
Abstract
The phenomenon of cryptic species is widespread among various fungal lineages. Fomes inzengae (Ces. & De Not.) Cooke has been recently recognized as a South European kin of wood-decay basidiomycete F. fomentarius (L.) Fr. due to the problematic morphological identification of both species, their taxonomic status has been disputed. The aim of this research is to examine the distribution, host preferences, morphological characters, and phylogenetic relationships between F. fomentarius and F. inzengae in the South Moravian region in Czechia (Central Europe), where both species occur sympatrically. The results revealed the ecological preferences of Fomes spp. along an altitudinal gradient, while F. inzengae is a lowland taxon, F. fomentarius dominates at higher altitudes in forests with abundant Fagus sylvatica. The main contact zone of the two taxa is located in the upper-colline vegetation belt (elevation ca. 400‒550 m a.s.l.). The morphological analysis revealed that the basidiospore size, the width of skeletal hyphae in basidiomes, and the linear density of pores of both taxa are almost identical and can not be used for the identification of the two species. Multigene sequence analyses of ITS, LSU, RPB1, RPB2, and TEF1 markers confirmed that F. fomentarius and F. inzengae are phylogenetically distinct species. The relationship of F. inzengae and F. fomentarius to Globifomes graveolens and Hexagonia spp. is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Species identification and delimitation are challenging for morphologically similar and geographically overlapping fungal species which can be distinguished by DNA barcoding and phylogenetic analyses. Especially in the case of biotechnologically valuable species, the exact identification of species is necessary. Fomes fomentarius (L.) Fr. (Polyporales, Polyporaceae) belongs to a remarkable wood-decaying fungi because of its perennial, ungulate (hoof-shaped) basidiomes. The fungus was historically used as an important source of tinder, in handicrafts and European traditional medicine (Papp et al. 2017; Peintner et al. 2019). Also, the enzymatic activities of Fomes fomentarius s.l. are a subject of research (Větrovský et al. 2013).
Fomes (Fr.) Fr. is a small genus characterized by perennial basidiomes with a mycelial core consisting of distinct sclerids and large basidiospores. Although several hundred historic names of Fomes spp. have been published, only two species, F. fomentarius and F. fasciatus (Sw.) Cooke are currently widely accepted (Dai 2012; McCormick et al. 2013a, 2013b; Ryvarden and Melo 2014; Rivoire 2020).
Recent studies revealed a remarkable variability in DNA sequences of the ITS region between the ribosomal RNA gene (ITS) of F. fomentarius (Judova et al. 2012; Gáper et al. 2013) in Slovakia (Central Europe). The studies revealed two different genotypes with distinct host and habitat preferences. The variability between the genotypes can be quantified as 10 changes within 650 nucleotide positions of the ITS region or 97% sequence similarity between the respective genotypes (Judova et al. 2012). The two genotypes were later delimited as two different species F. fomentarius s.s. and F. inzengae (Ces. & De Not.) Cooke (Peintner et al. 2019). F. inzengae is a historical name—the respective basionym Polyporus inzengae Ces. & De Not. was published in 1861, and its combination in Fomes in 1885, but the species was for a long time treated as a synonym or a form of F. fomentarius. Badalyan et al. (2022) and Zhuykova and Mukhin (2022) discussed whether genetic divergence between F. fomentarius s.s. and F. inzengae within the ITS region (9–11 bp or 1.85% of nucleotide substitutions per site on average) is sufficient to treat both genetic lineages as separate species. DNA sequences of other gene regions have not been sufficiently applied for species delimitation in F. fomentarius s.l., but Pristas et al. (2013) confirmed that phylogenetic analyses based on DNA sequences of partial translation elongation factor 1-alpha gene (TEF1) and the large subunit (25S) of ribosomal RNA gene (LSU) clearly separated the two cryptic species, and topologies of phylograms based on different genetic markers were in agreement. The standard for species delimitation in fungi is the genealogical concordance method which uses multiple genetic loci to assess the limits of recombination among different genetic lineages by multigene phylogeny (Taylor et al. 2000). The topologies of phylograms based on either ITS, LSU, and TEF1 sequences of F. fomentarius and F. inzengae seem to be in agreement (Judova et al. 2012; Pristas et al. 2013), so the two lineages could be treated as separate species. Another genetic marker useful for species delimitation in the Fomes sp. is the RNA polymerase II, the second largest subunit (McCormick et al. 2013a), but this has not been applied yet for the study of genetic variability in European F. fomentarius or F. inzengae.
