
Abstract
The xylariaceous genus Dematophora has recently been resurrected and segregated from Rosellinia based on a molecular phylogeny and morphological characters. This was an important taxonomic change because Dematophora in the current sense contains several important pathogens, while Rosellinia is limited to mainly saprotrophic species that have an endophytic stage in their life cycle and may even have beneficial effects on the host plants. During our ongoing work on the functional biodiversity of the Xylariales, we have encountered new strains of rosellinoid Xylariaceae from Iran and have studied their mycelial cultures for secondary metabolites in an attempt to establish further chemotaxonomic affinities. In the process, we isolated and identified 13 compounds, of which rosellisteroid (1), the cichorine derivative 2, and the alkaloid 3 are new. Out of these, nine were tested for their antimicrobial affinities with cytochalasin E (6) exhibiting weak activity against Schizosaccharomyces pombe. The cytotoxicity of three cytochalasin derivatives was examined and their effects on the F-actin cytoskeletal organization studied by fluorescence microscopy using fluorescent phalloidin. Cytochalasin E (6) and Δ6,12-cytochalasin E (7) showed strong and irreversible action on actin, while cytochalasin K (8) exhibited weaker, reversible effects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Xylariaceae is one of the largest families of the ascomycete class Sordariomycetes and is phenotypically characterized by geniculosporium-like anamorphs and the absence of stromatal pigments according to the recent classification by Wendt et al. (2018). Most species in this family are saprobes or endophytes, while some species are well-known plant pathogens (Helaly et al. 2018; Becker and Stadler 2021). Hyde et al. (2020) accepted 32 genera; however, it can be expected that this number will increase until a conclusive classification system has been established (e.g., Konta et al. 2020; Heredia et al. 2020). Furthermore, ongoing large-scale phylogenomic studies based on 3rd-generation genome sequencing techniques will strengthen future classification frameworks. This work has stabilized the phylogenetic backbone and has revealed that the genomes of the Xylariales contain a complex and extensive diversity of biosynthetic gene cluster encoding for secondary metabolites (Wibberg et al. 2021; Kuhnert et al. 2021). Especially taxa from tropical and subtropical areas are known to produce a high diversity of secondary metabolites, many of which are biologically active (Kuephadungphan et al. 2021). For instance, these fungi produce volatile antibiotics (Samarakoon et al. 2020), cytotoxic agents (Becker et al. 2021), and even inhibitors of biofilm formation in bacterial pathogens (Yuyama et al. 2017). In the sister family Hypoxylaceae, the use of chemotaxonomic methodology such as HPLC profiling has contributed greatly to the current, polyphasic taxonomic concepts, since secondary metabolite production was found to be widely in agreement with concurrent multi-locus genealogies and even phylogenomic reconstructions (Wendt et al. 2018; Wibberg et al. 2021).
One of the largest, but poorly studied, genera of the Xylariales is Rosellinia, which belongs to the Xylariaceae (Hyde et al. 2020). Recently, the fungus producing PF1022A, a cyclooctadepsipeptide, which is the precursor of the anthelmintic drug emodepside, was assigned to Rosellinia (Wittstein et al. 2020). Based on a multi-locus molecular phylogeny that was conducted in the same study, the genus Dematophora was resurrected for those Rosellinia species that have a characteristic synnematal anamorph. Dematophora now includes all important plant pathogens that were formerly placed in Rosellinia. However, the species segregation in these genera is based on morphology, and more than 100 of the ca. 160 morphologically described species remain uncultured and unassessed for secondary metabolite production or their phylogenetic affinities.
In the present study, we studied the secondary metabolites of Rosellinia and Dematophora species from Iran for previously undescribed secondary metabolites, complementing previous work by Wittstein et al. (2020). This work was also conducted as a starting point to obtain standards of the prevailing secondary metabolites for a subsequent chemotaxonomic study of these genera. Moreover, we aimed to isolate cytochalasins, F-actin-binding molecules, which are known to disrupt the eukaryotic actin cytoskeleton (Cooper 1987) to study their poorly understood structure activity relationships (Scherlach et al. 2010).
Materials and methods
Fungal sources/morphological observations
The fungal specimens were collected in Northern Iran (Guilan, Mazandaran, and Golestan provinces) and Isfahan Province. Dried specimens were deposited in the University of Guilan Mycological Herbarium (GUM). Living cultures were deposited in the culture collection MUCL (Louvain-la Neuve, Belgium) and Iranian Research Institute of Plant Protection (IRAN C), Tehran, Iran. For light microscopy, fresh collections were examined according to Petrini (2013). Single ascospore isolation and examination of culture macro-morphology were examined according to Pourmoghaddam et al. (2020). Observations and taxonomical diagnoses can be found in the SI.
DNA extraction, PCR, and sequencing
DNA extraction of fresh cultures and amplification of the ITS (nuc rDNA internal transcribed spacer region containing ITS1-5.8S-ITS2), LSU (5′ 1200 bp of the large subunit nuc 28S rDNA), RPB2 (partial second largest subunit of the DNA-directed RNA polymerase II), and TUB2 (partial β-tubulin) loci were carried out as described by Wendt et al. (2018). DNA sequences were generated by an in-house Sanger capillary sequencing solution and processed with Geneious® 7.1.9 (http://www.geneious.com).
Molecular phylogenetic analyses
Alignments and phylogenetic analyses of the combined multilocus matrix of ITS, LSU, TRPB2, and TUB2 DNA sequences using maximum parsimony and maximum likelihood were constructed as described in Lambert et al. (2021). For the combined matrix used for phylogenetic analyses, 59 accessions of 45 species of Xylariaceae and four outgroup taxa from Graphostromataceae (Biscogniauxia nummularia, Graphostroma platystomum) and Hypoxylaceae (Hypoxylon fragiforme, H. howeanum) were included. A table summarizing all used strains, their corresponding sequences, and GenBank accession numbers can be found in the SI (Table S1).
Cultivation, extraction, and isolation procedures
The fungal strains (see Table S1 in the SI, marked in bold) were grown on CS and CSA (cotton seed with and without agar supplement) (maltose 40 g/L, cotton seed flour 20 g/L, CaCO3 3 mg/L, soy peptone 2 g/L, MgSO4 × 7 H2O 2 g/L, NaCl 2 g/L, agar 20 g/L, pH adjusted to 6.3) media for 7 d at 23 °C in darkness. Fungal colonies grown on 90-mm agar plates were cut into pieces (1 × 1 cm) and regularly transferred to new CSA plates during the chemotaxonomic study. To compare their secondary metabolite production, the strains were inoculated in a 500-ml Erlenmeyer flask containing 200 ml of CS medium and shaken at 150 rpm in the dark until the glucose level was depleted (measured by using glucose test stripes) (Wittstein et al. 2020). In addition, the strains were inoculated on CSA plates and incubated in the dark at 23 °C until the mycelia covered two-thirds to the full agar plate (14–21 d). Extraction of the secondary metabolites followed Wittstein et al. (2020) with minor adaptations; briefly, the supernatant and biomass of submerged cultures were separated by vacuum filtration instead of gauze filtration, while the supernatant-ethyl acetate and solid-phase extraction (biomass harvested from submerged cultures, acetone; agar plates, ethyl acetate) procedures remained unchanged. The crude extract was analyzed by high-performance liquid chromatography coupled with diode array detection and mass spectrometry (HPLC-DAAD/MS) as described previously (Wittstein et al. 2020). The resulting chromatograms were analyzed using the Bruker Compass Data Analysis 4.4 software (Bruker Daltonik GmbH) and evaluated for the presence of known and unknown secondary metabolites.
Isolation of compounds 1 and 2
The crude extract derived from the supernatant produced by strains of Rosellinia corticium (MUCL 57712, MUCL 57715, IRAN 3731C, IRAN 3732C, GUM 1640, GUM 1641) and Rosellinia cf. akulovii (MUCL 57710) was pooled to afford 282 mg of combined crude extract. This crude extract was subjected to normal-phase flash chromatography (GRACE Reveleris X2 flash system) using a Reveleris 40 g silica cartridge (100% acetonitrile (ACN) for 6 min, 80:20 ACN/MeOH for 30 min, 60:40 ACN/MeOH for 6 min, 20:80 ACN/MeOH for 6 min, 100% MeOH for 6 min, UV detection at 220 nm, 255 nm and 360 nm), and two fractions (F1-F2) were selected for further purification. F1 (24.55 mg) was purified by NP-TLC (pre-coated TLC plates Silgur-25 UV 254, 20 × 20 cm, Macherey-Nagel, Düren, Germany) using 90% dichloromethane (DCM) and 10% acetone as mobile phase, yielding 1.46 mg of 1 and 0.35 mg of compound 2, which was then pooled with 0.3 mg of 2 obtained from purification of F2 (31.25 mg) by a PLC 2250 preparative HPLC system (Gilson, Middleton, WI, USA), using a Waters X-Bridge C18 column (250×19 mm, 5 μm) and in the following conditions: solvent A, H2O; solvent B, ACN; gradient, 0% B to 5% B for 5 min; 5–10% B in 5 min, followed by an increase to 23% B in 10 min, then increase to 45% B in 30 min, 45–60% B in 10 min, thereafter increase to 100% B in 5 min at a flow rate 15 mL/min.
Isolation of compounds 3–13
The cultivation of Rosellinia cf. akulovii (MUCL 57710) on 230 CSA plates provided 2.12 g of crude extract. One hundred twelve milligrams of the crude extract were subjected to normal-phase flash chromatography (GRACE Reveleris X2 flash system; Reveleris silica cartridge 12 g, solvent A: dichloromethane, solvent B: acetone; gradient 0% B to 100% B in 40 min, followed by 100% MeOH in 5 min). UV-Vis detection was carried out at 210 nm, 265 nm, and 365 nm. Fractions were combined according to the major UV-absorbing peaks to yield 1.3 mg of 4.
