
Abstract
Potato is one of the most important crops worldwide. Genetic resources of potato (Solanum tuberosum L. ssp. tuberosum) and related cultivated species are conserved through storage of tubers, in vitro plants and in cryopreservation. Cryopreservation, storage in or above liquid nitrogen, is the best option to maintain vegetatively propagated plants in the long term. The present review gives comprehensive information about various cryopreservation techniques for potato published from 1977 until the present. It discusses factors that affect the process and success of cryopreservation, such as donor culture conditions, preculture, cooling, warming and post-culture treatments. Studies are presented that analyse the histological and ultrastructural changes after different cryopreservation steps and the morphological pathways during regeneration of plants after rewarming. The maintenance of genetic stability in potato after cryopreservation has also been demonstrated by various phenotypic and molecular methods. The first thermal analyses on potato shoot tips are presented using differential scanning calorimetry to analyse the state of water during cooling and warming. Biochemical analyses of different compounds, such as soluble sugars and proteins, have been performed to understand and improve existing cryogenic methods. Potato is an example where successful virus elimination has been obtained via cryopreservation of shoot tips (cryotherapy). There are already cryopreserved collections of potato shoot tips in Germany, Peru, Czech Republic, South Korea and USA, but additional experiments on fundamental aspects of potato cryopreservation will help to improve understanding of the different cryopreservation methods, start new collections in other countries and also build up existing cryocollections of potato.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Potato belongs to the botanical family Solanaceae and within it to the genus Solanum, which consists of more than 2,000 species (Hawkes 1978). Apart from the cultivated potato Solanum tuberosum L. ssp. tuberosum, seven other cultivated and 228 wild potato species have been identified. Taxonomically, they all belong to the section Petota containing the tuber-bearing potatoes (Hawkes 1990). Spooner and Salas (2006) have recently revised the taxonomy of potato species by combining the seven cultivated species into one single potato species (S. tuberosum) with eight cultivar groups. The cultivated potato originated from wild Solanum species in the Andes of South America and the coastal strip of central to southern Chile (Hawkes 1978). After the first potatoes were brought to Europe in the late sixteenth century, this crop plant was rapidly distributed and is now utilised all over the world, mostly in temperate regions (Hawkes 1990).
Today, there are more than 4,500 varieties of S. tuberosum ssp. tuberosum (Hils and Pieterse 2009). Potato is known to have the richest genetic diversity of any cultivated plant because of its high number of varieties and related species (FAO 2007; Messer 2000). To prevent the loss of potato genetic resources, long-term conservation of plant material is accomplished in genebanks, genetic resource centres and private collections worldwide. For example in the IPK genebank (Satellite Collections North, Groß Lüsewitz, Germany), about 5,900 accessions are conserved comprising 2,700 cultivated accessions and more than 130 wild species (3,200 accessions; IPK 2007). Conservation of genetic resources in genebanks is important, because of their high value as breeding material and for research, industry and private needs.
Potato is highly heterozygous, and the sexually produced seeds are, therefore, not true to type. Thus, maintenance of cultivated potato accessions is not possible by true potato seeds (Dodds et al. 1991). Genebank stocks are therefore derived from vegetative material. To preserve the genetic resources of this crop plant for the long term, cryopreservation is the best current option. Cryopreservation is the storage of biological material at −196 °C in liquid nitrogen (LN) in such a way that viability is maintained and plant regeneration is obtained after rewarming.
Potato cryopreservation was started in 1977 with two-step cooling procedures and ultra-rapid freezing techniques (Bajaj 1977; Grout and Henshaw 1978; Towill 1981a; Towill 1981b). The first protocols were time consuming because of the need to use programmable freezers and resulted in low survival. Later on protocols were refined and new methods developed, such as the dimethyl sulfoxide (DMSO) droplet method (Schäfer-Menuhr et al. 1994), vitrification (Golmirzaie and Panta 1997), encapsulation/dehydration (Fabre and Dereuddre 1990), encapsulation/vitrification (Hirai and Sakai 1999) and droplet vitrification methods (Halmagyi et al. 2005). These protocols improved the results in potato cryopreservation in terms of survival and regeneration frequencies and they are now routinely applied in several institutes to build up cryopreserved potato collections (Table 1).
Recent reviews by Keller et al. (2008), Wang et al. (2008a) and Gonzalez-Arnao et al. (2008) have concentrated primarily on currently used potato cryopreservation protocols. In this review, we illustrate in detail (a) historically important, (b) currently used and (c) the most recent advances in potato shoot tip cryopreservation with various potato species and varieties. We compare and contrast evolving trends in potato cryogenic research based on analysis of original and contemporary techniques, such as proteomics and differential scanning calorimetry (DSC) and explain how advances in understanding of cryogenic science have shaped improvements in techniques. We conclude with a discussion as to how the most recent advances (e.g., proteomics and DSC) may shape the future of potato cryogenic research.