Both, Fomes fomentarius and F. inzengae are characterized by perennial hoof-shaped (ungulate) basidiomes which can reach a width of up to 60 cm and a weight of several kilograms. The upper surface of the basidiome is smooth, zonate and sulcate, glabrous with a thick and hard crust, pale brown, reddish brown to gray, and cracked when old. The hymenophore is poroid, the pore surface is ochraceous to grey, and pores are small with thick, entire tomentose dissepiments. The context is brownish, tough-fibrous, thick, and homogenous, black with KOH. The mycelial core of varying size is developing at the upper part of the context next to the substrate composed of white hyphae and brown tissue. Hyphal system is trimitic, generative hyphae thin-walled, colorless, branched, with clamps, inconspicuous; skeletal hyphae thick-walled, aseptate, pale yellowish brown, binding hyphae yellowish brown, thick-walled, frequently branched, aseptate. The mycelial core is composed of a mixture of skeletal binding hyphae and irregularly shaped thick-walled sclerids. Cystidia are absent, and cystidioles are present. Basidia are cylindric, with four sterigmata, enlarged base, and a basal clamp. Basidiospores are cylindric, thin-walled, smooth, and negative in Melzer’s reagent. Spore powder is whitish, apparent in spring during a short sporulation period.
F. fomentarius s.l. is growing on living and dead broadleaved trees, rarely on conifers (Peintner et al. 2019) causing heart rot of the simultaneous white rot decay type (Schwarze 2007). The latent symptomless presence of the fungi has also been repeatedly confirmed in the functional sapwood of intact trees (Baum et al. 2003; Parfitt et al. 2010; own observation).
The information about the local distribution and host preferences of the two species is infrequent (Badalyan et al. 2022; Zhuykova and Mukhin 2022). Peintner et al. (2019) also proposed several microscopic characters (basidiospore size, linear density of pores, diameter of skeletal hyphae in pure culture and basidiomes) for the identification of F. fomentarius and F. inzengae, but the respective dimensions were not in agreement with our preliminary data.
The aims of this work are (A) to obtain the information about distribution, host, and habitat preferences of Fomes fomentarius and F. inzengae in the South Moravian Region (South-East part) of Czechia where the occurrence of both species could be expected, (B) the critical evaluation of morphological characters possibly applicable for morphological identification of the two Fomes spp. using the statistically relevant data, and (C) multigene phylogeny of the two species using standard markers: LSU, TEF1, the largest subunit of RNA polymerase II (RPB1), and the second largest subunit of RNA polymerase II (RPB2).
Material and methods
The basidiomes of Fomes spp. were collected during 2021–2022 in various woodland habitats (natural and managed forests, parks, and urban greenery). The research was focused on species distribution in diverse woodland habitats along the elevational gradient in the South Moravian Region of Czechia with several reference specimens from other regions of Czechia and Slovakia.
Morphological characters
The comparison of the linear density of pores (pores per cm on the hymenophore surface) was done under a stereomicroscope (Olympus SZX12). The hymenophore was photographed with an enclosed piece of millimeter paper, and the values were read from the photos in the Inscape v. 0.92.1 program. In total, 15 values per specimen were measured.
The spore prints were obtained after incubation of basidiomes on microscope slides for 1–3 days at room temperature. To avoid contamination by spores from the air, each basidiome was covered by a beaker or a bell jar. The basidiospores were measured under the microscope, mounted in water or Melzer’s reagent, 30 basidiospores per specimen were measured. For basidiospores, the factors E (the ratio of the spore length to its width for each spore) and Q (the mean of E values for each specimen) were determined. The skeletal hyphae were prepared from the context tissues, and the diameters of 30 different hyphae per specimen were measured. The microscopic measurements were made using the Olympus BX50 microscope (magnification 1000 ×), with a camera and the QuickPhoto Micro program.
The specimens with a sufficient number of spores were deposited in the herbarium of the Moravian museum in Brno (BRNM).
Morphometric charecteristics were analysed with R 4.1.2 (https://www.R-project.org/). For each parameter, the difference between the species (i.e. the effect of species on the respective parameter) was tested by means of comparing the full model with the corresponding null model (without the respective parameter) using the likelihood-ratio test. All traits except for Q were analysed by means of generalized linear mixed models (GLMM) implemented in the lme4 package (Bates et al. 2015) with individual specimen treated as random factor. Q was modelled by the Gamma generalized linear model (GLM) with the inverse link function (package stats; https://www.R-project.org/). Since Q is calculated from all specimen per species, it was not possible to account for the differences between specimen by including the random factor. The linear density of pores was modelled by the GLMM with the Poisson error structure. The presence of overdispersion was tested by a function created and recommended by Bolker, one of the authors of the lme4 package (https://bbolker.github.io/mixedmodels-misc/glmmFAQ.html#overdispersion). This is based on Pearson residuals that are less biased than deviance residuals for this type of computation (McCullagh and Nelder 2019). The skeletal hyphae width and spore length and width were analysed using the Gamma GLMM with the logarithmic link function. In the case of E, the Gamma GLMM with the inverse link function was used.