In a second attempt, 1.96 g of crude extract was subjected to normal-phase flash chromatography (GRACE Reveleris X2 flash system), using a Reveleris 24 g silica cartridge with the same elution gradient to afford eight main fractions (F1-F8). The resulting fractions were further subjected to preparative HPLC (GX-271/172 DAD/pumps 305/306, Gilson Inc., Middleton, USA). Deionized water (Milli-Q, Millipore, Schwalbach, Germany) with 0.1% formic acid (solvent A) and acetonitrile with 0.1% formic acid (solvent B) were used as mobile phase. Prior to the preparative HPLC, the fractions were filtered using a SPME Strata-X 33 u Polymeric RP cartridge (Phenomenex, Inc., Aschaffenburg, Germany). UV spectra of eluents were monitored at 210 nm, 254 nm, and 280 nm.
Compound 3 (3.6 mg) was isolated from fraction F5 (132 mg), using a VP Nucleodur 100-5 C18 ec column (150 × 40 mm, 7μm, Macherey-Nagel), elution gradient: 5% solvent B isocratic for 3 min, 5–100% solvent B in 50 min, thereafter isocratic conditions at 100% solvent B for 5 min at a flow rate 33 mL/min.
Using the same column and elution gradient, fraction F4 (94 mg) was further purified to give 1.6 mg of 5.
The cultivation of D. necatrix (MUCL 57709) on 260 CSA plates resulted in 2.98 g of crude extract. This crude extract was first subjected to normal phase flash chromatography (GRACE Reveleris X2 flash system) [Reveleris cartridge 24 g, solvent A: dichloromethane; solvent B: acetone (0% B to 100 B in 40 min) and 100% MeOH in 5 min]. UV detection was conducted at 210 nm, 265 nm, and 365 nm. Compound 11 (15.7 mg) and seven fractions (F1–F7) were collected according to the observed peaks.
These fractions were further separated via preparative HPLC (GX-271/172 DAD/pumps 305/306, Gilson). Deionized water (Milli-Q, Millipore, Schwalbach, Germany) with 0.1% formic acid (solvent A) and acetonitrile with 0.1% formic acid (solvent B) were used as mobile phase. Prior to the preparative HPLC, the fractions were filtered using a SPME Strata-X 33 μm Polymeric RP cartridge (Phenomenex, Inc., Aschaffenburg, Germany). UV detection was conducted at 210 nm, 254 nm, and 280 nm.
Fraction F3 (121 mg) was then purified with an elution gradient: 10% B isocratic for 3 min, followed by 10–80% for 40 min, thereafter increased to 100% B in 5 min, and finally isocratic conditions of 100% B for 5 min at a flow rate 33 mL/min, using a VP Nucleodur 100-5 C-18 ec column (150 × 40 mm, 7 μm: Macherey-Nagel), yielding 23 mg of subfraction F13.
Compound 10 (5.1 mg) was obtained from purification of subfraction F13 with the elution gradient 5% B isocratic for 5 min, 5–100% B in 45 min, followed by isocratic elution with 100% solvent B for 5 min, using a VP Nucleodur 100-5 C18 ec column (250 × 21 mm, 5μm, Macherey-Nagel; 12 ml/min flow rate).
Compound 9 (2.3 mg) was purified from fraction F4 (47.8 mg) with the following gradient: 30% solvent B for 5 min, 30–100% B solvent B for 45 min, and a final isocratic elution of 100% B for 5 min with a VP Nucleodur 100-5 C18 ec column (250 × 21 mm, 5μm, Macherey-Nagel; 12 ml/min flow rate).
Fraction F6 (208 mg) was then fractionated by preparative reverse-phase liquid chromatography, using a VP Nucleodur 100-5 C-18 ec column (150 × 40 mm, 7 μm; Macherey-Nagel). Elution gradient was used as 5% B isocratic for 5 min, 5–100% B in 45 min, followed by isocratic elution with 100% solvent B for 10 min at a flow rate 33 mL/min, yielding 48.3 mg of subfraction F16. Subfraction F16 was further fractionated by preparative HPLC (solvent A [deionized water with 0.05% TFA] and solvent B [acetonitrile with 0.05% TFA]) with an elution gradient 15% B isocratic for 5 min, 15–80% B in 35 min, using a VP Nucleodur 100-5 C18 ec column (250 × 21 mm, 5 μm, Macherey-Nagel; 12 ml/min flow rate) to yield 40.8 mg of fraction F26. This fraction was further purified using a PLC 2250 preparative HPLC system (Gilson, Middleton, WI, USA), with a XBridge® Prep C18 5 μm OBDTM column (250 × 19 mm, 5 μm, Waters, Milford, MA, USA) as the stationary phase (gradient: 5% B for 5 min, 5–30% B in 5 min, 30% B isocratic for 30 min, 30–70% in 20 min, flow rate: 15 ml/min) to give 7 (1.6 mg), 8 (1.6 mg), and 35.4 mg of cytochalasin E (6).
The ethyl acetate extracts of strains R. corticium (MUCL 57712 and MUCL 57713), derived from 50 CSA plates from each strain, were pooled to afford 670 mg of crude extract. This extract was then subjected to reversed-phase flash chromatography (GRACE Reveleris X2 flash system), using a Reveleris 80 g C18 cartridge (solvent A: H2O, solvent B: ACN, gradient: 5–100% B in 50 min, 100% B isocratic for 10 min, and finally isocratic conditions for 100% MeOH for 10 min) to afford two fractions (F1, 12.4 mg; F2, 7.77 mg), which were subsequently brought to purity by NP-TLC resulting in 1.23 mg of 12 (mobile phase: 80% EtAc, 20% n-heptane) and 0.9 mg of 13 (mobile phase: 90% DCM, 10% acetone, adjusted to 1% formic acid concentration), respectively.
Marfey’s analysis
Preparation of authentic amino acid–derived d/l-FDVA
Authentic amino acid–derived d-/l-FDVA were prepared according to Primahana et al. (2021). In detail, d-, l-, or d-/l-mixtures of authentic amino acids dissolved in H2O were prepared with a concentration of 50 mM. An aliquot of the respective amino acids (25 μL) was taken and dried under N2 stream. Afterwards, the dried authentic amino acids were added with NaHCO3 (1m, 10 μL) and d- or l-FDVA (Nα-(2,4-dinitro-5-fluorophenyl)-d-/l-valinamide) (50 μL) and heated at 40 °C for 1 h. After 1 h, the mixtures were neutralized with HCl (1n, 10 μL), diluted with ACN (405 μL) and subjected for LC-MS analysis.
Marfey analysis on C4 column (C4 Marfey analysis)
Analyses of isoleucine (Ile) stereoisomers (isoleucine and allo-isoleucine) were performed using a previously described method (Primahana et al. 2021).
Determination of absolute amino acid stereochemistry in xylarotide (4)
Amino acid absolute stereochemistry of xylarotide A (4) was determined according to Primahana et al. (2021). Briefly, xylarotide A (4; 100 μg) was acid hydrolyzed using HCl (100 μL, 6n) at 110 °C for 14 h. The acid hydrolysate was dried under N2 stream and divided into two separated vials. The hydrolysates were treated with Marfey reagent (d or l FDVA) as described above.
Instrumentation for spectral data
An Agilent 1200 series HPLC-UV system (Santa Clara, CA, USA) with an ESI-TOF-MS (Maxis, Bruker, Bremen, Germany) (column 2.1 × 50 mm, 1.7 μm, C18 Acquity UPLC BEH) (Waters, Eschborn, Germany), with deionized water + 0.1% formic acid (solvent A) as well as acetonitrile + 0.1% formic acid (solvent B) and a gradient of 5% B for 0.5 min increasing to 100% B in 19.5 min, maintaining 100% B for 5 min, flow rate 0.6 mL/min, UV detection 200–600 nm, was used to obtain ESI-HR-MS mass spectra. NMR spectra were recorded with an Avance III 700 spectrometer with a 5-mm TCI cryoprobe (1H 700 MHz, 13C 175 MHz) and an Avance III 500 spectrometer (1H 500 MHz, 13C 125 MHz; both Bruker, Billerica, MA/USA). NMR data were referenced to selected chemical shifts of acetone-d6 (1H: 2.05 ppm, 13C: 29.32 ppm), chloroform-d (1H: 7.27 ppm, 13C: 77.00 ppm), and methanol-d4 (1H: 3.31 ppm, 13C: 49.15 ppm), respectively.
Spectral data
Rosselisteroid (1): colorless oil; 1H NMR (700 MHz, acetone-d4): see Table 1; 13C NMR (175 MHz, acetone-d4): see Table 1; HR-ESI-MS: m/z 629.3662 (M + Na)+ (calculated for C34H54O9Na, 629.3660), 607.3842 (M + H)+ (calculated for C34H55O9, 607.3841), 589.3736 (M + H–H2O)+ (calculated for C34H53O8, 589.3735), 445.3312 (M + H–C6H10O5)+ (calculated for C28H45O4, 445.3312).
Cichorine derivative (2): colorless, amorphous solid; 1H NMR (700 MHz, CHCl3-d): δH 6.12 (s, 2–H), 4.65 (dd, J = 11.4, 1.9 Hz, H–1’a), 4.39 (s, 3–H2), 4.08 (dd, J = 11.4, 9.2 Hz, H–1’b), 3.95 (dd, J = 9.2, 1.9 Hz, H–2’), 3.79 (s, 4–OMe), 2.24 (s, 5–Me), 1.43 (s, 5’–CH3), 1.35 (s, 4’–CH3); 13C NMR (175 MHz, CHCl3-d): δC 170.1 (C, C–1), 146.6 (C, C–4), 141.9 (C, C–6), 137.5 (C, C–7), 128.2 (C, C–3a), 124.4 (C, C–5), 117.0 (C, C–7a), 78.9 (CH, C–2’), 70.8 (C, C–3’), 65.0 (C, CH2–1’), 60.4 (CH3, 4–OMe), 42.7 (CH2, C–3), 26.2 (CH3, C–5’), 25.2 (CH3, C–4’), 9.4 (CH3, 5–Me); HR-ESI-MS: m/z 609.2424 (2M + Na)+ (calculated for C30H38N2O10Na, 609.2419), 587.2605 (2M + H)+ (calculated for C30H39N2O10, 587.2599), 316.1156 (M + Na)+ (calculated for C15H19NO5Na, 316.1155), 294.1337 (M + H)+ (calculated for C15H20NO5, 294.1336), 276.1230 (M + H–H2O)+ (calculated for C15H18NO5, 276.1230).
