
Abstract
The need of quick testing of nicotine (NIC) is vital because of its hurtful impacts on human. NIC detection has been successfully developed using a novel sensitive simple electrochemical sensor. The sensor system is based on modifying a carbon paste electrode (CPE) using Ni and Cu nanoparticles to develop the new nickel/copper nanoparticle–modified carbon paste electrode (NCNMCPE) with low-cost and simple procedure. The sensor showed excellent electrocatalytic activity for NIC oxidation in phosphate buffer solution (pH 7.0) at a lower voltage of 1.3 V, with a linear response from 6.0 to 1000.0 µM. The detection limit (LOD) was estimated to be 0.14 nM. The sensing performance of the NCNMCPE for the electro-oxidation of NIC was examined utilizing cyclic voltammetry (CV), chronoamperometry (CA), and electrochemical impedance spectroscopy (EIS). Scanning electron microscopy (SEM) and energy-dispersive X-ray analysis (EDX) techniques were used to achieve surface characteristics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nicotine (3-(1-methyl-2-pyrrolidinyl) pyridine) is a sort of addictive chemical (alkaloid) derived from tobacco leaves, which is primarily employed in the production of cigarettes [1]. NIC use through tobacco products is dangerous to both active and passive smokers and can result in diseases such as cardiovascular, pulmonary, and central nervous system problems as well as cancer [2,3,4,5,6]. In the realms of medicine, toxicology, and the tobacco industry, determining of NIC is critical. However, traditional analytical procedures such as gas chromatography [7], high-performance liquid chromatography [8, 9], and spectrophotometry [10] can be employed to quantify NIC. These procedures have some drawbacks such as the fact that they take longer time and involve complex sample preparation stages, expensive gear, and highly skilled staff [11]. Electrochemical detection is an alternative technology with practical advantages such as high sensitivity, selectivity, cost-effectiveness, quick reaction, and easier electrode fabrication process [12,13,14]. Electrochemical biosensors based on carbon electrodes for analytical and sensing purposes have attracted great attention. These materials present noticeable electroanalytical properties both for the direct detection of electroactive species and being functionally tailored to develop specific and sensitive electrochemical sensors and biosensors. Carbon paste electrode (CPE) is one of the most popular carbon electrodes due to its low background current, wide anodic potential range, ease of fabrication, and low-cost preparation [15]. Recently, a few investigations have been conducted on the detection of NIC utilizing CPEs modified with nanomaterial particles such as cerium nanoparticles [16], nano-TiO2 [17], and multi-wall carbon nanotubes [18]. M.A. Ameer et al. [19] developed a highly sensitive NIC detection method using a sensor based on Ag nanoparticle–modified carbon paste electrode. M. Shehata et al. [17] developed a modified carbon paste sensor with TiO2 nanoparticles for NIC detection in urine samples with good sensitivity. E. Molaakbari et al. [20] found that carbon paste electrode modified with ZnO nanoparticles and 3-(4′-amino-3′-hydroxy-biphenyl-4-yl)-acrylic acid gave accurate detection of norepinephrine, tyrosine, and NIC in mixtures without substantial interferences. Recently, the use of copper nanoparticles as an inhibitor [21] and in fuel cells [22], catalysis [23], and electrochemical sensors [24] has gained importance because of its increased efficiency due to increased surface to volume ratio. Nanostructured materials possess unique mechanical, electronic, physical, chemical, and physiochemical properties due to their high surface area which in turn increases the active centers [25, 26]. Nickel nanoparticles “Ni-NPs” have received great interest to use in electrochemical sensing because of their excellent electrocatalytic activity for oxidation of many compounds. Several researches have been reported for utilizing Ni-NPs in electrochemical applications such as glucose [27], amino acids [28], DNA [29], and myoglobin [30] sensors. In this study, a sensitive electrochemical technique using Ni/Cu-NP-modified carbon paste electrode (NCNMCPE) has been developed. The oxidation response of NIC at NCNMCPE was used as the indicator. The results show that this method is simple and very sensitive in detecting NIC without the use of expensive and complicated instruments.
Experimental
Reagent and chemicals
NIC standard samples (99%) were provided by the Egyptian Eastern Company for Smoking and were utilized without prior purification. NIC stock solutions of about 1.62 g L−1 were freshly prepared in water and maintained in a dark container because the compound is light sensitive. The standard NIC solutions were prepared using a supporting electrolyte, namely phosphate buffer solutions of pH 7.0 (Na2HPO4.7H2O + NaH2PO4.H2O). pH values were adjusted using 0.2 M NaOH and 0.2 M HCl. Analytical grade chemicals and triple-distilled water were used to make all of the solutions. All experiments were repeated twice or three times at room temperature and gave reproducible results.