DNA sequencing
Cultures were isolated from the context tissue of the fresh basidiomes on Petri dishes with malt extract agar (Himedia, India) for 1–2 weeks at 21 °C. The small piece of mycelium of fresh culture was used as a template for PCR using Phire Plant Direct PCR Master Mix (Thermo Scientific). The amplifications of ITS, LSU, TEF1, RPB1, and RPB2 were conducted according to standard protocols (Westphalen et al. 2022; Antonín et al. 2022).
Phylogeny
The ITS sequences were supplemented with those of F. fomentarius, F. inzengae and Fomes fasciatus published by Badalyan et al. (2022), Gáper et al. (2013), Judova et al. (2012), McCormick et al. (2013a), Náplavová et al. (2020), Peintner et al. (2019), and Zhuykova and Mukhin (2022). Moreover, the sequences of Globifomes graveolens (Schwein.) Murrill retrieved from the Genbank were added to the ITS dataset. Except the main ITS datasets including the large sampling of new previously published sequences, five regions were used, ITS, LSU, RPB1, RPB2, and TEF1, for the phylogenetic analysis with ten isolates available for each region, five per species. The phylogenetic trees were inferred for each of these regions individually as well as for their concatenation. Several closely related species pairs of Polyporaceae were added to all datasets of DNA sequences. Always, only specimens who have sequences of all five regions available were selected: Funalia gallica (Fr.) Bondartsev & Singer and Funalia trogii (Berk.) Bondartsev & Singer; Hexagonia apiaria (Pers.) Fr. and Hexagonia glabra Lév.; Perenniporia tephropora (Mont.) Ryvarden and Perenniporia subtephropora B.K. Cui & C.L. Zhao (Ji et al. 2023; Justo et al. 2017; Li et al. 2014; Zhao and Cui 2013). Grifola frondosa (Dicks.) Gray was selected as an outgroup of all datasets.
The main ITS dataset and multigene concatenated dataset were analysed by means of maximum likelihood (ML) and Bayesian inference (BI) algorithms, the single gene datasets were analyzed by BI only. The best-fitting partitioning schemes were found via PartitionFinder 2 (Lanfear et al. 2016) based on the corrected Akaike Information Criterion (AICc) with each codon being used as a separate data block to account for differences between the individual codon positions. For all the datasets, the analysis was done using both linked and unlinked branch lengths, with identical results obtained. The set of all partitioning schemes was evaluated (the option search = all;). The resulting partitioning schemes are listed in Supplementary Table 1.
The ML phylogenetic analysis was carried out with RAxML-NG 1.1.0 (Kozlov et al. 2019). The evolutionary model best describing the data was selected by PartitionFinder 2. All 84 models were used, including those with base frequencies estimated by ML (the parameter models = allx;). The MRE-based bootstopping test was applied to find out the necessary number of bootstrap replicates. The cutoff value was set to 0.01 (the option --bs-cutoff 0.01). Transfer Bootstrap Expectation (Lemoine et al. 2018) was used as a branch support measure. The presented phylogenetic trees are the best-scoring trees with the bootstrap support values mapped onto them.
The ML phylogenetic analysis was conducted within the BEAST 2 platform (Bouckaert et al. 2014). For all the datasets, the parameters were set up in the same way. The uncorelated log-normal relaxed molecular clock was used (Drummond et al. 2006). The best-fit evolutionary models for individual partitions were determined through model averaging implemented in the bModelTest package (Bouckaert and Drummond 2017). All the analyses utilized the Metropolis-coupled MCMC (MC3) alrogithm implemented in the CoupledMCMC package (Müller and Bouckaert 2019).Three heated and one cold chains were used. The chain length was always set to 20,000,000 (60,000,000 for concatenated multigene dataset) and every 5000th generation was sampled. Target switch probability was set to 0.234 (Kone and Kofke 2005; Atchadé et al. 2011). The burn-in was set to 25%.
The posterior parameter estimates were summarized with Tracer 1.7.1 (Rambaut et al. 2018). To assess the accuracy of the posterior estimates, the trace plots were inspected for the presence of a “hairy caterpillar” pattern indicating that the chains have mixed properly and reached a stationary distribution and the ESS values were checked, with the values ≥ 200 considered as indicative of good mixing of the MCMC (standard approach). Parameter estimates were summarized with TreeAnnotator 2.6.0 (part of BEAST 2) and mapped onto the 50% majority-rule consensus tree created with SumTrees 4.4.0 (Sukumaran and Holder 2010). Edge lengths were calculated as mean lengths of the corresponding edges in the input array of trees.