Cryopreservation Techniques
Two-Step Cooling
The first cryopreservation studies on potato were carried out by Bajaj (1977; Table 2), who used a two-step cooling procedure. In this method, cell water content was reduced by freeze-induced dehydration. Firstly, tuber sprouts and axillary buds were cryoprotected with various glycerol and/or sucrose solutions. Then they were cooled slowly in the vapour phase of LN and later directly in LN. Bajaj (1977) obtained between 7% and 18% survival of tuber sprouts and axillary buds with this protocol. Regeneration percentages of up to 21% and 33% were obtained for ‘Norland’ and ‘Red Pontiac’, applying two-step cooling (Towill 1983; Table 2). Explants were cooled slowly with a speed of 0.2–0.3 °C min−1 until −35 °C followed by immersion in LN (Towill 1983). Henshaw et al. (1985) applied quite a similar protocol using 0.3 °C min−1 until −30 °C and then direct cooling in LN. Shoot regeneration after rewarming ranged between 4% and 85% depending on species (Henshaw et al. 1985; Table 2). Overall, the method described is, however, rather complicated and time consuming (Schäfer-Menuhr et al. 1997) and needs special equipment (Kartha and Engelmann 1994).
Ultra-Rapid Cooling
In comparison to the previously mentioned two-step cooling method, ultra-rapid cooling is easier in handling (Grout and Henshaw 1978). In ultra-rapid cooling technique, shoot tips were isolated, precultured and cryoprotected with MS medium (Murashige and Skoog 1962) containing sucrose and 10% v/v DMSO. Subsequently, each single shoot tip was directly plunged into LN on a tip of a hypodermic needle. Rewarming was carried out quickly through transferring the needles directly into a vial of MS medium with benzylaminopurine (BAP) at 35 °C. After that shoot tips were removed from solution and were transferred to the regeneration medium. Grout and Henshaw (1978) presented one of the first successful cryopreservation protocols for potato shoot tips with as high survival as 20% and shoot regeneration capacity of 10% (Table 2). A significant step in this method is the quick cooling, so that water is transformed directly to amorphous ice, and the reverse occurs with rapid rewarming, so that devitrification does not occur on return to room temperature regime (Grout and Henshaw 1978; Benson et al. 2006).
Another ultra-rapid cooling method (developed for the potato cryopreservation collection in the Czech Republic by Zámečník et al. 2007) involved culturing in vitro donor plants on semi-solid hormone-free MS medium containing a reduced amount of nitrogen (Grospietsch et al. 1999). Four-day-old nodal cuttings were incubated in 2 M sucrose for 5 days; then shoot tips were excised and put on sterile filter paper moistened with 0.7 M sucrose for 20 h. Shoot tips were then transferred to aluminium foils and desiccated over silica gel for 1.5 h with a subsequent direct cooling in LN. Shoot tips were thawed in sterile water at room temperature and transferred to regeneration medium with growth regulators (0.5 mg l-1 indole acetic acid (IAA), 0.5 mg l-1 kinetin and 0.2 mg l-1 gibberellic acid (GA3)). The average regeneration percentage was 23% in the 35 accessions tested (Table 2). The main difference between this method compared to other rapid cooling protocols is the avoidance of DMSO and the use of aluminium foils for supporting the shoot tips.