Preparation of various electrodes
A CPE with a diameter of 3 mm was prepared by mixing drops of paraffin oil with (0.5 g) graphite powder and the mixture was blended thoroughly to acquire a uniform paste [31]. The resulting mixture was utilized to fill into a Teflon tube and pushed tightly. The CPE was polished with ultrafine emery paper until it was completely smooth.
-
1-
The Cu nanoparticle–modified CPE was obtained by electrodeposition onto the bare CPE at a fixed potential 0.17 V for 50 s from 0.1 M H2SO4 solution containing 0.02 M CuSO4 [32].
-
2-
The Ni nanoparticle–modified CPE Ni nanoparticles were electrodeposited on CPE surface by immersing the clean CPE into 5 ml NiSO4 (0.1 M) and 20 ml sodium–potassium tartrate (C4H4KNaO6) (0.5 M). The deposition condition was made by utilizing a current density 0.24 mA cm−2 for 5 min.
-
3-
For simultaneous deposition of both Cu and Ni nanoparticles over CPE, the clean CPE was dipped into 2.5 ml NiSO4 (0.1 M), 2.5 ml CuSO4 (0.1 M), and 20 ml sodium–potassium tartrate (C4H4KNaO6) (0.5 M). The deposition conditions were formed by using a current density of 0.24 mA cm−2 for 12 min [33]. The electrode was left to completely dry in air to obtain NCNMCPE.
Cell and apparatus
A three-electrode cell of 25 ml volume enclosing a platinum rod as a counter electrode (CE), saturated calomel electrode (SCE) as a reference electrode (RE), and NCNMCPE as the working electrode (WE) was utilized. Cyclic voltammetry (CV), chronoamperometry (CA), and electrochemical impedance spectroscopy (EIS) measurements are performed by SP-150 potentiostat supplied with EC-Lab® software package. EIS measurements are performed in the frequency range of 1.0 mHz to 100 kHz with a 10-mV ac amplitude. EC-Lab® software is used to analyze and fit the experimental spectra using the best equivalent circuit model. For pH readings, a microprocessor digital pH meter (Hanna Instruments, Italy) is employed. SEM measurements are performed using SEM Model Quanta 250 FEG (field emission gun) coupled to an EDX Unit (energy-dispersive X-ray analyses) (FEI business, Netherlands).
Analysis of urine
Standard NIC from the Egyptian Eastern Company was mixed in urine to make a stock solution (diluted 400 times using 100 ml of phosphate buffer pH 7.0). NIC standard samples were used for the additions. Standard additions of urine were prepared in the voltammetric cell from a solution containing NIC in 23 ml of phosphate buffer pH 7.0 and assessed under the same conditions as the calibration graph [16].
Analysis of cigarette samples
The cigarettes were taken out of their rolling papers and dried for 30 min in a 40 °C oven. A total of 0.1 g of tobacco was recovered from a mixture of 10 cigarettes obtained from two packs of the same brand and put to 10 ml water and then, the contents of the vial were sonicated for 3 h in an ultrasonic water bath and filtered. The filtrate was mixed with the phosphate buffer (pH 7.0) in the appropriate volume (100 ml) and analyzed under the identical conditions as the calibration graph [34].
Results and discussion
Characterization of the various electrode morphologies
The SEM picture of bare CPE (Fig. 1D) was as expected, showing rather uniform surface owing to the high compatibility of the paraffin oil with graphite particles and the formation of a compact, tight, and homogenous paste. The surface of the modified electrode was significantly different, exhibiting additional crystalline shapes of well-electrodeposited Ni nanoparticles over the CPE to give the new NNMCPE in Fig. 1C. In the same manner, a colony of Cu nanoparticles electrodeposited over the carbon paste layers formed the modified CNMCPE as displayed in Fig. 1B. Figure 1A shows the SEM image of the Ni-Cu nanoparticle–modified carbon paste electrode (NCNMCPE). The electrode surface revealed agglomeration of Cu nanoparticles at various places throughout the CPE surface which are grouped as nanosheets to form coralline-structure morphology and uniform distribution over a vast surface area and cover in between its layers the Ni nanoparticles as represented in the inset of Fig. 1. The high surface energy and surface tension of Cu-NPs cause them to aggregate together [35]. The existence of both Cu and Ni-NPs was confirmed by EDX data of the NCNMCPE surface as shown in Fig. 1E, indicating that Ni nanoparticles were coated on the CPE surface by the layers of Cu-NPs according to their peaks and its corresponding quantitative weight percent, which were 55.78% for oxygen, 18.31% for carbon, 16.99% for Ni, 6.91% for Cu, and 1.98% for K.