Genetic differentiation
The extent of genetic differentiation between Fomes fomentarius and F. inzengae was expressed by the fixation index (FST) defined by Weir and Cockerham (1984) and formally proven by Michalakis and Excoffier (1996) using the Arlequin 3.5.2.2 software (Excoffier and Lischer 2010).
Maps of distribution
The spatial distribution maps of Fomes specimens (Figs. 1, 2 and 3) were constructed with Quantum GIS, v. 3.6.2, using a digital elevation model and a base map of Czechia in 1:100 000 scale available from the WMS server (https://geoportal.cuzk.cz), using the data of EuroGeographics© for the administrative boundaries. The borders of natural reserves were vectorized from a base map of Czechia in 1:25 000 scale, obtained from the WMS server.

Distribution maps of Fomes spp. specimens in Czechia

Distribution maps of Fomes spp. specimens in the South Moravian region of Czechia

Co-occurrence of F. fomentarius and F. inzengae in the northern part of the South Moravian region (vicinity of Adamov town), Czechia
Results
Distribution, host spectrum, and habitat preferences of F. fomentarius and F. inzengae
We analysed 48 specimens of Fomes spp. from Czechia and two specimens from Slovakia. The ITS sequencing revealed 31 specimens of F. inzengae and 19 of F. fomentarius (Table 1, Figs. 1, 2, and 3). Results clearly delimited the ecological preferences of F. fomentarius and F. inzengae in the surveyed area. While F. fomentarius is distributed mainly at higher altitudes (400‒970 m a.s.l.) with beech (Fagus sylvatica) and birch (Betula spp.) as dominant hosts, F. inzengae is a lowland species (154–490 m a.s.l.) with a wide spectrum of hosts (Acer, Aesculus, Alnus, Betula, Fagus, Fraxinus, Quercus, Platanus, Populus, Salix, Sorbus, and Tilia). The contact zone of the two Fomes spp. seems to take place in the upper-colline vegetation belt (elevation ca 400–550 m a.s.l.) within the Mesophyticum phytogeographical region comprising mainly various types of mesic beech or hornbeam (Carpinus betulus) forests (Chytrý et al. 2017). The center of distribution of F. fomentarius continues at higher altitudes in forests with predominant beech (submontane and montane belts).
In contrast, F. inzengae is distributed at altitudes up to ca. 500 m a.s.l. in the Thermophyticum phytogeographical region. Thermophyticum includes warm areas of lowland and colline belts characterized by the occurrence of basiphilous oak and oak-hornbeam forests or softwood (dominant Salix and Populus) and hardwood (frequent Quercus) floodplain forests in lowland basins. The town parks and other urban areas sampled in this study are also located in Thermophyticum. The beech forests, a common habitat of F. fomentarius, are almost absent in Thermophyticum.
While F. fomentarius was collected mostly at higher altitudes, here we noticed a few specimens of this species at lower altitudes (< 400 m a.s.l.). This can be explained by either topographic shading of deep valleys which contain patches of vegetation resembling to vegetation belts of higher altitudes (F38 and F42) or association to an exotic host in an arboretum (specimen F1; unfortunately the exact identification of the Betula sp. at species level could not be done because the specimen was sampled from dead wood). Another exception is the locality of the specimen F36; despite the low elevation (273 m a.s.l.), this area is located in upper-colline vegetation belt of Mesophyticum (Chytrý et al. 2017) and references therein. Most of the localities of F. fomentarius at lower altitudes are protected areas characterized by natural forest vegetation with dominant beech trees and a certain amount of dead wood.
Morphometric charecteristics
The size of 967 basidiospores was measured (F. fomentarius: 270 spores/12 specimens; F. inzengae: 697 spores/26 specimens). Unfortunately, less than the planned 30 spores were measured at some specimens, due to their low abundance in hymenium. The basidiospore size of F. fomentarius (14.2)15.3–20.6(22.9) × (4.3)5.3–6.9(7.9) µm and F. inzengae (14.0)15.3–19.7(22.6) × (4.0)5.1–7.4(8.0) µm resulted almost identical and differences between their length, widths, and Q were non-significant (Table 2, Fig. S1). The diameters of skeletal hyphae (F. fomentarius: 155 values/5 specimens; F. inzengae: 151 values/5 specimens) were higher in basidiomes of F. inzengae, but the differences were not significant (p = 0.085). The detected dimensions were larger than those presented by Peintner et al. (2019), some measured hyphae of F. inzengae were > 7 µm, especialy those of large basidiomes. The only significant difference between the two Fomes spp. was detected in the case of linear density of pores (p = 0.009), but the respective values overlap: F. fomentarius: (13) 15–25 (27) pores per cm; F. inzengae: (13) 16–27 (32) pores per cm. In total, 255 values/17 specimens for F. fomentarius and 450 values/30 specimens for F. inzengae were measured.