2-Formyl-5-(methoxymethyl)-1H-pyrrole-1-pentanoic acid (3): colorless, amorphous solid; 1H NMR (700 MHz, CH3OH-d4): δH 9.44 (s, 7–H), 6.96 (d, J = 4.1, H–3), 6.26 (d, J = 4.1, H–4), 4.49 (s, H2–6), 4.34 (m, H2–1’), 3.36 (s, 6–OMe), 2.18 (t, J = 7.6, H2–4’), 1.74 (m, H2–2’), 1.63 (quin, J = 7.6, H2–3’); 13C NMR (175 MHz, CHCl3-d): δC 184.4 (C, C–5’), 181.1 (CH, C–7), 141.2 (C, C–5), 133.9 (C, C–2), 125.7 (CH, C–3), 112.8 (CH, C–4), 66.7 (CH2, C–6), 58.4 (CH3, 6–OMe), 46.8 (CH2, C–1’), 38.9 (CH2, C–4’), 32.7 (CH2, C–2’), 25.1 (CH2, C–3’); HR-ESI-MS: m/z 262.1049 (M + Na)+ (calculated for C12H17NO4Na, 262.1050), 240.1226 (M + H)+ (calculated for C12H18NO4, 240.1230), 222.1120 (M + H–H2O)+ (calculated for C12H16NO3, 222.1125), 208.0963 (M + H–H2O–OCH3)+ (calculated for C11H14NO3, 208.0968), 194.1170 (M + H–CH2O2)+ (calculated for C11H16NO2, 194.1176), 180.1012 (M + H–C2H4O2)+ (calculated for C10H14NO2, 180.1019).
Xylarotide A (4): colorless, amorphous solid; 1H NMR (500 MHz, CHCl3-d): δH 6.57 (br d, J = 8.5 Hz, 18-NH), 5.17 (d, J = 7.8 Hz, 2–H), 4.86 (dt, J = 9.0, 7.5 Hz, 24–H), 4. 57 (t, J = 7.8 Hz, 7–H), 4.20 (br t, J = 7.5 Hz, 18–H), 4.07 (br t, J = 10.0 Hz, 13–H), 3.74 (br t, J = 9.5 Hz, 5–Ha), 3.54 (m, 5–Hb), 3.17 (s, 29–H3), 2.15–2.27 (m, 14–H, 3–Ha), 1.80–2.00 (m, 3–Hb, 4–Ha, 4–Hb), 1.39–1.75 (m, 20–H, 9–H, 26–H, 25–H2, 8–H2, 19–H), 1.12 (quin, 20–H2), 0.87–0.99 (m, 22–H3, 27–H3, 28–H3, 21–H3, 11–H3, 15–H3, 16–H3, 10–H3); HR-ESI-MS: m/z 572.3785 (M + Na)+ (calculated for C29H51N5O5Na, 572.3782), 550.3966 (M + H)+ (calculated for C29H52N5O5, 550.3963), data is in good aggreement with those of Li et al. (2011).
Nodulisporisteroid C (5): colorless, amorphous solid; 1H NMR (700 MHz, CH3OH-d4): δH 3.98 (qd, J = 6.2, 3.4 Hz, 4–H), 2.73 (t, J = 8.7 Hz, 17–H), 2.37 (m, 14–H), 2.35 (m, 2–Ha), 2.22 (m, 16–Ha), 2.19 (m, 11–Ha), 2.14 (s, 21–H3), 2.13 (m, 11–Hb), 2.07 (m, 12–Ha), 2.03 (m, 7–Ha), 1.99 (m, 1–Ha), 1.96 (m, 2–Hb), 1.94 (m, 7–Hb), 1.82 (m, 1–Hb), 1.75 (m, 12–Hb), 1.74 (m, 15–Ha), 1.74 (m, 6–Ha), 1.72 (m, 16–Hb), 1.59 (dt, J = 12.2, 3.1 Hz, 5–H), 1.43 (m, 15–Hb), 1.38 (m, 6–Hb), 1.16 (d, J = 6.2 Hz, 22–H3), 0.97 (s, 19–H3), 0.57 (s, 18–H3); 13C NMR (175 MHz, CH3OH-d4): δC 212.5 (C, C–20), 179.1 (C, C–3), 133.6 (C, C–8), 133.0 (C, C–9), 68.2 (CH, C–4), 63.5 (CH, C–17), 53.6 (CH, C–14), 47.6 (CH, C–5), 45.0 (C, C–13), 41.2 (C, C–10), 37.4 (CH2, C–12), 34.1 (CH2, C–1), 31.7 (CH3, C–21), 30.9 (CH2, C–2), 28.9 (CH2, C–7), 25.3 (CH2, C–15), 24.2 (CH2, C–16), 23.9 (CH2, C–11), 22.6 (CH3, C–19), 21.1 (CH3, C–22), 20.2 (CH2, C–6), 13.5 (CH3, C–18); HR-ESI-MS: m/z 385.2347 (M + Na)+ (calculated for C22H34O4Na, 385.2349), 363.2531 (M + H)+ (calculated for C22H35O4, 363.2530), 345.2429 (M + H–H2O)+ (calculated for C22H33O3, 345.2424), 327.2319 (M + H–2 H2O)+ (calculated for C22H31O2, 327.2319), 309.2209 (M + H–3 H2O)+ (calculated for C22H29O, 309.2213), data is in good agreement woth those of Zhao et al. (2015).
Cytochalasin E (6): colorless, amorphous solid; 1H NMR (500 MHz, acetone-d6): δH 7.56 (br s, 2–NH), 7.29 (br t, J = 7.5 Hz, 3’–H, 5’–H), 7.20–7.25 (m, 2’–H, 4’–H, 6’–H), 6.36 (d, J = 11.8 Hz, 20–H), 5.92 (ddd, J = 15.0, 10.0, 1.8 Hz, 13–H), 5.79 (d, J = 11.8 Hz, 19–H), 5.24 (ddd, J = 15.0, 10.8, 4.2 Hz, 14–H), 4.40 (br s 18–OH), 3.92 (m, 3–H), 3.19 (dqd, J = 10.5, 6.9, 2.5 Hz, 16–H), 3.04 (dd, J = 5.2, 2.4 Hz, 4–H), 2.89 (dd, J = 13.5, 6.0 Hz, 10–Ha), 2.84 (dd, J = 13.5, 5.3 Hz, 10–Hb), 2.51 (m, 15–Ha), 2.51 (m, 8–H), 2.15 (m, 15–Hb), 2.08 (m, 5–H), 1.46 (s, 23–H3), 1.22 (s, 12–H3), 1.12 (d, , J = 6.9 Hz, 22–H3), 0.94 (d, J = 7.3 Hz, 11–H3); 13C NMR (125 MHz, acetone-d6): δC 213.0 (C, C–17), 170.3 (C, C–1), 150.2 (C, C–22), 142.5 (CH, C–20), 137.6 (C, C–1’), 132.2 (CH, C–14), 131.2 (2× CH, C–2’, C–6’), 129.6 (CH, C–13), 129.2 (2× CH, C–3’, C–5’), 127.6 (CH, C–4’), 121.8 (CH, C–19), 87.9 (C, C–9), 77.6 (C, C–18), 61.3 (CH, C–7), 57.8 (C, C–6), 53.7 (CH, C–3), 47.7 (CH, C–4), 47.1 (CH, C–8), 44.7 (CH2, C–10), 41.2 (CH, C–16), 40.1 (CH2, C–15), 37.0 (CH, C–5), 25.2 (CH3, C–24), 20.5 (CH3, C–23), 19.7 (CH3, C–12); HR-ESI-MS: m/z 518.2145 (M + Na)+ (calculated for C28H33NO7Na, 518.2149), 496.2324 (M + H)+ (calculated for C28H34NO7, 496.2330), 478.2220 (M + H–H2O)+ (calculated for C28H32NO6, 478.2224), 434.2324 (M + H–H2O–CO2)+ (calculated for C27H32NO4, 434.2326), 416.2216 (M + H–2 H2O–CO2)+ (calculated for C27H30NO3, 416.2220); data are in good agreement with those reported by Takamatsu et al. (2002).