A SEM image of Ni/Cu-NP-modified carbon paste electrode; inset shows the electrode morphology at higher magnification. B Electrodeposited Cu nanoparticles on CPE. C Electrodeposited Ni nanoparticles on CPE. D SEM image for bare CPE. E EDX spectra of the NCNMCPE surface
Electrochemistry of NIC
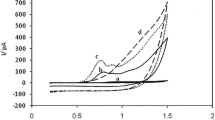
Using the CV approach, the interaction of 1.0 × 10−3 M NIC with various modified electrodes (CNMCPE, NNMCPE, and NCNMCPE) and unmodified bare electrode (BCPE) was investigated. At a scan rate of 50 mV/s, Fig. 2 reveals the four CVs of the BCPE and modified electrodes using phosphate buffer solution (pH = 7.0). NIC oxidized at the surface of the different modified electrodes at a potential of around + 1.3 V during anodic scanning, resulting in numerous and high oxidation peak currents due to improved electrocatalytic oxidation of NIC on modified electrodes. Running CVs in the same condition with BE presented very poor catalytic activity toward NIC. The response of CNMCPE to NIC was obviously around 46 µA and improved the detection of NIC nearly 4.5 times higher than that of BCPE, showing that CPE has been effectively improved by Cu-NPs as shown in Fig. 2. On using NNMCPE, its response to NIC was higher by about 6.0 times than that of BCPE, ensuring the enhancement of NIC detection through modifying CPE with Ni-NPs. The simultaneous electrodeposition of both Ni-NPs and Cu-NPs to produce NCNMCPE showed high efficiency toward the oxidation of NIC compared with the BCPE, which nearly reached to about 14.0 times higher than the oxidation peak of NIC over the BCPE. The existence of numerous catalytic centers on the Ni-NPs and Cu-NPs structure as well as topological defects on the electrode surface may explain the improved electrocatalytic activity of NIC on NCNMCPE [18, 36, 37]. During a cathodic scan, no voltammetric peak was seen that corresponded to the reduction of NIC. As a result, the irreversible NIC electrode oxidation at NCNMCPE has been confirmed.

CVs of NIC at bare CPE, NNMCPE, CNMCPE, and NCNMCPE using phosphate buffer (pH 7.0) at a scan rate of 50 mV/s
Optimization of detection test conditions
Effect of pH
Cyclic voltammetry (CV) technique was evaluated to estimate the impact of pH using phosphate buffer solution (pH 2.0–9.0) at BE and NCNMCPE (Fig. 3). The effect of pH on the peak current and the peak potential for the oxidation of NIC is demonstrated in Fig. 3 (insets (A) and (B), respectively). As the pH increased, the anodic peak potentials moved negatively, indicating that the electrocatalytic oxidation of NIC was a pH-dependent reaction including proton participation in the electrode reaction [38, 39]. As can be observed, the produced complex’s peak current increased somewhat as pH go from 2.0 to 5.0. The peak current’s highest intensity was at pH = 7.0 and it gradually declined up to pH 9.0. Consequently, the pH 7.0 value was utilized as the ideal pH in all of the preceding investigations. NIC is a diacidic weak base with pKa1 = 8.02 and pKa2 = 3.12. At pH 7.0–8.0 (near to pKa1), the monoprotonated form predominated while the deprotonated form predominated at pH 2.0–2.7. Accordingly, the pH value of the detecting system influences NIC response enabling the predictable performance of a proton-dependent approach with an irreversible chemical reaction [40, 41].

CVs of NIC in different pH values using NCNMCPE at a scan rate of 50 mV/s. Inset: (A) Variation of anodic peak current with pH at bare CPE and NCNMCPE. (B) Variation of anodic peak potential with pH at NCNMCPE
Effect of scan rate and electroactive area calculation
The influence of scan rate on the anodic peak current of 1.0 × 10−3 M NIC was investigated using the cyclic voltammetry technique (Fig. 4). As shown in the inset of Fig. 4, the relationship between anodic peak current (Ipa) and the square root of the scan rate v1/2 (v ranging from 50 to 200 mV s−1) yielded a straight line, implying that the NIC oxidation process is a diffusion-controlled mass transfer process for NIC on the NCNMCPE [42, 43]. Furthermore, increasing the scan rate causes the NIC peak potentials to shift to the positive direction without any cathodic peak current, demonstrating reaction irreversibility [28]. The Randles–Sevcik Eq. (1) was also used to explore the active surface area of the NCNMCPE and the diffusion coefficient D of NIC [44,45,46].