Vitrification
The process of vitrification is described as solidification of liquids without crystallisation. When the solutions within the cells are vitrified, they are in a “glassy state” and amorphous. They lack organised structures but possess the mechanical and physical properties of a solid (Benson 2008a; Taylor et al. 2004). Vitrification is one of the main and most widely applied plant cryopreservation methods. Most vitrification protocols apply the plant vitrification solution 2 (PVS2). It is a mixture of cryoprotectants containing 30% w/w glycerol, 15% w/w ethylene glycol, 15% w/w DMSO in MS medium supplemented with 0.4 M sucrose (Sakai et al. 1990). Sarkar and Naik (1998) published the first vitrification protocol for S. tuberosum cultivars ‘Kufri Badsha’, ‘Kufri Chandramukhi’, ‘Kufri Lalima’, ‘Kufri Lauvkar’ and ‘Kufri Sindhuri’. In this vitrification method, apical shoot tips of 0.5–0.7 mm in size were first isolated from 30-day-old plantlets and precultured for 2 days on filter paper discs over half strength liquid MS medium supplemented with 8.7 μM GA3 and different combinations of sucrose (0.3, 0.5 and 0.7 M) and mannitol (0, 0.2 and 0.4 M) under a 16 h photoperiod with approx. 40 μmol m−2 s−1 light intensity, at 24 °C. Shoot tips were then loaded in 20% PVS2 (30 min, 24 °C), afterwards in 60% PVS2 (15 min, ice bath) followed by 100% ice cold PVS2 (5 min) or directly in 100% PVS2 (5 min). Five shoot tips each were transferred into one cryovial filled with 0.7 ml of PVS2 and finally plunged into LN. After quick rewarming at 35 °C in a water bath for 1 min, cryovials were flushed with dilution medium (MS with 1.2 M sucrose) and incubated for 30 min. Then shoot tips were cultivated on semi-solid regeneration medium [MS with 0.2 M sucrose, 5.8 μM GA3, 1.0 μM BAP and 6 g/l Nobel agar (HiMedia, India)] and cultured under diffuse light (16 h photoperiod, approx. 6 μmol m−2 s−1) at 24 °C for 1 week. Subsequently, shoot tips were transferred to standard medium (MS medium with 0.09 M sucrose, 2.9 μM GA3 and 6 g l-1 agar) under light conditions of approximately 40 μmol m−2 s−1 with 16 h photoperiod, at 24 °C. For successful regeneration, preculture with sucrose in combination with mannitol was necessary. Concentrations of mannitol above 0.2 M were detrimental. The highest response was found applying a preculture with 0.3 M sucrose and 0.2 M mannitol followed by sequential vitrification with highest shoot regeneration rate of 56% (Sarkar and Naik 1998).
In the International Potato Center (CIP), cryopreservation research was initiated in 1995 using the vitrification method developed by Steponkus and co-operators (Golmirzaie and Toledo 1997; Golmirzaie and Panta 1997). In this method, the vitrification solution contained ethylene glycol: sorbitol: BSA (bovine serum albumin) in the ratio of 50:15:6% w/w and samples were cooled in polypropylene straws. A number of genotypes (197) were cryopreserved with an average regeneration rate of 46% (Golmirzaie and Panta 2000). Using this protocol, approximately 400 potato landraces were tested with highly variable responses. Of the different genotypes, 30% did not show any survival, and in 30% of the genotypes, the regeneration capacity was less than 15%. In 2004, this protocol was further optimised, and CIP (in cooperation with the Laboratory of Tropical Crop Improvement, Katholieke Universiteit Leuven, Belgium) adopted the droplet vitrification method (see below) initially designed for cryopreserving banana shoot tips also for potato (Gonzalez-Arnao et al. 2008; Panis et al. 2005). Kryszczuk et al. (2006) applied the vitrification method to four S. tuberosum cultivars (‘Ackersegen’, ‘Blaue Schweden’, ‘Carnea’ and ‘Désirée’). In their study, one-week-old nodal cultures were used to isolate apical explants from the sprouted nodal buds (1 mm in length). Material was incubated overnight in MS medium with 30 g/l sucrose. Next day shoot tips were placed in loading solution (MS medium with 0.4 M sucrose and 2 M glycerol) for 20 min followed by incubation in PVS2 for 30 min. Cryovials filled with 1.5 ml of PVS2 together with ten shoot tips each were directly cooled in LN. Rewarming was performed in a water bath at 38 °C for 2 min. Then explants were washed three times with MS solution containing 1.2 M sucrose and transferred to regeneration medium (MSTo (Towill 1983), solidified with 0.8% agar). Average shoot regeneration capacity of the described original protocol was 58.0%, which was higher compared to experiments applying cold preculture at 21/8 °C for 7 days (45.9% regeneration capacity), liquid regeneration medium (21.3% regeneration capacity) or filter-sterilised DMSO (57.5% regeneration capacity). Using vitrification, significantly higher shoot tip regeneration frequency was obtained (58.0%) in comparison with DMSO droplet method (13.8%, Kryszczuk et al. 2006; Table 2). In contrast to this, treatment at 10 °C for 3 weeks and application of the ice-blocking agent Supercool X1000 within the PVS2 resulted in regeneration of 71% and 55% in S. tuberosum cultivars ‘Atlantic’ and ‘Superior’, respectively (Zhao et al. 2005; Table 2).
DMSO Droplet Method (Droplet Freezing)
DMSO droplet method is an optimisation of the ultra-rapid cooling method (Schäfer-Menuhr et al. 1994; Table 2). The expression ‘droplet’ refers to droplets of cryoprotectant on an aluminium foil, onto which the shoot tips are placed for cooling and rewarming. Also droplets of agarose together with a single shoot tip each and liquid medium have been used for regeneration. The innovative idea of using aluminium foils originated from Kartha et al. (1982), who cryopreserved cassava shoot tips on foils using a two-step cooling method. Schäfer-Menuhr et al. (1994) adopted the idea of a fast cooling method also for potato shoot tips. The foils are a good carrier in transferring a large number of shoot tips at once quickly into and out of LN compared with using hypodermic needles, where only one shoot tip at a time can be transferred. Furthermore, aluminium is a very good heat conductor, which is significant for quick cooling and also for rewarming of explants. Schäfer-Menuhr et al. (1994) used foils of 20 × 7 × 0.03 mm in size, so that two foils fit well into one cryovial. Regarding the state of water during cooling, no information about the state of the water (ice or glass) in the droplets was given in their study (Schäfer-Menuhr et al. 1994). However, as observed by Benson et al. (1992) using cassava, it is highly probable that vitrification may occur (Benson et al. 2006).