Δ6,12-cytochalasin E (7): colorless, amorphous solid; 1H NMR (700 MHz, CH3OH-d4): δH 8.44 (br s, 2–NH), 7.28 (br t, J = 7.5 Hz, 3’–H, 5’–H), 7.22 (br t, J = 7.5 Hz, 4’–H), 7.14 (br d, J = 7.5 Hz, 2’–H, 6’–H), 6.29 (d, J = 11.8 Hz, 20–H), 5.79 (d, J = 11.8 Hz, 19–H), 5.56 (ddd, J = 15.1, 9.7, 1.1 Hz, 13–H), 5.33 (br s, 12–Ha), 5.24 (ddd, J = 15.1, 10.8, 3.8 Hz, 14–H), 5.13 (br s, 12–Hb), 3.71 (br d, J = 10.5 Hz, 7–H), 3.43 (m, 3–H), 3.20 (m, 5–H), 3.07 (dqd, J = 10.8, 6.9, 2.5 Hz, 16–H), 3.03 (dd, J = 4.8, 3.2 Hz, 4–H), 2.92 (dd, J = 14.0, 4.8 Hz, 10–Ha), 2.89 (br t, J = 10.5 Hz, 8–H), 2.68 (dd, J = 14.0, 5.2 Hz, 10–Hb), 2.49 (ddd, J = 13.6, 11.6, 10.8 Hz, 15–Ha), 2.12 (ddt, J = 13.6, 3.8, 2.0 Hz, 15–Hb), 1.45 (s, 24–H3), 1.10 (d, J = 6.9 Hz, 23–H3), 1.03 (d, J = 6.9 Hz, 11–H3); 13C NMR (175 MHz, CH3OH-d4): δC 213.5 (C, C–17), 172.3 (C, C–1), 151.3 (C, C–6), 151.0 (C, C–21), 142.5 (CH, C–20), 137.7 (C, C–1’), 133.5 (CH, C–14), 131.6 (2× CH, C–2’, C–6’), 130.2 (CH, C–13), 129.6 (2× CH, C–3’, C–5’), 128.0 (CH, C–4’), 122.8 (CH, C–19), 114.4 (CH2, C–12), 88.3 (C, C–9), 78.7 (C, C–18), 71.3 (CH, C–7), 54.5 (CH, C–3), 49.5 (CH, C–8), 47.2 (CH, C–4), 43.5 (CH2, C–10), 42.2 (CH, C–16), 40.6 (CH2, C–15), 33.3 (CH, C–5), 24.7 (CH3, C–24), 20.5 (CH3, C–23), 14.7 (CH3, C–11); HR-ESI-MS: m/z 518.2143 (M + Na)+ (calculated for C28H33NO7Na, 518.2149), 496.2322 (M + H)+ (calculated for C28H34NO7, 496.2330), 434.2334 (M + H–H2O–CO2)+ (calculated for C27H32NO4, 434.2326), data are in good agreement with those reported by Takamatsu et al. (2002).
Cytochalasin K (8): colorless, amorphous solid; 1H NMR (700 MHz, CH3OH-d4): δH 7.32 (br t, J = 7.5 Hz, 3’–H, 5’–H), 7.24 (br t, J = 7.5 Hz, 4’–H), 7.18 (br d, J = 7.5 Hz, 2’–H, 6’–H), 6.52 (d, J = 11.7 Hz, 20–H), 5.98 (ddd, J = 15.2, 10.0, 1.7 Hz, 13–H), 5.87 (d, J = 11.7 Hz, 19–H), 5.24 (ddd, J = 15.2, 10.8, 3.8 Hz, 14–H), 3.80 (br s, 4–H), 3.77 (br d, J =10.0 Hz, 7–H), 3.50 (ddd, J = 8.3, 4.9, 0.8 Hz, 3–H), 3.07 (dqd, J = 10.8, 6.9, 2.5 Hz, 16–H), 2.94 (dd, J = 13.4, 4.8 Hz, 10–Ha), 2.73 (m, 10–Hb), 2.73 (m, 8–H), 2.57 (ddd, J = 13.6, 11.6, 10.8 Hz, 15–Ha), 2.14 (m, 15–Hb), 1.64 (m, 12–H3), 1.48 (m, 24–H3), 1.36 (m, 11–H3); HR-ESI-MS: m/z 518.2145 (M + Na)+ (calculated for C28H33NO7Na, 518.2149), 496.2325 (M + H)+ (calculated for C28H34NO7, 496.2330), 434.2322 (M + H–H2O–CO2)+ (calculated for C27H32NO4, 434.2326), 416.2217 (M + H–2 H2O–CO2)+ (calculated for C27H30NO3, 416.2217); data are in good agreement with those reported by Liu et al. (2006).
Bioactivity, cytotoxicity assessment, and F-actin disruption assay
Antimicrobial activities were measured by serial dilution (minimal inhibitory concentration (MIC)) in a 96-well format for the following organisms: Schizosaccharomyces pombe (DSM 70572), Pichia anomala (DSM 6766), Mucor hiemalis (DSM 2656), Candida albicans (DSM 1665), Rhodotorula glutinis (DSM 10134), Acinetobacter baumanii (DSM 30008), Escherichia coli (DSM 1116), Bacillus subtilis (DSM 10), Mycobacterium smegmatis (ATCC 700084), Staphylococcus aureus (DSM 346), Pseudomonas aeruginosa (PA14), and Chromobacterium violaceum (DSM 30191) as described previously (Mulwa et al. 2018). Cytotoxic effects of compounds 6, 7, and 8 were evaluated indirectly by measuring the decline of metabolic activity using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) against various human cancer cell lines (KB 3.1, PC-3, MCF-7, SKOV-3, A431, and A549) and a mouse fibroblast (L929) cell line as described previously (Sandargo et al. 2020). These standard tests for initial assessment of antimicrobial and cytotoxic effects were carried out using duplicates of each concentration tested for all compounds and the appropriate standard antibiotics as positive controls. The cell lines were purchased from DSMZ (Braunschweig, Germany).
Subsequently, the compounds were examined in a eukaryotic F-actin network disruption assay using adherent mammalian osteosarcoma (U2-OS, ATCC HTB-96) cells. This cell line has already been used to describe effects of cytochalasins on the actin organization in previous publications (Kretz et al. 2019; Moussa et al. 2020; Lambert et al. 2021). Briefly, 20,000 cells of a maintained cell culture were sown on fibronectin-coated glass coverslips and expanded overnight. The growth medium for the assay was equilibrated in an incubator overnight. Cells were treated with full medium supplied with a concentration gradient ranging from 0.01 to 10 μg/ml (0.01 μg/ml, 0.03 μg/ml, 0.1 μg/ml, 0.3 μg/ml, 1 μg/ml, 3 μg/ml, 10 μg/ml) and subsequently, varying concentrations for compounds 6 (0.13 μg/ml, 0.65 μg/ml) and 7 (0.084 μg/ml, 0.42 μg/ml) and compound 8 (7.2 μg/ml, 36 μg/ml) dissolved in DMSO. Cells treated with 10 μl/ml DMSO served as a vehicle control. After 1 h, cells were washed once with PBS and fixed with PBS containing 4% para-formaldehyde (PFA). In a separate experiment, the reversibility of the cytochalasins’ impact on the F-actin organization was examined by wash-out after 1 h of treatment with PBS (3×) and addition of equilibrated full medium, allowing cells to recover for 1 h. Cells were then fixed using 4% PFA dissolved in PBS. After permeabilization with 0.1% Triton X-100 at room temperature for 1 min, all cells fixed on cover slips were washed with PBS and stained for the actin cytoskeleton by administering phalloidin coupled to a fluorescent probe (ATTO-594, ATTO-Tec, Siegen, Germany) for 1 h. The cover slips were embedded using Pro-long Diamond Antifade (Invitrogen, Carlsbad, CA, USA) mountant supplied with DAPI to stain for the nucleus. Epifluorescence was detected and images captured using either an inverted microscope (Axio Vert 135 TV, Zeiss, Jena, Germany) with a 63× Zeiss oil immersion objective (Neofluar, 1.25 NA) equipped with a PCOedge 4.2 (sCMOS, PCO, Germany) camera operated by the Metamorph software package (molecularDevices, San Jose, CA, USA) or a Nikon Ti eclipse 2 microscope with a 60× Nikon oil immersion objective (plan apo, NA 1.4) equipped with a pE-4000 LED light source (CoolLED, Andover, UK) and PCOedge 4.2 camera and operated by NIS elements (Nikon, Düsseldorf Germany). Images were processed by Image J (NIH, Bethesda, MD, USA).
Results
Morphological analysis and molecular identification
The specimens were identified by comparison of their morphological characteristics and sequencing of four loci (ITS, LSU, RPB2, and TUB2) (see molecular phylogenetic reconstruction and taxonomy, Figure S1, Figure S2, Figure S3, Figure S4, and Table S1 in Supplementary Information (SI)). Two well-known widely distributed species (Dematophora necatrix and Rosellinia corticium) were identified from Iran. Moreover, another taxon was found that may either constitute an undescribed taxon or be conspecific with Rosellinia akulovii. However, the clarification of the identity and status of the latter specimen will require additional work that is unrelated to the main objective of this manuscript. Additional specimens need to be made available and studied. For the time being, this fungus is referred to R. cf. akulovii, and the preliminary data are being reported in the Supplementary Information.
Secondary metabolites
All strains were screened for secondary metabolite production using submerged shake cultures in cotton seed medium and in static condition on cotton seed medium supplemented with agar (Wittstein et al. 2020). Submerged cultures were harvested after glucose was consumed and plates after the fungal strains covered approximately the whole plate. Shake cultures were divided into supernatant and mycelial biomass. Extracts were screened for peaks potentially representing novel compounds and cytochalasins. Briefly, compounds 1 and 2 were isolated from pooled extracts derived from the supernatant of R. corticium and R. cf. akulovii, compounds 12 and 13 from combined agar extracts of R. corticium (MUCL 57712 and MUCL 57713), compounds 3–5 from agar extracts derived from strain R. cf. akulovii (MUCL 57710), and compounds 6–11 from a D. necatrix (MUCL 57709)–derived extract grown on agar plates. A complete list of the occurrence of each isolated metabolite in all strains is given in the SI (Table S2).