Effect of changing the scan rate from 50 to 200 mV/s on the anodic peak response of NIC. Inset: Relation between anodic peak current and the square root of the scan rate
(1) For a given concentration of K4[Fe(CN)6], the active surface area of the modified electrode was determined, where Ip is the peak current (A), n is the number of electrons transferred, v is the scan rate (V s−1), D is the diffusion coefficient, A is the electrode area, and C is the concentration of K4[Fe(CN)6]. For 1.0 mmol L−1 K4[Fe(CN)6] in 0.10 mol L−1 KCl electrolyte with n = 1 and D = 7.6106 cm2 s−1 [47], the bare surface area is 0.0706 cm2, and the NCMCPE electroactive surface area is 0.181 cm2.
(2) Based on the Randles–Sevcik equation, the diffusion coefficient Dapp for NIC in phosphate buffer (pH 7.0) was derived from the relationship between anodic peak current Ip (A) and square root of scan rate v1/2 (Fig. 4 inset).
In this equation, Ipa is the maximum current (A), n is the number of electrons transferred in the redox event, C0 is the analyte concentration (1 × 10−6 mol cm−3), A is the electroactive area of the electrode which previously calculated (0.181 cm2), D is the electroactive species diffusion coefficient (cm2 s−1), ν is the scan rate in V s−1, F is Faraday constant in C mol−1, T is the temperature in K, and R is the gas constant in J mol−1 K−1. Thus, the apparent diffusion coefficient, Dapp, of NIC was found to be 1.692 × 10−5cm2 s−1.
Chronoamperometry
The chronoamperometric measurements of NIC (Fig. 5) were done at a constant potential (+ 1300 mV vs. SCE) using NCNMCPE in phosphate buffer (pH = 7.0) containing different NIC concentrations. Using the Cottrell equation [48], the diffusion coefficient can be calculated
where i = current, in A, n = number of electrons, F = Faraday constant, 96,485 C/mol, A = electrode area in cm2, c0 = bulk analyte concentration in mol/cm3, D = species diffusion coefficient in cm2/s, and t = time in s.

Chronoamperograms obtained at NCNMCPE in phosphate buffer (pH 7.0) using different concentrations of NIC (20, 80, 200, 600, 800, and 1000 μM). Insets: (A) plot of I vs. t.−1/2 obtained from chronoamperogram of 1000 μM. (B) Calibration curve for different concentrations of NIC at a fixed time of 1 s. Error bars represent the standard deviations of three repetitive tests
The slope derived from the relationship between the anodic peak current and t−1/2 (inset) equals nFAco (D/π) 1/2; hence, the diffusion coefficient of NIC was determined to be 2.782 × 10−5 cm2s−1 using the Cottrell equation. As a result, the diffusion coefficient values derived using chronoamperometric data are equivalent to the diffusion coefficient values computed using CV measurements. This demonstrates that the analyte species undergoes a diffusion-controlled redox reaction.
The calibration curve is shown as a linear relationship between different NIC concentrations (20 to 1000 M) and the current at a given time (1 s) in Fig. 5 inset (B) [49]. Ip(A) = 0.034 C + 9.74, with a correlation coefficient of 0.994, was the linear equation. The steady-state oxidation current increases as NIC concentration rises [50].
Effect of repeated CVs
CVs of 1.0 × 10−3 M NIC in phosphate buffer (pH 7.0) were repeated in different time intervals (Fig. 6), with the peak current value decreasing (inset) from the first scan to the remaining scans. This might be owing to oxidation product adsorption as an adsorbed coating on the NCNMCPE surface, which deactivates the sensor and lowers its electroactive surface area [51]. NCNMCPE’s repeatability and precision investigations are carried out by repeating the measurements five times, yielding a relative standard deviation (RSD) of 1.58% for the first anodic scan. To achieve novel sensitivity and reproducibility, a first anodic peak current must be recorded for all the analysis in the coming studies.