In the original droplet freezing protocol, shoot tips were isolated from 10 cm long in vitro plantlets to explants of 2–3 mm in length and 0.5–1 mm width, and then incubated in MS medium with 30 g/l-1 of sucrose, 0.5 mg/l-1 zeatin riboside, 0.2 mg/l-1 GA3 and 0.5 mg/l-1 IAA (MSTo, Towill 1983) overnight. On the second day, shoot tips were placed in cryoprotectant solution (MSTo medium with 10% DMSO) at room temperature for 2 h. They were then transferred into droplets of 2.5 μl cryoprotectant solution and placed onto small heat-sterilised aluminium foils and directly cooled in LN. Shoot tips were rewarmed quickly by putting aluminium foils in liquid MS medium at room temperature. For regeneration, shoot tips were placed in drops of warmed agarose in Petri dishes. After solidification of drops, liquid MSTo solution was added (Schäfer-Menuhr et al. 1994). The average survival of the cryopreserved 219 accessions was 80%, and plant regeneration capacity was 40% (Schäfer-Menuhr et al. 1997; Table 2). Barandalla et al. (2003) analysed the influence of phytohormones (0.5 mg/l-1 zeatin riboside, 0.2 mg/l-1 GA3 and 0.5 mg/l-1 IAA) in the agarose drops on plant regeneration for 10 potato accessions. The original protocol according to Schäfer-Menuhr et al. (1994) without any phytohormones in the agarose drops showed less plant regeneration frequency in comparison with the application of agarose drops having phytohormones (Barandalla et al. 2003). Plant regeneration was between 2.5% and 22% with a mean of 13.2% (Table 2). By applying the DMSO droplet method, Kryszczuk et al. (2006) compared the original DMSO droplet protocol with slightly modified versions for S. tuberosum cultivars ‘Ackersegen’, ‘Blaue Schweden’, ‘Carnea’ and ‘Désirée’. In their study, no survival and regeneration was seen at all, if autoclaved DMSO was used instead of filter-sterilised DMSO. Application of solid medium during regeneration enhanced regeneration rate to an average of 15% in comparison with the original protocol using liquid medium (average regeneration rate of 13.8%). The best results with an average of 29.6% plant regeneration (Table 2) were given as a result of applying cold preculture of the potato donor plants (21/8 °C day/night temperature, 8 h photoperiod, 7 days) prior to shoot tip isolation (Kryszczuk et al. 2006). Similar results were subsequently observed by Kaczmarczyk et al. (2008b). Improvements in regeneration results from 34.6% to 45.2% were found, when an alternating temperature preculture (22/8 °C day/night temperature, 8 h photoperiod, 7 days) was applied for the donor in vitro plants prior to shoot tip isolation (Kaczmarczyk et al. 2008b). Kaczmarczyk et al. (2008b) carried on by a comparison between the original method of Schäfer-Menuhr et al. (1994) by applying agarose drops and liquid medium for regeneration and by using solid regeneration medium (MSTo with 10 g/l-1 agar). It was found that the regeneration frequency was increased on solid medium in explants of S. tuberosum ‘Désirée’ and ‘King Edward’ and the regenerated shoots showed better plantlet structure with less callus formation (Kaczmarczyk et al. 2008b). The combination of alternating temperature preculture and solid regeneration medium caused the highest average regeneration rate of 58.7% (Kaczmarczyk et al. 2008b). As a result of those positive findings in the IPK Germany, the DMSO droplet method, which is the routine cryopreservation method, was improved by the application of alternating temperature preculture and solid medium for regeneration. In the IPK, up to date 1,119 accessions have been stored (Table 1, Kaczmarczyk et al. 2009) with a mean regeneration percentage of 46%. This collection includes the accessions formerly stored at German Collection of Microorganisms and Cell Cultures (DSMZ, Germany) and the Institute of Crop Science of the Federal Agricultural Research Centre (FAL, Braunschweig, Germany; Schäfer-Menuhr et al. 1997) which have subsequently been integrated into the IPK collection (Keller and Dreiling 2003).