Phylogeny
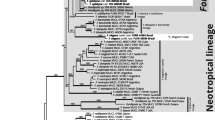
All 50 specimens of F. fomentarius and F. inzengae were provided with ITS sequences. The 10 selected specimens (5 of F. fomentarius and 5 of F. inzengae) were subjected to multigene sequence analyses of ITS, LSU, TEF1, RPB1, and RPB2 markers. The selected ITS sequences and all sequences of other markers are deposited in the GenBank database (Table 3), some of the ITS sequences were published by Cristini et al. (2023). The results of phylogenetic analyses of individual markers clearly delimited F. fomentarius and F. inzengae (Fig. S2). The resulting single-gene phylograms had identical topologies as the ITS phylogram of our dataset completed with previously published sequences (Fig. 4) and the phylogram of the multigene concatenated dataset (Fig. 5). Because incongruences among gene trees were not found and the two lineages are completely sorted, F. fomentarius and F. inzengae are distinct, well delimited species. The specimens of F. fomentarius from North America (Fig. 4) can belong to a geographically separated species (F. fomentarius aff. USA). Similarly, two ITS sequences of F. inzengae from China and Korea differ from the others and may form a separate genetically distinct lineage close to Globifomes graveolens which resulted as a sister species of F. inzengae. Hexagonia apiaria resulted as closely related to Fomes fasciatus and Fomes sp. from Mexico and unrelated to H. glabra. The DNA sequence datasets of this study are available within the Supplementary Information files (SI1a-c).

The phylogenetic tree of F. fomentarius and F. inzengae specimens based on the Bayesian analysis of the ITS region. Numbers at branches indicate maximum likelihood bootstrap proportion and Bayesian posterior probability values. For legend to specimen codes, see “Table 1”. The asterisks (*) mark low support (< 75 in maximum likelihood; < 0.9 in Bayesian analysis). The bar indicates the number of expected substitutions per position

The concatenated phylogenetic tree of F. fomentarius and F. inzengae specimens based on the maximum likelihood analysis of the combined ITS-LSU-RPB1-RPB2-TEF1 dataset. Numbers at branches indicate maximum likelihood bootstrap proportion and Bayesian posterior probability values. For legend to specimen codes, see “Table 1”. The bar indicates the number of expected substitutions per position
Genetic differentiation
The fixation index (FST) calculated for the 50 ITS sequences (31 sequences of F. inzengae and 19 thats of F. fomentarius) resulted FST = 0.991, showing a very high genetic differentiation (Wright 1978).
Discussion
F. fomentarius and F. inzengae are undoubtedly genetically separated species. Even though the ranges of the two species overlap (the populations are sympatric), the DNA sequences support their complete genetic separation and a gene flow between them is improbable. Co-occurrence of F. fomentarius and F. inzengae at one locality is possible, but infrequent. Judova et al. (2012) revealed both species (referred to as genotypes A and B) at some localities in East Slovakia. We confirmed both Fomes species (specimens F01 and F02, Fig. 3) at only one locality, which is an arboretum, where the fungal diversity can be influenced by cultivation of non-native tree species.
Our results confirmed the geographical distribution pattern of F. fomentarius and F. inzengae in Czechia is in agreement with those of the previous studies (Peintner et al. 2019; Badalyan et al., 2022; Zhuykova and Mukhin 2022). The two species are ecologically segregated by altitudinal and latitudinal vegetation zonation. F. inzengae has a southern, Mediterranean distribution (Peintner et al. 2019, Badalyan et al., 2022) extending its range to warmer areas in Central Europe, while F. fomentarius inhabits colder temperate woodlands. Apparently F. fomentarius is distributed mainly in mesic and (sub)montane beech forests in Central Europe. The range and distribution pattern of F. fomentarius and F. inzengae are similar to those of Armillaria cepistipes and A. gallica (Antonín et al. 2009) in the respective habitats. Náplavová et al. (2020) hypothesized the southern border of the occurrence of F. fomentarius s.s. (sublineage A2) in Europe being associated with the distribution of Betula pendula and the absence of F. inzengae on this host. Nevertheless, we confirmed B. pendula also as a host of F. inzengae (specimen F18).
F. fomentarius s.l. is reported not only from the Mediterranean and temperate biomes but also from subalpine birch forests in Fennoscandia (Ryvarden and Melo 2014). However, the identity of Fomes specimens in such a habitat is unclear. These may belong to F. fomentarius s.s. or a separate cryptic species. Nevertheless, the American lineage of F. fomentarius s.l. (named F. fomentarius aff. USA in this study—Fig. 4, or F. fomentarius II by McCormick et al. 2013a) is also confirmed from boreal forests in Alaska (McCormick et al. 2013b).