Effect of repeated CVs for 1.0 mM NIC in phosphate buffer (pH 7.0) using NCNMCPE, at a scan rate of 50 mV/s. Inset: Relation between stirring time in minutes and the anodic peak current of 1.0 mM NIC
EIS measurements
Impedance plots for the bare and NCNMCPE electrodes are given as Nyquist plots (Fig. 7 inset). At both high and low frequencies, a semicircle represents a charge transfer resistance whereas a small line as a spike represents a diffusion process. A one-time constant model with Rs (solution resistance), RCT (charge transfer resistance), Zw (Warburg impedance), and CPE (constant phase element of capacitance) [52] is the best model that fits the experiments (Fig. 8). This similar circuit model was used to simulate the data. RCT is due to the semicircle at high frequency (charge transfer resistance) while ZW is related to the small linear region at low frequency (diffusion process) [16, 53]. To account for surface non-ideality and heterogeneity, a constant phase element (CPE) [52] was developed with the impedance ZCPE = [C (jω)α] −1, where α is due to the surface inhomogeneity (− 1 ≤ α ≤ 1), j is the imaginary number (j2 = − 1), ω = 2πf (rad s−1), and f (Hz) is the frequency. As a result, both charge transfer and diffusion play a role in the reaction mechanism [54]. The fitting is performed using EC-Lab® software provided with SP-150 workstation. The NCNMCPE electrode has a higher CPE (14.2 µF cm−2) and Warburg impedance (1440 Ω cm2 s−1/2) and lower charge transfer resistance (1713 Ω cm2), suggesting a greater conductivity compared to the bare electrode with CPE = 2.1 µF cm−2, W = 325 Ω cm2 s−1/2, and RCT = 8059 Ω cm2. As a result, both charge transfer and diffusion play a role in the reaction mechanism [54]. Furthermore, Nyquist plots (Fig. 7) of NIC in different pH values were measured using NCNMCPE at peak potential vs. SCE and gave the same order of pH as the CV results. The impedance value decreases in the same order as the current increases suggesting an increase in the conductivity. This shows that pH 7.0 has the lowest impedance value with smallest semicircle diameter when compared to the bare electrode (inset) and the maximum conductivity as obtained from CV results with highest current [17]. The best fitting values for the impedance data of each pH value for the NCNMCPE are listed in Table 1.

Nyquist plots of NIC in different pH values using NCNMCPE at peak potential. Inset: Nyquist plots of NIC at bare CPE and NCNMCPE using phosphate buffer (pH 7.0), at peak potential

The equivalent circuit used in the fitting experiments of the EIS
Calibration curve and detection limit
Figure 9 illustrates differential pulse voltammograms (DPVs) for the effect of varying NIC concentrations (6.0 to 1000 µM) in phosphate buffer (pH 7.0) at a scan rate of 10.0 mV/s utilizing NCNMCPE. Figure 9 (inset) depicts a linear relationship using the equation: Ip(µA) = 0.0208 C + 19.650. According to the IUPAC recommendation, the limits of detection (LOD) and quantification (LOQ) were determined [17, 55] to be 1.44 × 10−8 M and 4.8 × 10−7 M, respectively.

Differential pulse voltammograms for successive addition of NIC in phosphate buffer (pH 7.0) using NCNMCPE at step potential of 4.0 mV, modulation amplitude of 25.0 mV, and scan rate 10.0 mV/s. Inset: Calibration curve of NIC using NCNMCPE. Error bars represent the standard deviations of three repetitive tests
Comparisons of the results for NIC detection obtained by several methods are shown in Table 2. Compared with several current techniques for the assay of NIC, the NCNMCP sensor has some advantages, obtaining higher specific selectivity with excellent stability and cheap reagents. The instruments of the NCNMCP sensor are simpler and cheaper than those of HPLC, GC, SPE, and spectrometry. The results are fairly satisfactory and the background causes no significant effects.
Sample analysis
Application of NCNMCPE sensor in urine
Figure 10 shows the calibration plot obtained from DPV for the use of the NCNMCPE sensor on actual samples such as urine, which showed a straight line across the concentration range of 1 × 10−8 M to 8 × 10−7 M. The NIC concentration in urine samples was determined using the calibration curve equation Ipa (A) = 0.0231 C + 8.9041. LOD = 7.4 × 10−7 M and LOQ = 2.46 × 10−6 M were also estimated. Table 3 shows the precision and accuracy of the suggested technique for NIC detection in urine samples for four different concentrations on the calibration curve that are repeated five times, with the recovery ranging from 98.0 to 100.4.

Calibration curve of NIC in urine
Analysis of real cigarette brand samples
Two products from distinct cigarette brands (L&M and Marlboro) were evaluated for a practical application to ensure that the proposed method was validated with real samples [38]. The NIC content of the tobacco sample was determined by adding the proper volume of standard NIC solution produced in supporting electrolyte to the NIC content previously prepared. Because NIC quantities may be quantitatively recovered by the suggested method, the recovery data confirm the accuracy of the voltammetric detection of NIC in tobacco which is represented in Table 4.
Interference study reproducibility and long-term stability of NCNMCPE
Because of the low concentrations of other minor alkaloids (0.2–0.5% of total alkaloids), they cannot affect the accuracy of NIC detection at the sensitivity level of voltammetric measurements; thus, the interference of alkaloids which may be present in tobacco was not performed in this study.
To determine the influence of interfering compounds on NIC detection by NCNMCPE, a constant concentration of NIC (500 µM) was spiked with similar and two-fold concentrations of the NIC biomarker “cotinine.” Cotinine is structurally similar to NIC and its usage with NIC would determine the selectivity of the investigated sensor perfectly. By using the same experimental conditions, CV measurements were performed and the obtained recovery data between 99.3 and 100% ensures the high sensor selectivity to NIC.