The DMSO droplet method is a simple freezing protocol, which is now applicable to a wide variety of potato accessions. The method is quick and easy and the procedure is also relatively cheap (Schäfer-Menuhr 1996). Problematic is still the low regeneration frequency of certain potato genotypes. Therefore, additional studies are needed to improve the results for accessions showing low regeneration capacity (Keller and Dreiling 2003; Schäfer-Menuhr 1996, 1997).
Droplet Vitrification
Droplet vitrification, which combines elements of the DMSO droplet method and vitrification, is a relatively new method in potato cryopreservation (Halmagyi et al. 2005, Kim et al. 2006; Panta et al. 2006, 2009a, b; Zhao et al. 2005; Table 2). In the droplet vitrification method, shoot tips are plated on aluminium foils during cooling, like in the DMSO droplet method, but other types of cryoprotectants, such as PVS2 are used. The use of aluminium foils makes the transfer of shoot tips into and out of LN easier and faster. This can be important during application of PVS2, because slightly longer incubation times can be toxic for shoot tips. Using the droplet vitrification, first successful results were presented by Halmagyi et al. (2005) in three potato cultivars (‘Désirée’, ‘Ostara’ and ‘Santé’). The plants were multiplied on hormone-free MS medium and maintained at 24 °C with photosynthetically active radiation of 39.06 μmol m−2 s−1 and 16 h photoperiod. Shoot tips with different sizes (1–6 mm in length) were isolated with hypodermic needles from four- to eight-week-old plants and incubated on filter paper with liquid MS medium containing 0.4 mg/l-1 of GA3, 0.5 mg/l-1 of zeatin and 0.2 mg/l-1 of IAA at 24 °C for 24 h. Afterwards various types of precultures with different sugar or sugar alcohols (sucrose, glucose, mannitol and sorbitol) were performed at 24 °C for 24 h. Then shoot tips were dehydrated in droplets of 4 μl with PVS2 on aluminium foil (0.6 × 1.5 cm) at room temperature for 10–30 min, before their transfer into pre-cooled cryovials and then into LN. Rapid warming was carried out by shaking aluminium foils in liquid medium at room temperature. For regeneration shoot tips were put onto semi-solid medium with 3.5 g/l-1 agar at 24 °C for 20 days. Afterwards elongated shoots were transferred to solidified medium with 7 g/l-1 of agar (Halmagyi et al. 2005). The highest shoot regeneration frequencies were 55% in ‘Désirée’, 51% in ‘Ostara’ and 46% in ‘Santé’.
Panta et al. (2006) used plantlets that were precultured at 6 °C on MS medium with 0.07 M sucrose for 3 weeks. Afterwards shoot tips were isolated (2–3 mm in size) and incubated in loading solution (MS with 2 M glycerol and 0.4 M sucrose) for 20 min. Shoot tips were transferred to PVS2 at 0 °C for 50 min, plated on aluminium foil strips, put into cryovials and plunged into LN. Rewarming was done rapidly in MS medium enriched with 1.2 M sucrose for 20 min. Explants were then cultivated on MS medium with 0.04 mg/l-1 kinetin and 0.1 mg/l-1 GA3 and 0.3–0.1 M sucrose concentrations, reducing the sucrose level by 0.1 M per day. Regeneration capacity of shoot tips was genotype-dependent and ranged between 8% in S. tuberosum ‘Wila Yari’ to 47% in ‘Désirée’ (Panta et al. 2006; Table 2). Panta et al. (2009a) also analysed certain biochemical compounds, such as phospholipids, glycolipids, aromatic amines and polyamines to find links to cryopreservation ability. They found that frost resistant genotypes had significantly higher regeneration rates after cryopreservation and that the linoleic acid content was positively correlated with tolerance to cryopreservation. It was also concluded that addition of putrescine to preculture medium can further enhance recovery rates for poorly regenerating genotypes (Panta et al. 2009a).
In recent years, the number of species successfully cryopreserved using the combined droplet vitrification protocol with rapid cooling and rewarming is continuously increasing (Gonzalez-Arnao et al. 2008; Halmagyi et al. 2005; Kim et al. 2006; Leunufna and Keller 2005; Panta et al. 2006; Sakai and Engelmann 2007; Sant et al. 2008; Senula et al. 2007; Yoon et al. 2006), whereas two-step cooling methods are now used less often and then in most cases only for callus and suspension cultures (Gnanapragasam and Vasil 1992; Mikula et al. 2005). The reason for the increasing use of droplet vitrification lies in its fast and easy application without expensive cooling device which is necessary for two-step cooling.