Especially when Fomes spp. can be dispersed with nursery stock due to its latent presence in the sapwood of healthy trees, its spread to an arboretum with a planted tree can not be excluded.
The most typical hosts of F. fomentarius s.s. are Fagus and Betula, less common Acer, Alnus, Picea, and Populus in Europe (Peintner et al. 2019); Alnus, Betula, Prunus, Salix, and Sorbus in the Ural regions in Russia and North Kazakhstan (Zhuykova and Mukhin 2022) and Fagus and Quercus in Armenia (Badalyan et al. 2022). Hosts of F. inzengae are Quercus, Carpinus, Castanea, Cerasus, Platanus, Populus, and Abies in Europe (Peintner et al. 2019), Acer, Populus, Salix, Tilia in the Ural regions in Russia and North Kazakhstan (Zhuykova and Mukhin 2022), and Carpinus, Juglans, Fagus, Populus, and Salix in Armenia (Badalyan et al. 2022). Most of these host genera of F. inzengae were also confirmed by our results. Obviously the host spectrum of Fomes spp. follows the local diversity of tree species including both native and non-native tree species (e.g. Aesculus hippocastanum and Platanus × hispanica are non-native hosts of F. inzengae revealed in this study), so the occurrence of Fomes spp. is not determined by specific host species, but by environmental conditions. Surprisingly, F. inzengae was also isolated from roots of Festuca paniculata in the alpine grassland in France (Mouhamadou et al. 2011), but its possibility to form basidiomes in such a habitat is questionable.
Identification of F. fomentarius and F. inzengae according to morphological characters of basidiomes is hardly possible. Because the morphological characters such as linear density of pores, spore size, and skeletal hyphae diameter overlap between F. fomentarius and F. inzengae, these characters can not be used for their reliable identification. Spore size of the F. inzengae detected by us do not correspond with those by Peintner et al. (2019), who published values of (9.0) 10–12 (12.5) × (2.8) 3.0–3.5 (3.8) µm, Q = (2.8) 3.0–3.6 (3.7), which are markedly smaller. Our results are closer to the values of F. fomentarius s.l. stated by different authors (Ryvarden and Melo 2014; Bernicchia 2005; Niemelä, 2005; Rivoire 2020).
Unfortunately, the only reliable method for identification of the two Fomes spp. is the DNA barcoding and ITS region is sufficiently informative to distinguish the species. Nevertheless, the ITS-RFLP analysis proposed by Judova et al. (2012) can help decrease the costs of DNA sequencing.
Our results also revealed that Globifomes graveolens seems to be closely related to F. inzengae. Although the more comprehensive phylogeny of this American species is needed, results indicate that even though the morphology of its basidiome composed of small petaloid pilei differs from Fomes spp., this apomorphic character is not in agreement with phylogenetic position of the species. Another phylogenetic problem that was revealed in Hexagonia. H. apiaria is likely to be closely related to F. fasciatus than to H. glabra. Therefore, a critical revision of the genus including the type species H. crinigera Fr. is desirable.
Conclusions
The results of our survey confirm that Fomes inzengae and F. fomentarius are well delimited species, according to Genealogical Concordance Phylogenetic Species Recognition, and gene flow between them is unlikely. In contrast, morphological characters proposed for the identification of the two species by Peintner et al. (2019) resulted as unreliable. Either basidiospore size, diameter of skeletal hyphae, or linear density of pores can not help to distinguish Fomes inzengae from F. fomentarius. Nevertheless, the trends of geographic distribution of the two species. in different phytogeographical regions is obvious, although coocurrence can not be excluded in some habitats. In conclusion, for correct identification of F. inzengae and F. fomentarius, DNA sequencing (ITS region is sufficiently informative) is necessary.
Data availability
DNA sequence data are freely available in the Genbank. The DNA sequence datasets generated during the study are available as Supplementary information files; the morphometric data are available from the corresponding author on request.