To verify the reproducibility of NCNMCPE in terms of RSD, 5 sequential voltammetric determinations for 50 μM of NIC were performed without obvious alteration in NIC peak current with an RSD of 1.7%, which ensures the precision of the electrode under study. The long-term stability of the introduced method was assessed by keeping the NCNMCPE in a refrigerator (4 °C) for 7 days. Then, a single voltammetric measurement for NIC was performed, disclosing a current response at 95% of the value measured directly after fresh preparation, denoting good storage constancy of the electrode.
Conclusions
New NCNMCPE sensor based on deposition of Ni and Cu-NPs on the surface of CPE has been presented and experimentally tested for the selective detection of NIC. The experimental results demonstrate that NCNMCPE sensor exhibits good performances for the electrochemical determination of NIC, indicating that Ni nanoparticles improved the sensitivity of NIC. The detection limit for NIC was found to be 0.14 nM. Real sample analysis was also carried out successfully in urine and tobacco sample using a newly prepared sensor, which detected NIC with great accuracy. We believe that this methodology may be useful for a simple and effective electrochemical sensor for NIC detection.
References
Lin B, Chen J, Zeng Y, Li L, Qiu B, Lin Z, Guo L (2019) A facile approach for on-site evaluation of nicotine in tobacco and environmental tobacco smoke. ACS sensors 4:1844–1850
Domino EF (1998) Tobacco smoking and nicotine neuropsychopharmacology: some future research directions. Neuropsychopharmacology 18:456–468
Heeschen C, Jang JJ, Weis M, Pathak A, Kaji S, Hu RS, Tsao PS, Johnson FL, Cooke JP (2001) Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med 7:833–839
Lloyd GK, Williams M (2000) Neuronal nicotinic acetylcholine receptors as novel drug targets. J Pharmacol Exp Ther 292:461–467
Jha P (2009) Avoidable global cancer deaths and total deaths from smoking. Nat Rev Cancer 9:655–664
Khlifi R, Hamza-Chaffai A (2010) Head and neck cancer due to heavy metal exposure via tobacco smoking and professional exposure: a review. Toxicol Appl Pharmacol 248:71–88
Acosta MC, Buchhalter AR, Breland AB, Hamilton DC, Eissenberg T (2004) Urine cotinine as an index of smoking status in abstinent smokers: comparison of GC/MS with immunoassay test strip. Nicotine Tob Res 6:615–620
Abu-Qare AW, Abou-Donia MB (2001) Quantification of nicotine, chlorpyrifos and their metabolites in rat plasma and urine using high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 757:295–300
Vindatiche I, Roche D, Callais F, Lequang N, Labrousse F (2000) Analytical improvements in Barlow reaction coupled to HPLC detection of nicotine and its metabolites. J Liq Chromatogr Relat Technol 23:1423–1437
Al-Tamrah S (1999) Spectrophotometric determination of nicotine. Anal Chim Acta 379:75–80
Ray M, Ray A, Dash S, Mishra A, Achary KG, Nayak S, Singh S (2017) Fungal disease detection in plants: traditional assays, novel diagnostic techniques and biosensors. Biosens Bioelectron 87:708–723
Kumar TV, Yadav SK, Sundramoorthy AK (2018) Electrochemical synthesis of 2D layered materials and their potential application in pesticide detection. J Electrochem Soc 165:B848
Sundramoorthy AK, Gunasekaran S (2014) Applications of graphene in quality assurance and safety of food. TrAC, Trends Anal Chem 60:36–53
Sims MJ, Rees NV, Dickinson EJ, Compton RG (2010) Effects of thin-layer diffusion in the electrochemical detection of nicotine on basal plane pyrolytic graphite (BPPG) electrodes modified with layers of multi-walled carbon nanotubes (MWCNT-BPPG). Sens Actuators, B Chem 144:153–158
Teradal NL, Kalanur SS, Prashanth S, Seetharamappa J (2012) Electrochemical investigations of an anticancer drug in the presence of sodium dodecyl sulfate as an enhancing agent at carbon paste electrode. J Appl Electrochem 42:917–923
Fekry A, Azab S, Shehata M, Ameer M (2015) A novel electrochemical nicotine sensor based on cerium nanoparticles with anionic surfactant. RSC Adv 5:51662–51671
Shehata M, Azab S, Fekry A, Ameer M (2016) Nano-TiO2 modified carbon paste sensor for electrochemical nicotine detection using anionic surfactant. Biosens Bioelectron 79:589–592