Encapsulation/Dehydration
In 1990, a new cryopreservation technique for plants called encapsulation/dehydration was developed by Fabre and Dereuddre (1990) using potato species Solanum phureja. In this method, donor plants were subcultured on medium A (MS minerals with Morel and Wetmore vitamins (Morel and Wetmore 1951), Na-Fe-EDTA, 30 g/l-1 sucrose, 8 g/l-1 agar, pH 5.85). Apical sections of eight- to ten-week-old plantlets were transferred to medium A for 7 days. Shoot tips with three to four leaf primordia (approximately 0.5 mm in length) were excised and incubated on medium B (Morel macronutrients (Morel and Muller 1964), Heller micronutrients (Heller 1953), vitamins, Na-Fe-EDTA, 8 g/l-1 agar, 0.1 M sucrose and growth regulators (O’Hara et al. 1985), pH 5.8). Donor plants and shoot tips were cultured at 20 °C with 70% relative humidity and light intensity of 50 μmol m−2 s−1. Afterwards shoot tips were suspended in liquid calcium-free medium B supplemented with 3% (w/v) sodium alginate. This mixture was dropped with the explants into liquid medium B containing 100 mM calcium chloride to form alginate beads. Beads of approximately 3 mm in diameter containing one or two shoot tips inside each bead were precultured in liquid medium B supplemented with sucrose. The beads were then dehydrated under sterile air flow at room temperature for 4 h. Encapsulated shoot tips were cooled directly by LN or they were progressively cooled from 20 to −40 °C before immersion into LN. Beads were slowly warmed in air at room temperature and transferred to medium B for further regeneration. The best results (9.3% regeneration) were found using a combination of preculture with 0.5 M sucrose and two-step freezing (Table 2).
Optimization of the above described protocol was made by Bouafia et al. (1996) using three different diploid accessions of S. phureja and two tetraploid S. tuberosum cultivars (‘Charly’ and ‘Hertha’). Donor materials were cultured at temperature of 21 ± 1 °C with a photoperiod of 16 h per day and light intensity of 25–30 μmol m−2 s−1. Shoot tips were excised from nodal cuttings, grown for 3, 7, 14 and 21 days, and then incubated on culture medium. After encapsulation, preculture in liquid medium was applied during the next 2 days with stepwise increasing sucrose (0.3, 0.5, 0.75 and 1 M) to obtain a final concentration of 1 M. Beads were kept in 1 M sucrose medium for 1–7 days. In addition, direct preculture of encapsulated shoot tips in sucrose-enriched medium (0.1, 0.3, 0.5, 0.75, 1, 1.25 and 1.5 M) was tested for 2 days. After preculture, shoot tips were dried and dehydrated for 4.5 h in airtight boxes over silica gel to reach a water content of 0.20–0.22 g g-1 dry weight. Encapsulated shoot tips were transferred to cryovials and directly immersed in LN. Rewarming was done slowly in air at room temperature. For regeneration, shoot tips were plated on medium with growth regulators for 1 week and then later plated on medium supplemented only with GA3 to avoid callus formation (Bouafia et al. 1996). The highest response was found with direct preculture of encapsulated shoot tips in 0.75 M sucrose medium for 2 days prior to cooling in LN. Regeneration was between 50% and 64% in two S. tuberosum accessions tested, whereas diploid S. phureja was more tolerant to dehydration and had higher or similar shoot regeneration rates (64%, 69%, and 78%; Bouafia et al. 1996). Applying encapsulation/dehydration to six different Solanum species, Benson et al. (1996) found considerable variation in early and late recovery responses and between experimental repetitions. However, plant regeneration was attained for all cryopreserved species with the highest average regeneration percentages in Solanum acaule (32%) and the lowest in S. iopetalum (7%, Table 2). Differences in regeneration between diploid (2n = 2x = 24) (S. phureja) and tetraploid (2n = 4x = 48) species (S. tuberosum, S. acaule) were not found in contrast to the observations of Bouafia et al. (1996). Only hexaploid (2n = 6x = 72) species showed reduced percentages in regeneration capacity (between 7% and 19%) in comparison to diploid and tetraploid accessions (29–31%). Variations between experiments were also found in S. tuberosum ‘Brodick’ and ‘Golden Wonder’, when the encapsulation/dehydration method was applied (Harding and Benson 2001). Shoot regeneration was between 7% and 41% (‘Brodick’) and 0–61% (‘Golden Wonder’) in six repetitions. Different preculture treatments were performed to increase plant regeneration after cryopreservation using encapsulation/dehydration by Grospietsch et al. (1999). These precultures were: (1) sucrose preculture of isolated shoot tips [1 day on MS-P medium (MS without casein and myo-inositol, and with reduced N content 25% NH4NO3 and 50% of original KNO3) with 0.7 M sucrose], (2) sucrose-glycerol preculture of isolated shoot tips (1 day on MS-P medium with 0.3 M sucrose and 0.5 M glycerol), (3) drought hardening of donor plants (4 ml of 2 M sucrose solution were added to donor plants for 5 days followed by shoot tip isolation) and (4) drought hardening plus sucrose preculture (as in case 3 and additional shoot tip preculture for 1 day in MS-P medium with 0.7 M sucrose). The highest survival (79%) and shoot regeneration responses (59%) were found in S. tuberosum ‘Désirée’ as a result of drought hardening plus sucrose preculture. Those results can be explained by the function of sucrose as cryoprotectant to stabilise membranes, to decrease the phase transition temperature of dehydrated phospholipid bilayer and to prevent protein denaturation during dehydration (Grospietsch et al. 1999). Overall, a clear progress in optimising the encapsulation/dehydration method can be seen from the first experiments with two-step cooling and limited growth recovery (Fabre and Dereuddre 1990) to more recent experiments with high survival after rapid cooling (Grospietsch et al. 1999; Benson et al. 1996).