References
Antonín V, Ďuriška O, Jančovičová S, Para R, Kudláček T, Tomšovský M (2022) Multilocus phylogeny and taxonomy of European Melanoleuca subgenus Melanoleuca. Mycologia 114:114–143. https://doi.org/10.1080/00275514.2021.1966246
Antonín V, Tomšovský M, Sedlák P, Májek T, Jankovský L (2009) Morphological and molecular characterization of the Armillaria cepistipes ‒ A. gallica complex in the Czech Republic and Slovakia. Mycol Prog 8:259–271. https://doi.org/10.1007/s11557-009-0597-1
Atchadé YF, Roberts GO, Rosenthal JS (2011) Towards optimal scaling of metropolis-coupled Markov chain Monte Carlo. Stat Comput 21:555–568. https://doi.org/10.1007/s11222-010-9192-1
Badalyan S, Zhuykova E, Mukhin V (2022) The phylogenetic analysis of Armenian collections of medicinal tinder polypore Fomes fomentarius (Agaricomycetes, Polyporaceae). Ital J Mycol 51:23–33. https://doi.org/10.6092/issn.2531-7342/14474
Bates D, Mächler M, Bolker BM, Walker SC (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48. https://doi.org/10.18637/jss.v067.i01
Baum S, Sieber TN, Schwarze FWMR, Fink S (2003) Latent infections of Fomes fomentarius in the xylem of European beech (Fagus sylvatica). Mycol Prog 2:141–148
Bernicchia A (2005) Polyporaceae s.l. Edizioni Candusso, Alassio
Bouckaert R, Drummond A (2017) bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol Biol 17:42
Bouckaert R, Heled J, Kühnert D et al (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537
Chytrý M, Danihelka J, Kaplan Z, Pyšek P (2017) Flora and vegetation of the Czech Republic. Springer, Cham
Cristini V, Nop P, Zlámal J et al (2023) Fomes fomentarius and F. inzengae ‒ a comparison of their decay patterns on beech wood. Microorganisms 11:679. https://doi.org/10.3390/microorganisms11030679
Dai YC (2012) Polypore diversity in China with an annotated checklist of Chinese polypores. Mycoscience 53:49–80. https://doi.org/10.1007/s10267-011-0134-3
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:699–710. https://doi.org/10.1371/journal.pbio.0040088
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Gáper J, Pristaš P, Gáperová S, Maliničová L (2013) Molecular identification of Fomes fomentarius in hosts from urban and suburban areas in Slovakia. Folia Oecol 40:22–27
Ji X, Sun YF, Wu DM et al (2023) An updated phylogenetic assessment and taxonomic revision of Perenniporia sensu lato (Polyporales, Basidiomycota). J Fungi 9:173. https://doi.org/10.3390/jof9020173
Judova J, Dubikova K, Gaperova S et al (2012) The occurrence and rapid discrimination of Fomes fomentarius genotypes by ITS-RFLP analysis. Fungal Biol 116:155–160. https://doi.org/10.1016/j.funbio.2011.10.010
Justo A, Miettinen O, Floudas D et al (2017) A revised family-level classification of the Polyporales (Basidiomycota). Fungal Biol 121(9):798–824. https://doi.org/10.1016/j.funbio.2017.05.010
Kone A, Kofke DA (2005) Selection of temperature intervals for parallel-tempering simulations. J Chem Phys 122:1–2. https://doi.org/10.1063/1.1917749
Kozlov A, Darriba D, Flouri T et al (2019) RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35(21):4453–4455. https://doi.org/10.1093/bioinformatics/btz305
Lanfear R, Frandsen PB, Wright AM et al (2016) PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34:772–773. https://doi.org/10.1093/molbev/msw260
Lemoine F, Domelevo Entfellner J, Wilkinson E et al (2018) Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 556:452–456. https://doi.org/10.1038/s41586-018-0043-0
Li HJ, Li XC, Vlasák J, Dai YC (2014) Neofomitella polyzonata gen. et sp. nov., and N. fumosipora and N. rhodophaea transferred from Fomitella. Mycotaxon 129:7–20. https://doi.org/10.5248/129.7
McCormick MA, Grand LF, Post JB, Cubeta MA (2013a) Phylogenetic and phenotypic characterization of Fomes fasciatus and Fomes fomentarius in the United States. Mycologia 105:1524–1534. https://doi.org/10.3852/12-336
McCormick MA, Cubeta MA, Grand LF (2013b) Geography and hosts of the wood decay fungi Fomes fasciatus and Fomes fomentarius in the United States. N Am Fungi 8:1–53. https://doi.org/10.2509/naf2013.008.002
McCullagh P, Nelder JA (2019) Generalized linear models, 2nd edn. Routledge, New York
Michalakis Y, Excoffier L (1996) A generic estimation of population subdivision using distances between alleles with special reference to microsatellite loci. Genetics 142:1061–1064
Mouhamadou B, Molitor C, Baptist F et al (2011) Differences in fungal communities associated to Festuca paniculata roots in subalpine grasslands. Fungal Divers 47:55–63. https://doi.org/10.1007/s13225-011-0091-3