Xiong H, Zhao Y, Liu P, Zhang X, Wang S (2010) Electrochemical properties and the determination of nicotine at a multi-walled carbon nanotubes modified glassy carbon electrode. Microchim Acta 168:31–36
Ameer M, Fekry A, Azab S, Shehata M (2018) Synthesis of a simply modified electrochemical nicotine sensor based on silver nanoparticles. Can J Chem 96:821–827
Molaakbari E, Mostafavi A, Beitollahi H (2014) First electrochemical report for simultaneous determination of norepinephrine, tyrosine and nicotine using a nanostructure based sensor. Electroanalysis 26:2252–2260
Saji VS, Cook R (2012) Corrosion protection and control using nanomaterials, 1st Edition
Kidwai M, Bhardwaj S, Mishra NK, Bansal V, Kumar A, Mozumdar S (2009) A novel method for the synthesis of β-enaminones using Cu-nanoparticles as catalyst. Catal Commun 10:1514–1517
Grecu R, Samide A, Iacobescu GE, Cioateră N, Popescu A (2019) Copper corrosion inhibitors based on polyvinyl alcohol and silver nanoparticles. Chem Ind Chem Eng Q 25:267–275
Jo S, Jeong HG, Kim YH, Neagu D, Myung J-H (2021) Stability and activity controls of Cu nanoparticles for high-performance solid oxide fuel cells. Appl Catal B 285:119828
Teng S, Siegel G, Prestgard MC, Wang W, Tiwari A (2015) Synthesis and characterization of copper-infiltrated carbonized wood monoliths for supercapacitor electrodes. Electrochim Acta 161:343–350
Manoj D, Saravanan R, Santhanalakshmi J, Agarwal S, Gupta VK, Boukherroub R (2018) Towards green synthesis of monodisperse Cu nanoparticles: an efficient and high sensitive electrochemical nitrite sensor. Sens Actuators, B Chem 266:873–882
Mu Y, Jia D, He Y, Miao Y, Wu H-L (2011) Nano nickel oxide modified non-enzymatic glucose sensors with enhanced sensitivity through an electrochemical process strategy at high potential. Biosens Bioelectron 26:2948–2952
Roushani M, Shamsipur M, Pourmortazavi SM (2012) Amprometric detection of glycine, l-serine, and l-alanine using glassy carbon electrode modified by NiO nanoparticles. J Appl Electrochem 42:1005–1011
Noorbakhsh A, Salimi A (2011) Development of DNA electrochemical biosensor based on immobilization of ssDNA on the surface of nickel oxide nanoparticles modified glassy carbon electrode. Biosens Bioelectron 30:188–196
Liu X, Ma L, Wang Z (2011) Focus on the nanomaterial-based biosensor papers in Chinese Journal of Analytical Chemistry of the year 2010. Sci China Chem 54:1365
Chaki NK, Vijayamohanan K (2002) Self-assembled monolayers as a tunable platform for biosensor applications. Biosens Bioelectron 17:1–12
Hosseini SR, Raoof J-B, Ghasemi S, Gholami Z (2015) Synthesis of Pt–Cu/poly (o-Anisidine) nanocomposite onto carbon paste electrode and its application for methanol oxidation. Int J Hydrog Energy 40:292–302
Eshghi A, Kheirmand M (2018) Surface modification of glassy carbon electrode by Ni-Cu nanoparticles as a competitive electrode for ethanol electro-oxidation. Iran J Chem Chem Eng (IJCCE) 37:1–8
Levent A, Yardim Y, Senturk Z (2009) Voltammetric behavior of nicotine at pencil graphite electrode and its enhancement determination in the presence of anionic surfactant. Electrochim Acta 55:190–195
Theivasanthi T, Alagar M (2012) Chemical capping synthesis of nickel oxide nanoparticles and their characterizations studies, arXiv preprint arXiv:1212.4595
Jerome R, Sundramoorthy AK (2020) Preparation of hexagonal boron nitride doped graphene film modified sensor for selective electrochemical detection of nicotine in tobacco sample. Anal Chim Acta 1132:110–120
Karthika A, Karuppasamy P, Selvarajan S, Suganthi A, Rajarajan M (2019) Electrochemical sensing of nicotine using CuWO4 decorated reduced graphene oxide immobilized glassy carbon electrode. Ultrason Sonochem 55:196–206
Suffredini H, Santos DM, De Souza D, Codognoto L, Homem-de-Mello P, Honório K, Da Silva A, Machado S, Avaca L (2005) Electrochemical behavior of nicotine studied by voltammetric techniques at boron-doped diamond electrodes. Anal Lett 38:1587–1599
Mehmeti E, Kilic T, Laur C, Carrara S (2020) Electrochemical determination of nicotine in smokers’ sweat. Microchem J 158:105155