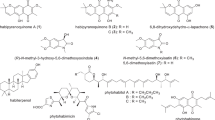
Rosselisteroid (1) in Fig. 1 was isolated as an oil by preparative HPLC and displayed a molecular ion sodium adduct at m/z 629.3662 in the HRESIMS spectrum, corresponding to a molecular formula of C34H54O9 and consequently eight degrees of unsaturation. 1H and HSQC spectra of 1 (Table 1) showed the presence of six methyls, eight methylenes, one olefinic methine, and eight oxy- and seven other sp3 hybridized methines. The 13C NMR spectrum indicated the further presence of a conjugated ketone and four other carbon atoms devoid bound protons. The planar structure of 1 was constructed utilizing COSY, TOCSY, and HMBC data. COSY and TOCSY NMR spectra provided the spin systems 1’–H–6’–H2, which was identified at an early stage as a sugar unit, 1–H2/2–H2, 6–H2/7–H, 11–H2/12–H2, and 14–H–28–H3. These spin systems were connected by HMBC correlations (Figure S1) and established the steroid backbone glycosylated with a pyranose. Subsequently, the stereochemistry was tentatively assigned by ROESY correlations, proton-proton-coupling constants, and comparison of 13C chemical shifts to structurally related compounds. The strong ROESY correlations between 1’–H and 3’–H/5’–H in combination with the absence of ROESY correlations of long range couplings from 4’–H indicated a β-d-mannose moiety (Agrawal et al. 1992), which was confirmed by characteristic 13C chemical shift data (observed δC = 102.2, 72.6, 75.8, 69.5, 77.5, 63.3 ppm; methyl pyranoside δC = 94.5, 72.1, 74.0, 67.7, 77.0, 62.0 ppm). The β-configuration was further confirmed by the 1JC1’,H1’ coupling constant of 156 Hz, observed in the HMBC NMR spectrum. Since 1’–H and 2’–H show ROESY correlations to 20–H/21–H3 and 17–H3, respectively, a 23S stereochemistry was concluded (Laskowski et al. 2019). Intraside chain ROESY correlations were specific for the 22R,23S,24R configuration (Stoldt et al. 1998; Drosihn et al. 1999). More precisely, the strong ROESY correlations between 20–H/22–H and 20–H/28–H3 are characteristic for this relative configuration, which was analyzed by molecular modeling by Drosihn et al. (1999). ROESY correlations between H–7 and H–15a and H–15b indicate the epoxide being in α orientation (Fig. S4). Because the configuration at the stereo centers C–9, C–10, C–13, C–14, and C–17 is the same for all steroids from natural sources, the stereochemistry of 1 is tentatively proposed to be 7R,8S,9R,10R,13R,17R,20S,22R,23S,24R.

Chemical structures of rosselisteroid (1); the new cichorine derivative (2); the pyrrol alkaloid (3); xylarotide A (4); nodulisporisteroid C (5); cytochalasin E (6); Δ6,12-cytochalasin E (7); cytochalasin K (8); 5-carboxymellein (9); methylparaben (10); 4-hydroxybenzamide (11); 1H-indole-3-carboxylic acid methylester (12); (10E,12Z)-9-hydroxyocata-10,12-dienoic acid (13). Spectral data are compiled in the SI (Figs. S5–S29 and Tables S4 and S5)
The HRESIMS data of metabolite 2 supported a molecular formula of C15H19NO5, implying 7 degrees of unsaturation. 1H and HSQC NMR data accounted for four methyls, two methylenes, and one oxymethine. The 13C NMR spectrum contained signals for six further aromatic carbons devoid bound protons and one carboxyl atom. HMBC correlations from 3–H2, 5–Me, and 4–OMe connected the indole core, which was connected to a 3-methylbutanetriolyl subunit (see Fig. S60 for details).
The molecular formula C12H17NO4 of the pyrrol alkaloid 3 was deduced from its HRESIMS data, implying five numbers of unsaturation. 1H and HSQC NMR data accounted for one oxymethyl, five methylenes, two olefinic methines, and a conjugated aldehyde. The 13C NMR spectrum showed signals for a carboxylic acid two additional aromatic carbons. COSY correlations connected the 1–H2–4–H2 side chain, whereas HMBC correlations assembled 3 as 2-formyl-5-(methoxymethyl)-1H-pyrrole-1-pentanoic acid (see Fig. S60 for details).
Determination of the absolute stereochemistry of xylarotide A (4)
Xylarotide A (4) was first isolated from an unidentified Xylaria sp. collected from Gaoligong Mountain (Li et al. 2011). This compound was allegedly assigned to Xylaria by ITS-nDNA sequence data. However, sequence information has not been deposited by the authors in the public domain, and such an “identification” procedure is imprecise, for Xylariales and many other Ascomycota (Hyde et al. 2020; Becker et al. 2021). The structure of xylarotide A (4) had been elucidated on the basis of 1D- and 2D-NMR spectroscopy and ESI-HR-MS, but the absolute configuration of the amino acids has never been described. The determination of amino acid absolute configuration of xylarotide A (4) using Marfey analysis and comparison to the authentic amino acid–derived d-/l-FDVA revealed the presence of l-proline (d-FDVA, tR = 7.2 min), l-valine (d-FDVA, tR = 8.9 min), l-leucine (d-FDVA, tR = 9.7 min), and N-Me-l-leucine (d-FDVA, tR = 9.6 min). Since isoleucine has four stereoisomers (d-/l-isoleucine and d-/l-allo-isoleucine), its absolute configuration was determined using the previously described method using the C4 column (Uhrinova et al. 1991) and revealed the presence of l-isoleucine (C4 Marfey analysis, tR = 22.01 min). Chromatographic and spectral data (AmaZon system) can be found in the Supplementary Information (Figs. S25-S29 and Tables S4 and S5).
Bioactivity assays and actin disruption assays
Compounds 1–13 were evaluated by a set of serial dilution assays to determine MIC that are employed as standard procedure for characterization growth inhibition. Not all compounds were tested because for some, only small quantities were available after structure elucidation. For compounds 3, 4, 6–11, and 13 (Supplementary Information, Table S3), only compound 6 inhibited the growth of Schizosacharomyces pombe at 16.8 μM. A summary of tested organisms and effects of compounds is given in the Supplementary Information, Table S3. Next, we determined 50% inhibition of cellular survival by assessing metabolic reduction of MTT (compounds 6–8) with a panel of tested eukaryotic cell lines.
The cytochalasins (6–8) belong to a class of molecules that are known as potent F-actin disrupting toxins (Scherlach et al. 2010) and were evaluated for their cytotoxicity against a panel of eukaryotic cell lines (Fig. 2A). All exhibited moderate cytotoxicity against all tested cell lines. Notably, compound 8 was about 80 times less toxic than its close derivatives 6 and 7, which only differ by the presence of a double bond in the 6-ring system (7), putting the attached methyl groups into a planar position and an epoxide at C-12 (compound 6). As cytochalasans are well known F-actin network disruptors, the cellular bioactivity against the cellular target actin was evaluated in a functional assay in U2-OS cells using fluorescence microscopy. We examined the effect of the three cytochalasins on the F-actin network in cells fixed and stained for F-actin using fluorescent phalloidin upon cytochalasin treatment and DMSO treatment as vehicle control. A titration experiment ranging from 0.01 to 10 μg/ml (Fig. 2B) showed different degrees of F-actin network disruption severity, which was accompanied by a substantial reduction of F-actin bundles in the cell body. Higher concentrations of compounds 6 and 7 (> 0.3 μg/ml) lead to a total collapse of the F-actin network, while the F-actin network antagonizing effect of compound 8 was only observed at a higher concentration (10 μg/ml). Interestingly, concentrations of compounds 6, 7, and 8 that lead to significant alterations in the F-actin network approximately corresponded to the determined IC50 value determined in mouse fibroblasts L929. This prompted us to test the toxins at a “low-dose” (1 × IC50) and a “high dose” (5 × IC50), resulting in partial (Fig. 2C, panels a, b, c) or total (panels d, e, f) actin cytoskeletal network disruption. Moreover, a wash-out experiment upon high-dose treatment demonstrated the irreversible impact of compounds 6 and 7 on the F-actin cytoskeletal organization after a recovery time of 1 h (Fig. 2C; compare d and g; e and h), while the F-actin network partially recovered from a high dosage of compound 8 (Fig. 2C; compare f and i).

A Table summarizing half-maximal inhibitory concentrations (IC50; in μM) of the cytochalasin derivatives 6, 7 and 8 measured by inhibititon of metabolic activity with MTT against various eukaryotic fibroblast and human cancer cell lines; B Overlay of the fluorescence signal of the F-actin network (pseudocoloured in red) and nuclear DNA (blue), of U2-OS cells treated for 1 h with increasing concentrations of compounds 6, 7, 8 and DMSO as vehicle control as indicated, stained with fluorescently tagged phalloidin- and DAPI, respectively. Arrowheads represent previously determined IC50 concentrations, corresponding with the onset of visible perturbations on the actin cytoskeletal organization; C Overlay images of recorded fluorescence signal of visualized F-actin and nuclear DNA as above. Images show U2-OS cells treated for 1 h with concentrations of compound 6, 7, 8 equaling their determined IC50 (low dose; 0.13 μg/ml, (a) 0.084 μg/ml, (b) 7.2 μg/ml, (c) or 5× the IC50 (high dose; 0.65 μg/ml, (d) 0.42 μg/ml, (e) 36 μg/ml, f). After 1 h of high-dose treatment, the test compounds were washed-out, the cells supplemented with fresh medium and given 1 h of recovery time prior to fixation and staining for F-actin reorganization (g, h, i). DMSO was used as a vehicle control in a 1 h treatment experiment (j) and 1 h treatment followed by 1 h recovery after washout (k). Scale bars represent 20μm
Discussion
With hyfraxins A, we recently isolated a related steroid to the novel compound 1 from the Ash dieback causal fungus Hymenoscyphus fraxineus (Surup et al. 2018). Rosselisteroid (1) shares the conjugated C–3 ketone and the β-d-mannose moiety attached to C–23. However, the stereo configuration at C–23 is contrary and the π-system of 1 is truncated by the C8/C9 epoxide.
The new metabolite 2 is closely related with cichorine, a phytotoxin isolated from Alternaria cichorii, a phytopathogen of Russian knapweed (Stierle et al. 1993). From Lycium chinense fruits, another chemically similar derivative has been isolated (Chin et al. 2003), which, however, bears a butanoic acid side chain instead of the pentanoic moiety of the novel pyrol alkaloid 3. This compound showed hepatoprotective effects comparable to the standard, silybin.