Müller N, Bouckaert R (2019) Adaptive parallel tempering for BEAST 2. bioRxiv. https://doi.org/10.1101/603514
Náplavová K, Gáper J, Gáperová S et al (2020) Genetic and plant host differences of Fomes fomentarius in selected parts of Southern Europe. Plant Biosyst 154:125–127. https://doi.org/10.1080/11263504.2019.1701129
Niemelä T (2005) Polypores, lignicolous fungi. Norrlinia 13:105–106
Papp N, Rudolf K, Bencsik T, Czégényi D (2017) Ethnomycological use of Fomes fomentarius (L.) Fr. and Piptoporus betulinus (Bull.) P. Karst. in Transylvania Romania. Gen Resour Crop Evol 64:101–111. https://doi.org/10.1007/s10722-015-0335-2
Parfitt D, Hunt J, Dockrell D, Rogers HJ, Boddy L (2010) Do all trees carry the seeds of their own destruction? PCR reveals numerous wood decay fungi latently present in sapwood of a wide range of angiosperm trees. Fungal Ecol 3:338–346. https://doi.org/10.1016/j.funeco.2010.02.001
Peintner U, Kuhnert-Finkernagel R, Wille V, Biasioli F, Shiryaev A, Perini C (2019) How to resolve cryptic species of polypores: an example in Fomes. IMA Fungus 10:17. https://doi.org/10.1186/s43008-019-0016-4
Pristas P, Gaperova S, Gaper J, Judova J (2013) Genetic variability in Fomes fomentarius reconfirmed by translation elongation factor 1-alpha DNA sequences and 25S LSU rRNA sequences. Biologia 68:816–820. https://doi.org/10.2478/s11756-013-0228-9
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard M (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904
Rivoire B (2020) Polypores de France et d´ Europe. Mycopolydev, Orliénas
Ryvarden L, Melo I (2014) Poroid Fungi of Europe, 1st edn. Fungiflora, Oslo
Schwarze FWMR (2007) Wood decay under the microscope. Fungal Biol Rev 21:133–170. https://doi.org/10.1007/s11557-006-0052-5
Sukumaran J, Holder MT (2010) DendroPy: A Python library for phylogenetic computing. Bioinformatics 26:1569–1571. https://doi.org/10.1093/bioinformatics/btq228
Taylor JW, Jacobson DJ, Kroken S, Kasuga T et al (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31:21–32
Větrovský T, Baldrian P, Gabriel J (2013) Extracellular enzymes of the white-rot fungus Fomes fomentarius and purification of 1,4-β-Glucosidase. Appl Biochem Biotechnol 169:100–109. https://doi.org/10.1007/s12010-012-9952-9
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Westphalen MC, Motato-Vásquez V, Rajchenberg M et al (2022) New insights on Flaviporus (Polyporales) in the neotropics. Mycol Prog 21:93. https://doi.org/10.1007/s11557-022-01845-6
Wright S (1978) Evolution and the genetics of populations, volume 4.Variability within and among natural populations. University of Chicago Press, Chicago
Zhao C, Cui B (2013) Morphological and molecular identification of four new resupinate species of Perenniporia (Polyporales) from southern China. Mycologia 105:945–958. https://doi.org/10.3852/12-201
Zhuykova EV, Mukhin VA (2022) Diversity and ecological features of phylogenetic lineages of tinder fungus in the Urals. Russ J Ecol 53:366–372. https://doi.org/10.1134/S1067413622050113
Acknowledgements
We thank our colleagues who provided their specimens of Fomes spp. or assisted us during the collecting of specimens: V. Antonín, J. Beneschová, V. Cristini, J. Hrabáková, D. Janda, J. Kašák, M. Kolényová, L. Kotyz, I. Milenković, D. Palovčíková, L. Praus, J. Rozsypálek, J. Salaš, H. Ševčíková, and M. Trifković.
Funding
Open access publishing supported by the National Technical Library in Prague. This research was funded by the European Regional Development Fund, Project Phytophthora Research Centre CZ.02.1.01/0.0/0.0/ 15_003/ 0000453.
Author information
Authors and Affiliations
Contributions
Michal Tomšovský and Tomáš Kudláček contributed to the study conception and design. Material preparation and data collection were performed by Michal Tomšovský and Sirapitcha Kaeochulsri. Data analyses were performed by Tomáš Kudláček and Sirapitcha Kaeochulsri. László Benedek Dálya was responsible for the construction of the maps of distribution. The first draft of the manuscript was written by Michal Tomšovský and all authors commented on the previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Section Editor: Yu-Cheng Dai
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tomšovský, M., Kaeochulsri, S., Kudláček, T. et al. Ecological, morphological and phylogenetic survey of Fomes fomentarius and F. inzengae (Agaricomycetes, Polyporaceae) co-occurring in the same geographic area in Central Europe. Mycol Progress 22, 79 (2023). https://doi.org/10.1007/s11557-023-01928-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11557-023-01928-y