Encapsulation/Vitrification
Encapsulation/vitrification methods combine certain elements of vitrification and encapsulation/dehydration. Hirai and Sakai (1999) analysed 14 potato cultivars applying this method. Plantlets were grown on solidified basal medium (MS medium with 0.5 g/l casamino acid, 30 g/l sucrose, 2.5 g/l gellan gum) at 23 °C and 16 h photoperiod with light intensity of 96 mol m−2 s−1. For cryopreservation, axillary meristems from nodal segments with 5 leaf primordia (approximately 1 mm in size) were isolated and precultured on basal medium with 0.3 M sucrose, 1 mg/l-1 GA3, 0.01 mg/l-1 BAP and 0.001 mg/l-1 NAA. Meristems were suspended in calcium-free MS medium supplemented with 2% (w/v) Na-alginate and 0.4 M sucrose. The mixture containing the explants was transferred into 0.4 M sucrose solution plus 0.1 M CaCl2 for 30 min, at 25 °C, to form beads. Beads were osmoprotected with MS medium with 0.4–1.6 M sucrose and 2 M glycerol and plant hormones as in basal medium on a rotary shaker (60 rpm) at 25 °C for 90 min. Encapsulated meristems were dehydrated with PVS2 at 0 °C for different lengths of time. Beads with PVS2 were shaken (45 rpm) in a water bath. In total, 10–15 beads were suspended in a volume of 1 ml PVS2 solution per cryovial and plunged directly in LN. For rewarming, cryovials were put into water bath at 38 °C for 3 min. After removing the PVS2 solution, rewarmed beads were washed with 1 ml of 1.2 M sucrose solution for 10 min. For regeneration, beads with shoot tips were plated on basal medium for 1 day. Then they were transferred to basal medium with 0.0005 mg/l-1 GA3. Regeneration capacity was determined for all 14 analysed potato accessions and it was the highest (70%) as a result of osmoprotection with 0.6 M sucrose and 2 M glycerol for 90 min, followed by 3 h incubation with PVS2 (Table 2). No improvement was found in cold treated nodal segments (4 °C, 3 weeks) in comparison with the control explants (without cold treatment). Rate of shoot formation and speed of growth were much higher and faster using encapsulation/vitrification in comparison with encapsulation/dehydration (Hirai and Sakai 1999, 2000). In conclusion, the encapsulation/vitrification method is easy to handle (less time for bead dehydration by PVS2 is required instead of placement in laminar airflow or silica gel) and a large number of explants can be cryopreserved in the same time. Therefore, it seems to be relatively suitable for cryopreservation even on a large scale (Hirai and Sakai 1999, 2000).
Parameters Affecting Cryopreservation
Many elements which encompass cryopreservation, such as condition of donor cultures, shoot tips, preculture etc. play a significant role in the success of potato shoot tip cryopreservation. Some of these elements and their influence on regeneration capacity and plant vigour with their flow-on effects on the success of cryopreservation are reviewed below.
