda earthquake. Bull Seismol Soc Am 95:1170–1180
Chen P, Jordan TH, Zhao L (2007) Full three-dimensional tomography: a comparison between the scattering-integral and adjoint-wavefield methods. Geophys J Int 170:175–181
Chen P, Jordan TH, Zhao L (2010) Resolving fault plane ambiguity for small earthquakes. Geophys J Int 181:493–501
Cockburn B, Karniadakis GE, Shu CW (2000) Discontinuous Galerkin methods, theory, computation and applications. LNC-SE, 11. Springer
Dumbser M, Käser M (2006) An arbitrary high-order discontinuous Galerkin method for elastic waves on unstructured meshes-II. The three-dimensional isotropic case. Geophys J Int 167(1):319–336. doi:10.1111/j.1365-246X.2006.03120.x
Dumbser M, Käser M, Toro EF (2007) An arbitrary high-order discontinuous Galerkin method for elastic waves on unstructured meshes-V. Local time stepping and p-adaptivity. Geophys J Int 171(2):695–717. doi:10.1111/j.1365-246X.2007.03427.x
Graves RW (1996) Simulating seismic wave propagation in 3D elastic media using staggered-grid finite differences. Bull Seismol Soc Am 86(4):1091–1106
Graves RW, Aagaard BT, Hudnut KW, Star LM, Stewart JP, Jordan TH (2008) Broadband simulations for Mw 7.8 southern San Andreas earthquakes: ground motion sensitivity to rupture speed. Geophys Res Lett 35:L22302. doi:10.1029/2008GL035750
Graves R, Jordan T, Callaghan S, Deelman E (2010) CyberShake: a physics-based seismic hazard model for Southern California. Pure Appl Geophys 168:367–381
Hughes TJR (1987) The finite element method—linear static and dynamic finite element analysis. Prentice Hall, Englewood Cliffs
Igel H (1999) Wave propagation in three-dimensional spherical sections by the Chebyshev spectral method. Geophys J Int 136:559–566
Igel H, Mora P, Riollet B (1995) Anisotropic wave propagation through finite-difference grids. Geophysics 60:1203–1216
Käser M, Dumbser M (2006) An arbitrary high-order discontinuous Galerkin method for elastic waves on unstructured meshes-I. The two-dimensional isotropic case with external source terms. Geophys J Int 166(2):855–877
Käser M, Dumbser M (2008) A highly accurate discontinuous Galerkin method for complex interfaces between solids and moving fluids. Geophysics 73(3):T23–T35
Käser M, Dumbser M, la Puente de J, Igel H (2007) An arbitrary high-order discontinuous Galerkin method for elastic waves on unstructured meshes-III. Viscoelastic attenuation. Geophys J Int 168(1):224–242. doi:10.1111/j.1365-246X.2006.03193.x
Käser M, Pelties C, Castro CE (2010) Wavefield modeling in exploration seismology using the discontinuous Galerkin finite-element method on HPC infrastructure. Lead Edge 29:76–84
Klöckner A, Warburton T, Bridge J, Hesthaven JS (2009) Nodal discontinuous Galerkin methods on graphics processors. J Comput Phys 228(21):7863–7882. doi:10.1016/j.jcp.2009.06.041
Komatitsch D, Tromp J (1999) Introduction to the spectral-element method for 3-D seismic wave propagation. Geophys J Int 139:806–822
Komatitsch D, Tromp J (2002) Spectral-element simulations of global seismic wave propagation-II. Three-dimensional models, oceans, rotation and self-gravitation. Geophys J Int 150(1):303–318
Komatitsch D, Vilotte JP (1998) The spectral-element method: an efficient tool to simulate the seismic response of 2D and 3D geological structures. Bull Seismol Soc Am 88:368–392
Komatitsch D, Liu Q, Tromp J, Suss P, Stidham C, Shaw J (2004) Simulations of ground motion in the Los Angeles basin based upon the spectral-element method. Bull Seismol Soc Am 94:187–206
Komatitsch D, Michéa D, Erlebacher G (2009) Porting a high-order finite-element earthquake modeling application to NVIDIA graphics cards using CUDA. J Parallel Distrib Comput 69(5):451–460
Komatitsch D, Göddeke D, Erlebacher G, Michéa D (2010) Modeling the propagation of elastic waves using spectral elements on a cluster of 192 GPUs. Comput Sci Res Dev 25(1):75–82