Shalaeva M, Kenseth J, Lombardo F, Bastin A (2008) Measurement of dissociation constants (pKa values) of organic compounds by multiplexed capillary electrophoresis using aqueous and cosolvent buffers. J Pharm Sci 97:2581–2606
Yang S, Smetena I (1995) Evaluation of capillary electrophoresis for the analysis of nicotine and selected minor alkaloids from tobacco. Chromatographia 40:375–378
Reddy SL, Arul C, Zhaoqi L, Lavanya N, Sekar C (2020) A novel electrochemical sensor based on Fe-doped MgNi2O3 nanoparticles for simultaneous determination of dopamine, uric acid, nicotine and caffeine over very wide linear ranges. J Electroanal Chem 878:114648
Li D, Zhang L, Chen H, Wang J, Ding L-X, Wang S, Ashman PJ, Wang H (2016) Graphene-based nitrogen-doped carbon sandwich nanosheets: a new capacitive process controlled anode material for high-performance sodium-ion batteries. J Mater Chem A 4:8630–8635
Kissinger P, Heineman WR (2018) Laboratory techniques in electroanalytical chemistry, revised and expanded, CRC press
Li X, Lin D, Lu K, Chen X, Yin S, Li Y, Zhang Z, Tang M, Chen G (2020) Graphene oxide orientated by a magnetic field and application in sensitive detection of chemical oxygen demand. Anal Chim Acta 1122:31–38
Bai L, Yuan R, Chai Y, Yuan Y, Wang Y, Xie S (2012) Direct electrochemistry and electrocatalysis of a glucose oxidase-functionalized bioconjugate as a trace label for ultrasensitive detection of thrombin. Chem Commun 48:10972–10974
Wang S, Li L, Jin H, Yang T, Bao W, Huang S, Wang J (2013) Electrochemical detection of hepatitis B and papilloma virus DNAs using SWCNT array coated with gold nanoparticles. Biosens Bioelectron 41:205–210
Bard AJ, Faulkner LR (2001) Fundamentals and applications. Electrochem Methods 2:580–632
Hemmateenejad B, Safavi A, Honarasa F (2015) Deriving calibration curves at early times of chronoamperograms using the chemometrically resolved net faradaic current. J Electroanal Chem 755:221–227
Baghayeri M, Amiri A, Farhadi S (2016) Development of non-enzymatic glucose sensor based on efficient loading Ag nanoparticles on functionalized carbon nanotubes. Sens Actuators, B Chem 225:354–362
Chen T-S, Kuo Y-M, Chen J-L, Huang K-L (2013) Anodic degradation of ofloxacin on a boron-doped diamond electrode. Int J Electrochem Sci 8:7625–7633
Fekry AM (2016) Electrochemical behavior of a novel nano-composite coat on Ti alloy in phosphate buffer solution for biomedical applications. RSC Adv 6:20276–20285
Fekry A (2010) Impedance and hydrogen evolution studies on magnesium alloy in oxalic acid solution containing different anions. Int J Hydrogen Energy 35:12945–12951
Fekry A, Ameer M (2011) Electrochemical investigation on the corrosion and hydrogen evolution rate of mild steel in sulphuric acid solution. Int J Hydrogen Energy 36:11207–11215
Meier PC, Zünd RE (2005) Statistical methods in analytical chemistry, John Wiley & Sons
CornazGudet A-L, Buri P (1998) A simple and rapid high-performance liquid chromatography method to quantify and evaluate the purity of nicotine in solution and in microspheres. STP Pharma Sci 8:139–144
Baskin LB, Anderson RW, Charlson JR, Hurt RD, Lawson GM (1998) A solid phase extraction method for determination of nicotine in serum and urine by isotope dilution gas chromatography/mass spectrometry with selected ion monitoring. Ann Clin Biochem 35:522–527
Lu GH, Ralapati S (1998) Application of high-performance capillary electrophoresis to the quantitative analysis of nicotine and profiling of other alkaloids in ATF-regulated tobacco products. Electrophoresis 19:19–26
Xu M, Qi R, Gao Q (1998) Photometric determination of nicotine in tobacco with bromocresol green. Physical testing and chemical analysis PART B CHEMICAL ANALYSIS 34:78–78
Garrigues JM, Pérez-Ponce A, Garrigues S, de La Guardia M (1999) Flow injection Fourier transform infrared determination of nicotine in tobacco. Analyst 124:783–786
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zaki, M., Shafie, E.E., Abdel-Gawad, S.A. et al. Sensitive detection for nicotine using nickel/copper nanoparticle–modified carbon paste electrode. Ionics 28, 4881–4891 (2022). https://doi.org/10.1007/s11581-022-04649-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-022-04649-6