The cyclopeptide 4 has been previously encountered in a Xylaria strain (Li et al. 2011), and we confirmed the report by these authors that lacks antimicrobial effects. The steroid 5 was previously isolated from a Nodulisporium sp. grown on rice medium and found to be devoid of cytotoxicity against a panel of human cancer cell lines (Zhao et al. 2015). Interestingly, nodulisporium-like anamorphs can be observed in all genera in the Hypoxylaceae and even in some Rosellinia species (Wittstein et al. 2020).
The cytochalasin 6 is well known to be produced by D. necatrix ((Aldridge et al. 1972), as Rosellinia necatrix) and discussed as a pathogenicity factor (Kanematsu et al. 1997), while the occurrence of 8 has been discussed to represent either a natural compound, or a degradation product of 6. The isomer 7 has so far only been obtained through acid rearrangement of 6 (Steyn and van Heerden 1982; Kajimoto et al. 1989). Since we used solvents supplemented with TFA, it cannot be formally excluded that 7 and 8 might have been isolated as artifacts. Besides their phytotoxic effect, cytochalasans are well known to interfere with F-actin polymerization (Cooper 1987) and actin cytoskeletal dependent biological phenomena such as cell adhesion (Takamatsu et al. 2002) and is discussed as an anti-angiogenetic and tumor growth suppressing agent (Udagawa et al. 2000). Since 6, 7, and 8 only differ by an epoxide (6 versus 7 and 8) and an endo- or exocyclic position of a double bond, influencing the stereochemistry of the methyl groups (compounds 7 and 8), the here isolated compounds are ideal for experiments to gain deeper insights into the structure activity relationship of cytochalasans. Interestingly, 6 and 7 share almost equal cytotoxic properties with comparable F-actin network disrupting activities, while the toxicity and effect of 8 appears strongly reduced. Liu et al. (2006) observed a similar phenomenon while testing the cytotoxicity of 6 and 8 in the P388D1 (mouse macrophage, ATCC: CCL-46) and A549 (human lung epithelial, ATCC: CCL-185) cell lines, suggesting the epoxide moiety to strongly increase the toxicity of 6. Since we observed 6 and 7 to exhibit a comparable cytotoxicity, this rather speaks for a role of the positioning of the substituents in the 6-ring system through the placing of the double bond. In a previous study, no correlations between differently positioned double bonds on the strength of F-actin network disruptive capabilities could be drawn (compare 19,20-epoxycytochalasin C and 19,20-epoxycytochalasin D, 18 and 19 in Kretz et al. (2019)). This observation highlights that a chemical change in the 6-ring system alone is not sufficient to influence its F-actin binding capabilities by such drastic extent as has been observed in this study. This leads us to the conclusion that the stereochemical properties of the 6-ring have to influence the cytotoxicity and F-actin network disrupting capabilities in the cytochalasin E backbone, if one excludes the possibility of a reduced membrane permeability due to the structural differences in the molecules. Additionally, the effect of the tested compounds on the F-actin organization did not appear to be reversible, in line with previous results suggesting the keto function to be an important structural parameter to influence reversibility (Lambert et al. 2021). It should be noted that a partial F-actin network re-organization after 1 h recovery time has been observed with compound 8, indicating its weaker affinity for actin.
Metabolite 9 is known from a static culture extract of Dematophora arcuata ((Oppong et al. 2009), as Rosellinia arcuata) and other fungi, including other species of Xylariaceae (Schmeda-Hirschmann et al. 2005; Chinworrungsee et al. 2011; Wu et al. 2011; Xu et al. 2020). It was reported to possess weak to moderate cytotoxic, antimicrobial and even antimalarial activities, indicating a weak, non-selective mode of action in biological systems. We could not reproduce the antimicrobial and cytotoxic effects reported in our standard assays, which may be due to the use of different parameters and strains.
Metabolite 10 has been isolated from other fungi of the Xylariales belonging to the Hypoxylaceae (Annulohypoxylon stygium (Cheng et al. 2014)) and Graphostromataceae (Biscogniauxia cylindrospora (Cheng et al. 2011)), with this study being the first record from Rosellinia spp. No bioactivity was reported in either of the above-cited papers, and we could also not detect any significant effect.
Metabolite 11 has been previously isolated from an endophytic isolate of Colletotrichum gloeosporioides and reported to exhibit antifungal activity against Cladosporium cladosporioides and Cladosporium sphaerospermum (Chapla et al. 2014; Kim and Shim 2019). The compound was inactive against the tested strains in this study. The small compound 12 has previously been isolated from a Xylaria strain and did not show any antimicrobial or cytotoxic effects (Sun et al. 2017). Like the fatty acid 13, it proved to be devoid of significant activities in our own bioassays, too.
Conclusion
The current study has investigated Dematophora and Rosellinia species from Iran. Representative strains of two known and one potentially new species were characterized taxonomically and intensively examined for secondary metabolites in their cultures including their isolation by means of preparative chromatography and their structure elucidation using NMR spectroscopy and other state-of-the-art methodology. Interestingly, three out of 13 compounds characterized turned out to be unprecedented natural products belonging to several different molecule classes, again indicating the creativity of the Xylariaceae with respect to secondary metabolite production. Most of the major components in the extracts did not show prominent biological activities in a panel of standard assays that is established as a routine procedure in our laboratory. They may possess other biological function that could not be detected by these methods and may be involved in intraspecific communication or the defense against nematodes or other organisms that the fungi are confronted with in their natural environments. The described compounds add towards the repertoire of chemodiversity of Rosellinia and Dematophora. The isolated compounds can serve as standards for our pending, comprehensive studies on the chemotaxonomy of the Xylariaceae as more strains become available. Moreover, the spectral data reported here will easily allow other scientists to compare the metabolite profiles of their strains. In addition, the characterization of three cytochalasins in cell-based assay provided important information on the structure-activity relationships of this compound class on the actin cytoskeleton.
Data availability
All data except for the DNA sequences, which are deposited in GenBank (https://www.ncbi.nlm.nih.gov/genbank/) are available in the Supplementary information.
References
Aldridge DC, Burrows BF, Turner WB (1972) The structures of the fungal metabolites cytochalasins E and F. JCS Chem Comm 3:148–149. https://doi.org/10.1039/C39720000148
Becker K, Stadler M (2021) Recent progress in biodiversity research on the Xylariales and their secondary metabolism. J Antibiotics 74:1–23. https://doi.org/10.1038/s41429-020-00376-0
Becker K, Pfütze S, Kuhnert E, Cox R, Stadler M, Surup F (2021) Hybridorubrins A−D, novel azaphilone heterodimers from stromata of Hypoxylon fragiforme and insights into the biosynthetic machinery for azaphilone diversification. Chem Eur J 27:1438–1450. https://doi.org/10.1002/chem.202003215
Chapla VM, Zeraik ML, Leptokarydis IH, Silva GH, Bolzani VS, Young MCM, Pfenning LH, Araújo AR (2014) Antifungal compounds produced by Colletotrichum gloeosporioides, an endophytic fungus from Michelia champaca. Molecules 19:19243–19252. https://doi.org/10.3390/molecules191119243
Cheng MJ, Wu MD, Hsieh SY, Chen IH, Yuan GF (2011) Secondary metabolites isolated from the fungus Biscogniauxia cylindrospora BCRC 33717. Chem Nat Comp 47:527. https://doi.org/10.1007/s10600-011-9988-z
Cheng MJ, Wu MD, Chen JJ, Cheng YC, Hsieh MT, Hsieh SY, Yuan GF, Su YS (2014) Secondary metabolites from the endophytic fungus Annulohypoxylon stygium BCRC 34024. Chem Nat Comp 50:237–241. https://doi.org/10.1007/s10600-014-0921-0
Chin YW, Lim SW, Kim SH, Shin DY, Suh YG, Kim YB, Kim YC, Kim J (2003) Hepatoprotective pyrrole derivatives of Lycium chinense fruits. Bioorg Med Chem Lett 13:79–81. https://doi.org/10.1016/S0960-894X(02)00846-6
Chinworrungsee M, Kittakoop P, Isaka M, Rungrod A, Tanticharoen M, Thebtaranonthm Y (2011) Antimalarial halorosellinic acid from the marine fungus Halorosellinia oceanica. Bioorg Med Chem Lett 11:1965–1969. https://doi.org/10.1016/S0960-894X(01)00327-4
Cooper JA (1987) Effects of cytochalasin and phalloidin on actin. J Cell Biol 105:1473–1478. https://doi.org/10.1083/jcb.105.4.1473
Drosihn S, Porzel A, Voigt B, Brandt W, Wagner C, Merzweiler K, Adam G (1999) Conformational studies of two new brassinosteroid analogues with a 22,23-trans diol function. J Chem Soc Perkin Trans 2:233–238. https://doi.org/10.1039/A807440B
Helaly SE, Thongbai B, Stadler M (2018) Diversity of biologically active secondary metabolites from endophytic and saprotrophic fungi of the ascomycete order Xylariales. Nat Prod Rep 35:992–1014. https://doi.org/10.1039/C8NP00010G