la Puente De J, Käser M, Dumbser M, Igel H (2007) An arbitrary high-order discontinuous Galerkin method for elastic waves on unstructured meshes-IV. Anisotropy. Geophys J Int 169(3):1210–1228
la Puente De J, Käser M, Cela JM (2009) SeisSol optimization, scaling and synchronization for local time stepping. Science and Supercomputing in Europe, pp 300–302
Lee E, Chen P, Jordan T, Wang L (2011) Rapid full-wave centroid moment tensor (CMT) inversion in a three-dimensional earth structure model for earthquakes in Southern California. Geophys J Int 186:311–330
Levander AR (1988) Fourth-order finite difference P-SV seismograms. Geophysics 53:1425–1436
Liu Q, Tromp J (2006) Finite-frequency kernels based on adjoint methods. Bull Seismol Soc Am 96:2383–2397
Lysmer J, Drake LA (1972) A finite element method for seismology. In: Alder B, Fernbach S, Bolt BA (eds) Methods in Computational Physics, vol 11. Academic Press, New York, Ch. 6, pp 181–216
Madariaga R (1976) Dynamics of an expanding circular fault. Bull Seismol Soc Am 65:163–182
Michéa D, Komatitsch D (2010) Accelerating a three-dimensional finite-difference wave propagation code using GPU graphics cards. Geophys J Int 182(1):389–402
Moczo P, Kristek J, Vavrycuk V, Archuleta RJ, Halada L (2002) 3D heterogeneous staggered-grid finite-difference modeling of seismic motion with volume harmonic and arithmetic averaging of elastic moduli and densities. Bull Seismol Soc Am 92:3042–3066
Mora P (1989) Modeling anisotropic seismic waves in 3-D, 59th Ann. Int. Mtg Exploration Geophysicists, expanded abstracts, pp 1039–1043
Mu D, Chen P, Wang L (2013) Accelerating the discontinuous Galerkin method for seismic wave propagation simulations using the graphic processing unit (GPU)—single-GPU implementation. Comput Geosci 51:282–292
Okamoto T, Takenaka H, Nakamura T, Aoki T (2010) Accelerating large-scale simulation of seismic wave propagation by multi-GPUs and three-dimensional domain decomposition. Earth Planet Space 62(12):939–942. doi:10.5047/eps.2010.11.009
Olsen K (2000) Site amplification in the Los Angeles basin from three-dimensional modeling of ground motion. Bull Seismol Soc Am 90:577–594
Tessmer E (1995) 3-D Seismic modelling of general material anisotropy in the presence of the free surface by a Chebyshev spectral method. Geophys J Int 121:557–575
Tessmer E, Kosloff D (1994) 3-D elastic modeling with surface topography by a Chebyshev spectral method. Geophysics 59(3):464–473
Toro EF (1999) Riemann Solvers and Numerical Methods for Fluid Dynamics. Springer, Berlin
Toro EF, Titarev VA (2002) Solution of the generalized Riemann problem for advection-reaction equations. Proc R Soc Lond 458:271–281
Tromp J, Komatitsch D, Liu Q (2008) Spectral-element and adjoint methods in seismology. Commun Comput Phys 3:1–32
Virieux J (1984) SH-wave propagation in heterogeneous media: velocity-stress finite-difference method. Geophysics 49:1933–1942
Virieux J (1986) P-SV wave propagation in heterogeneous media: velocity-stress finite-difference method. Geophysics 51:889–901
Wang Z, Peng S, Liu T (2010) Modeling seismic wave propagation using graphics processor units (GPU). The Second International Symposium on Networking and Network Security (ISNNS 2010), pp 129–132
Acknowledgments
We thank the “SeisSol” working group at the Ludwig-Maximilians University in Germany and Yifeng Cui at the San Diego Supercomputer Center for providing us the CPU code “SeisSol.” This study would not be possible without their help. The first author of this article is supported by the School of Energy Resources at the University of Wyoming. The GPU hardware used in this study was purchased using the NSF Grant EAR-0930040.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Mu, D., Chen, P. & Wang, L. Accelerating the discontinuous Galerkin method for seismic wave propagation simulations using multiple GPUs with CUDA and MPI. Earthq Sci 26, 377–393 (2013). https://doi.org/10.1007/s11589-013-0047-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11589-013-0047-7