Heredia G, Li DW, Wendt L, Réblová M, Arias RM, Gamboa-Angulo M, Štěpánek V, Stadler M, Castañeda-Ruíz RF (2020) Natonodosa speciosa gen. et sp. nov. and rediscovery of Poroisariopsis inornata: neotropical anamorphic fungi in Xylariales. Mycol Prog 19:15–30. https://doi.org/10.1007/s11557-019-01537-8
Hyde KD, Norphanphoun C, Maharachchikumbura SSN, Bhat DJ, Jones EBG, Bundhun D et al (2020) Refined families of Sordariomycetes. Mycosphere 11:305–1059. https://doi.org/10.5943/mycosphere/11/1/7
Kajimoto T, Imamura Y, Yamashita M, Takahashi K, Shibata M, Nohara T (1989) Nuclear magnetic resonance studies of cytochalasin E and its decomposition product. Chem Pharm Bull 37:2212–2213. https://doi.org/10.1248/cpb.37.2212
Kanematsu S, Hayashi T, Kudo A (1997) Isolation of Rosellinia necatrix mutants with impaired cytochalasin E production and its pathogenicity. Ann Phytopathol Soc Jpn 63:425–431. https://doi.org/10.3186/jjphytopath.63.425
Kim JW, Shim SH (2019) The fungus Colletotrichum as a source for bioactive secondary metabolites. Arch Pharm Res 42:735–753. https://doi.org/10.1007/s12272-019-01142-z
Konta S, Hyde KD, Phookamsak R et al (2020) Polyphyletic genera in Xylariaceae (Xylariales): Neoxylaria gen. nov. and Stilbohypoxylon. Mycosphere 11:2629–2651. https://doi.org/10.5943/mycosphere/11/1/17
Kretz R, Wendt L, Wongkanoun S, Luangsa-ard JJ, Surup F, Helaly SE, Noumeur SR, Stadler M, Stradal TEB (2019) The effect of cytochalasans on the actin cytoskeleton of eukaryotic cells and preliminary structure–activity relationships. Biomolecules 9:73. https://doi.org/10.3390/biom9020073
Kuephadungphan W, Macabeo APG, Luangsa-ard JJ, Stadler M (2021) Discovery of novel biologically active secondary metabolites from Thai mycodiversity with antiinfective potential. Curr Res Biotechnol 3:160–172. https://doi.org/10.1016/j.crbiot.2021.05.003
Kuhnert E, Navarro-Muñoz JC, Becker K, Stadler M, Collemare J, Cox RJ (2021) Secondary metabolite biosynthetic diversity in the fungal family Hypoxylaceae and Xylaria hypoxylon. Stud Mycol 99:100118. https://doi.org/10.1016/j.simyco.2021.100118
Lambert C, Pourmoghaddam MJ, Cedeño-Sanchez M, Surup F, Khodaparast SA, Krisai-Greilhuber I, Voglmayr H, Stradal TEB, Stadler M (2021) Resolution of the Hypoxylon fuscum complex (Hypoxylaceae, Xylariales) and discovery and biological characterization of two of its prominent secondary metabolites. J Fungi 7:131. https://doi.org/10.3390/jof7020131
Laskowski T, Szwarc K, Szczeblewski P, Sowiński P, Borowski E, Pawlak J (2019) Monosaccharides as potential chiral probes for the determination of the absolute configuration of secondary alcohols. J Nat Prod 79:2797–2804. https://doi.org/10.1021/acs.jnatprod.6b00471
Li YY, Hu ZH, Shen YM (2011) Two new cyclopeptides and one new nonenolide from Xylaria sp. 101. Nat Prod Comm 6:1843–1846. https://doi.org/10.1177/1934578X1100601214
Liu R, Gu Q, Zhu W, Cui C, Fan G, Fang Y, Zhu T, Liu H (2006) 10-Phenyl-[12]-cytochalasins Z7, Z8, and Z9 from the marine-derived fungus Spicaria elegans. J Nat Prod 69:871–875. https://doi.org/10.1021/np050201m
Moussa AY, Lambert C, Stradal TEB, Ashrafi S, Maier W, Stadler M, Helaly SE (2020) New peptaibiotics and a cyclodepsipeptide from Ijuhya vitellina: isolation, identification, cytotoxic and nematicidal activities. Antibiotics 9:132. https://doi.org/10.3390/antibiotics9030132
Mulwa LS, Jansen R, Praditya DF, Mohr KI, Okanya PW, Wink J, Steinmann E, Stadler M (2018) Lanyamycin, a macrolide antibiotic from Sorangium cellulosum, strain Soce 481 (Myxobacteria). Beilstein J Org Chem 14:1554–1562. https://doi.org/10.3762/bjoc.14.132
Oppong EK, Edwards RL, Maitland DJ, Whalley AJS, Ameyaw Y (2009) Secondary metabolites from static cultures of the fungus Rosellinia arcuata. Int J Appl Chem 5:145
Petrini LE (2013) Rosellinia—a world monograph. Bibliotheca Mycologica, Vo. 205. J. Cramer: Stuttgart, DE
Pourmoghaddam MJ, Lambert C, Surup F, Khodaparast SA, Krisai-Greilhuber I, Voglmayr H, Stadler M (2020) Discovery of a new species of the Hypoxylon rubiginosum complex from Iran and antagonistic activities of Hypoxylon spp. against the Ash Dieback pathogen, Hymenoscyphus fraxineus, in dual culture. MycoKeys 66:105–133. https://doi.org/10.3897/mycokeys.66.50946
Primahana G, Risdian C, Mozef T, Wink J, Surup F, Stadler M (2021) Amycolatomycins A and B, cyclic hexapeptides isolated from an Amycolatopsis sp. 195334CR. Antibiotics 10:261. https://doi.org/10.3390/antibiotics10030261
Samarakoon MC, Thongbai B, Hyde KD, Broenstrup M, Beutling U, Lambert C, Stadler M (2020) Elucidation of the life cycle of the endophytic genus Muscodor and its transfer to Induratia in Induratiaceae fam. nov., based on a polyphasic taxonomic approach. Fungal Divers 101:177–210. https://doi.org/10.1007/s13225-020-00443-9
Sandargo B, Michehl M, Stadler M, Surup F (2020) Antifungal sesquiterpenoids, rhodocoranes F-L from submerged cultures of the wrinkled peach mushroom, Rhodotus palmatus. J Nat Prod 83:720–724. https://doi.org/10.1021/acs.jnatprod.9b00871
Scherlach K, Boettger D, Remme N, Hertweck C (2010) The chemistry and biology of cytochalasans. Nat Prod Rep 27:869–886. https://doi.org/10.1039/B903913A
Schmeda-Hirschmann G, Hormozabal E, Astudillo L, Rodriguez J, Theoduloz C (2005) Secondary metabolites from endophytic fungi isolated from the Chilean gymnosperm Prumnopitys andina (Lleuque). World J Microbiol Biotechnol 21:27–32. https://doi.org/10.1007/s11274-004-1552-6
Steyn PS, van Heerden FR (1982) Cytochalasins E and K, toxic metabolites from Aspergillus clavatus. J Chem Soc Perkin Trans 1(1982):541–544. https://doi.org/10.1039/P19820000541
Stierle A, Hershenhorn J, Strobel G (1993) Zinniol-related phytotoxins from Alternaria chichorii. Phytochemistry 32:1145–1149. https://doi.org/10.1016/S0031-9422(00)95080-5
Stoldt M, Porzel A, Adam G, Brandt W (1998) Side chain conformation of the growth-promoting phytohormones brassinolide and 24-epibrassinolide. Magn Reson Chem 35:629–636. https://doi.org/10.1002/(SICI)1097-458X(199709)35:9<629::AID-OMR147>3.0.CO;2-E
Sun DW, Cao F, Liu M, Guan FF, Wang CY (2017) New fatty acid from a gorgonian-derived Xylaria sp. fungus. Chem Nat Comp 53:227–230. https://doi.org/10.1007/s10600-017-1958-7
Surup F, Halecker S, Nimtz M, Rodrigo S, Schulz B, Steinert M, Stadler M (2018) Hyfraxins A and B, cytotoxic ergostane-type steroid and lanostane triterpenoid glycosides from the invasive ash dieback ascomycete Hymenoscyphus fraxineus. Steroids 135:92–97. https://doi.org/10.1016/j.steroids.2018.03.007
Takamatsu S, Zhang Q, Schrader KK, ElSohly HN, Walker LA (2002) Characterisation of Mycotypha metabolites found to be inhibitors of cell adhesion molecules. J Antibiotics 55:585–592. https://doi.org/10.7164/antibiotics.55.585
Udagawa T, Yuan J, Panigrahy D, Chang YH, Shah J, D’Amato RJ (2000) Cytochalasin E, an epoxide containing Aspergillus-derived fungal metabolite, inhibits angiogenesis and tumor growth. J Pharmacol Exp Ther 294:421–427
Uhrinova S, Uhrin D, Liptaj T, Bella J, Hirsch J (1991) Measurement of one-bond proton-carbon coupling constants of saccharides. Magn Reson Chem 29:912–922. https://doi.org/10.1002/mrc.1260290909
Wendt L, Sir EB, Kuhnert E, Heitkämper S, Lambert C, Hladki AI, Romero AI, Luangsa-ard JJ, Srikitikulchai P, Peršoh D, Stadler M (2018) Resurrection and emendation of the Hypoxylaceae, recognized from a multigene phylogeny of the Xylariales. Mycol Prog 17:115–154. https://doi.org/10.1007/s11557-017-1311-3
Wibberg D, Stadler M, Lambert C, Bunk B, Spröer C, Rückert C, Kalinowski J, Cox RJ, Kuhnert E (2021) High quality genome sequences of thirteen Hypoxylaceae (Ascomycota) strengthen the phylogenetic family backbone and enable the discovery of new taxa. Fungal Divers 106:7–28. https://doi.org/10.1007/s13225-020-00447-5
Wittstein K, Cordsmeier A, Lambert C, Wendt L, Sir EB, Weber J, Wurzler N, Petrini LE, Stadler M (2020) Identification of Rosellinia species as producers of cyclodepsipeptide PF1022A and resurrection of the genus Dematophora as inferred from polythetic taxonomy. Stud Mycol 96:1–16. https://doi.org/10.1016/j.simyco.2020.01.001
Wu SH, Chen YW, Miao CP (2011) Secondary metabolites of endophytic fungus Xylaria sp YC-10 of Azadirachta indica. Chem Nat Comp 47:858–861. https://doi.org/10.1007/s10600-011-0086-z
Xu ZL, Zheng N, Cao SM, Li ST, Mo TX, Qin YY, Li J, Yang RY (2020) Secondary metabolites from the endophytic fungus Stemphylium lycopersici and their antibacterial activities. Chem Nat Comp 56:1162–1165. https://doi.org/10.1007/s10600-020-03256-z
Yuyama KT, Chepkirui C, Wendt L, Fortkamp D, Stadler M, Abraham WR (2017) Bioactive compounds produced by Hypoxylon fragiforme against Staphylococcus aureus biofilms. Microorganisms 5:80. https://doi.org/10.3390/microorganisms5040080
Zhao Q, Wang GQ, Chen GD, Hu D, Li XX, Guo LD, Li Y, Yao XS, Gao H (2015) Nodulisporisteroids C-L, new 4-methyl-progesteroid derivatives from Nodulisporium sp. Steroids 102:101–109. https://doi.org/10.1016/j.steroids.2015.08.004
Acknowledgements
Wera Collisi is thanked for technical assistance with biological assays. Silke Reinecke is thanked for continuous technical support in the natural product chemistry lab and Christel Kakoschke, Esther Surges and Aileen Gollasch are thanked for recording spectra. Moreover, Astri Septiar Fakhriyah is thanked for assistance during her BSc thesis work.
Funding
Open Access funding enabled and organized by Projekt DEAL. This research was supported by a grant from the Deputy of Research and Technology of the University of Guilan, and the Iran National Science Foundation (INSF) No. 99027605 to M.J.P. We are also grateful to the German Academic Exchange Service for a DAAD postdoc stipend for G.E., to the LifeScience Foundation (Munich) for a PhD stipend for C.L.. T.E.B.S. and M.S. would like to thank the Deutsche Forschungsgemeinschaft for a Research Unit grant “Cytolabs” (DFG FOR 5170).
Author information
Authors and Affiliations
Contributions
M.J.P.: conceptualization, isolation and identification of the producer strain, software, investigation, compound isolation, writing—original draft preparation; G.E.: culturing, compound isolation, structure elucidation and writing; C.L.: conceptualization, supervision, writing – original draft preparation, cell biological bioactivity assays, visualization. S.A.K.: resources, review and editing; M.S., T.E.B. S, H.V., and I.K.G.: supervision, review, and editing; G. P., F.S.: structure elucidation and writing, K.W.: compound isolation.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Disclaimer
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Additional information
Section Editor: Marco Thines
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
Molecular phylogenetic reconstruction, Figure S1. Phylogram of the best ML trees, Table S1. Isolation and accession numbers of sequences used in the phylogenetic analyses, Taxonomy (Rosellinia cf. akulovii L.E. Petrini (2013)), Figure S2. Rosellinia cf. akulovii (GUM 1633), Figure S3. Culture and anamorphic structures of Rosellinia spp. on OA, Taxonomy (Rosellinia corticium (Schwein.: Fr) Sacc., Syll. F. 1:253. 1882.), Taxonomy (Dematophora necatrix R. Hartig, Untersuch. Forstbot. Inst. München 3: 126. 1883.), Figure S4. Rosellinia corticium, Table S2. List of isolated secondary metabolite occurrence during screening experiments, Table S3. Minimal Inhibitory Concentration of selected compounds against a panel of microorganisms, Figure S5. HPLC-ESI-HR-MS data of rosellisteroid, Figure S6. 1H NMR spectrum (700 MHz, acetone-d6) of rosellisteroid (1), Figure S7. 13C NMR spectrum (175 MHz, acetone-d6) of rosellisteroid (1), Figure S8. HSQC NMR spectrum (700 MHz, acetone-d6) of rosellisteroid (1), Figure S9. COSY NMR spectrum (700 MHz, acetone-d6) of rosellisteroid (1), Figure S10. HMBC NMR spectrum (700 MHz, acetone-d6) of rosellisteroid (1), Figure S11. ROESY NMR spectrum (700 MHz, acetone-d6) of rosellisteroid (1), Figure S12. HPLC-ESI-HR-MS data of the cichorin derivative (2), Figure S13. 1H NMR spectrum (700 MHz, CHCl3-d) of the cichorin derivative (2), Figure S14. 13C NMR spectrum (175 MHz, CHCl3-d) of the cichorin derivative (2), Figure S15. HSQC NMR spectrum (700 MHz, CHCl3-d) of the cichorin derivative (2), Figure S16. COSY NMR spectrum (700 MHz, CHCl3-d) of the cichorin derivative (2), Figure S17. HMBC NMR spectrum (700 MHz, CHCl3-d) of the cichorin derivative (2), Figure S18. ROESY NMR spectrum (700 MHz, CHCl3-d) of the cichorin derivative (2), Figure S19. HPLC-ESI-HR-MS data of 2-formyl-5-(methoxymethyl)-1H-pyrrole-1-pentanoic acid (3), Figure S20. 1H NMR spectrum (700 MHz, CH3OH-d4) of 2-formyl-5-(methoxymethyl)-1H-pyrrole-1-pentanoic acid (3), Figure S21. 13C NMR spectrum (700 MHz, CH3OH-d4) of 2-formyl-5-(methoxymethyl)-1H-pyrrole-1-pentanoic acid (3), Figure S22. HSQC NMR spectrum (700 MHz, CH3OH-d4) of 2-formyl-5-(methoxymethyl)-1H-pyrrole-1-pentanoic acid (3), Figure S23. COSY NMR spectrum (700 MHz, CH3OH-d4) of 2-formyl-5-(methoxymethyl)-1H-pyrrole-1-pentanoic acid (3), Figure S24. HMBC NMR spectrum (700 MHz, CH3OH-d4) of 2-formyl-5-(methoxymethyl)-1H-pyrrole-1-pentanoic acid (3), Figure S25. HPLC-ESI-HR-MS data of xylarotide A (4), Figure S26. 1H NMR spectrum (500 MHz, CHCl3-d) of xylarotide A (4), Figure S27. HSQC NMR spectrum (500 MHz, CHCl3-d) of xylarotide A (4), Figure S28. HPLC-ESI-HR-MS data of nodulisporisteroid C (5), Figure S29. 1H NMR spectrum (700 MHz, CH3OH-d4) of nodulisporisteroid C (5), Figure S30. 13C NMR spectrum (175 MHz, CH3OH-d4) of nodulisporisteroid C (5), Figure S31. HPLC-ESI-HR-MS data of cytochalasin E (6), Figure S32. 1H NMR spectrum (500 MHz, acetone-d6) of cytochalasin E (6), Figure S33. 13C NMR spectrum (125 MHz, acetone-d6) of cytochalasin E (6), Figure S34. HPLC-ESI-HR-MS data of Δ6,12-cytochalasin E (7), Figure S35. 1H NMR spectrum (500 MHz, acetone-d6) of Δ6,12-cytochalasin E (7), Figure S36. 13C NMR spectrum (125 MHz, acetone-d6) of Δ6,12-cytochalasin E (7), Figure S37. HPLC-ESI-HR-MS data of cytochalasin K (8), Figure S38. 1H NMR spectrum (700 MHz, CH3OH-d4) of cytochalasin K (8), Figure S39. HMBC NMR spectrum (700 MHz, CH3OH-d4) of cytochalasin K (8), Figure S40. HPLC-ESI-HR-MS data of 5-carboxymellein (9), Figure S41. 1H NMR spectrum (700 MHz, CHCl3-d) of 5-carboxymellein (9), Figure S42. 1H NMR spectrum (175 MHz, CHCl3-d) of 5-carboxymellein (9), Figure S43. HPLC-ESI-HR-MS data of methylparaben (10), Figure S44. 1H NMR spectrum (500 MHz, CH3OH-d4) of methylparaben (10), Figure S45. 13C NMR spectrum (125 MHz, CH3OH-d4) of methylparaben (10), Figure S46. HPLC-ESI-HR-MS data of 4-hydroxybenzamide (11), Figure S47. 1H NMR spectrum (500 MHz, CH3OH-d4) of 4-hydroxybenzamide (11), Figure S48. 13C NMR spectrum (125 MHz, CH3OH-d4) of 4-hydroxybenzamide (11), Figure S49. HPLC-ESI-HR-MS data of 12, Figure S50. 1H NMR spectrum (700 MHz, acetone-d6) of 12, Figure S51. HMBC NMR spectrum (700 MHz, acetone-d6) of 12, Figure S52. HPLC-ESI-HR-MS data of 13, Figure S53. 1H NMR spectrum (700 MHz, acetone-d6) of 13, Figure S54. 13C NMR spectrum (175 MHz, acetone-d6) of 13, Figure S55. Marfey analysis of xylarotide A (4) derived d-FDVA, Figure S56. Authentic amino acids (d/l or dl mixtures) derived d-FDVA at 340 nm, Figure S57. Authentic amino acids (d/l or dl mixtures) derived l-FDVA at 340 nm, Figure S58. Authentic amino acids derived d-FDVA of l-allo-Ile (upper), mixtures of l-Ile and l-allo-Ile (middle), and l-Ile (bottom), Figure S59. Marfey analysis of xylarotide A derived d-FDVA on C4 column showing the presence of l-isoleucine, Table S4. Retention time of l or d authentic amino acid derived d-FDVA, Table S5. Retention time of l or d authentic amino acid derived l-FDVA, Figure S60. Key COSY (blue arrows), HMBC (green arrows) and ROESY correlations specifying the structures of 2 (left) and 3 (right), References to Table S1. (PDF 6.97 MB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pourmoghaddam, M.J., Ekiz, G., Lambert, C. et al. Studies on the secondary metabolism of Rosellinia and Dematophora strains (Xylariaceae) from Iran. Mycol Progress 21, 65 (2022). https://doi.org/10.1007/s11557-022-01816-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11557-022-01816-x