Donor Cultures
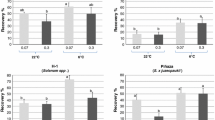
Cold preculture of donor plants prior to cryopreservation is known to improve results for woody (Niino and Sakai 1992; Reed et al. 2003; Chang and Reed 2000) and herbal (Seibert and Wetherbee 1977; Keller 2005) species, which are naturally able to cold acclimate at low temperatures. Also in other species like Zoysia matrella, Zoysia japonica and yam (Dioscorea spp.), which are usually not able to acclimate to cold temperatures, cryopreservation results could be improved after exposure to low temperatures (Chang et al. 2000; Leunufna and Keller 2005). In potato, low temperature preculture was applied before cryopreservation, when encapsulation/vitrification and droplet vitrification methods were used, but with weaker results when compared with the original protocols (Hirai and Sakai 2000; Halmagyi et al. 2005; Kryszczuk et al. 2006). Cold preculture at 4 °C for 3 weeks (Hirai and Sakai 2000), at 4 °C for 1 week (Halmagyi et al. 2005) or alternating temperature of 21/8 °C (day/night) for 1 week (Kryszczuk et al. 2006) have been applied. However, with the DMSO droplet method, Kryszczuk et al. (2006) used an alternating temperature preculture of 21/8 °C (day/night) for 1 week and were able to improve cryopreservation results in four potato accessions. In a similar experiment applying also the DMSO droplet method and alternating temperature preculture at 22/8 °C (day/night) for 1 week, Kaczmarczyk (2008) found significant improvement in regeneration ability of S. tuberosum ‘Désirée’, ‘King Edward’ and the wild species Solanum demissum using a solid regeneration medium. Based on the results of Kaczmarczyk (2008), the conclusion can be drawn that the improvement in regeneration after alternating temperature preculture is not simply based on cold acclimation. The increase of soluble sugars found in the tissue is surely a step towards an acclimation response. However, since no complete cold acclimation has been found in the analysed potato samples, this better response should rather be explained as the result of a general increase of vigour of the in vitro plantlets due to the alternating temperature regime. Improvement in regeneration rate was also found applying cold preculture at 10 °C for 3 weeks (Zhao et al. 2005) or at 6 °C for 3 weeks (Panta et al. 2006). Based on those results, it could be concluded that cold preculture conditions need to be warmer than 6 °C for potato in vitro cultures to receive better cryopreservation response. Alternating temperature during preculture can also enhance recovery. Sometimes the effect of cold acclimation can even be replaced by incubation in sucrose solutions (Dumet et al. 2000). Since the cultivated potato is not able to cold acclimate at all, drought hardening like incubation in sucrose-enriched media can replace the cold preculture (Grospietsch et al. 1999). In ‘Désirée’ using the encapsulation/dehydration method, Grospietsch et al. (1999) noticed that incubation of donor plants in medium with 2 M sucrose for 5 days followed by shoot tip preculture in medium with 0.7 M sucrose for 1 day together increased the plant regeneration levels after cryopreservation. Criel et al. (2008) observed an increase in shoot regeneration from 50% to 80% after preculture of donor plants in medium with 0.055 M up to as high as 0.22 M sorbitol for 21 days followed by droplet vitrification.
The subculture duration can also have a significant impact on cryopreservation results. Different subculture durations were tested by Yoon et al. (2006). They found that optimum subculture duration was genotype-dependent and the best recovery results were obtained with subcultures of 7 weeks in S. tuberosum ‘Dejima’ and 5 weeks in S. stenotomum ‘STN13’. Wide variations of subculture methods have been applied for different cryopreservation methods and genotypes. For example explants were taken 4 weeks after subculture (Kaczmarczyk 2008) or alternatively completely new cultures were established and explants were taken 1 week after each transfer onto fresh medium (Kryszczuk et al. 2006). Culture vessels and the place of cultivation can also have an influence on the cryopreservation response. Keller et al. (2006) showed that plants grown in certain incubators showed significantly higher survival and regeneration percentages in comparison to potato plants grown in culture rooms together with other in vitro cultures like garlic and shallot. Many other environmental subculture conditions, such as light intensity, aeration and also planting density significantly influence both, regeneration of non-cryopreserved and cryopreserved shoot tips. Yoon et al. (2006) attained the highest shoot regeneration percentages with a combination of high light intensity, ventilation of culture vessels and low planting density. Influence of culture vessel type for the donor plants was analysed by Keller et al. (2006). On average, plants grown in jars with cotton plugs in the lid (two similar types) gave best results in comparison with 1,000 ml WECK jars, 300 ml Erlenmeyer flasks or SIGMA tubes (Keller et al. 2006). Plant quality is also an important factor. Superficially clean looking plants can bear infections by endogenous bacteria which may break out after cryopreservation and thereby reduce regeneration levels (Senula et al. 2007; Kaczmarczyk 2008; Keller et al. 2008).
Shoot Tips
The physiological state of shoot tips can influence the regeneration results. Halmagyi et al. (2005) found higher regeneration levels in three S. tuberosum cultivars when apical shoot tips instead of axillary shoot tips were used. The regeneration rate was decreasing with bud position from apical to lower axillary buds. Similar results were found by Schäfer-Menuhr et al. (1994). Percentages of regeneration from apical vs. nodal shoot tips were compared in four S. tuberosum accessions resulting in four- to eightfold better results, when apical shoot tips were used (Schäfer-Menuhr et al. 1994). Yoon et al. (2006) also found that the apical shoot tips of S. tuberosum ‘Dejima’ or S. stenotomum ‘STN13’ subcultured every 3 or 4 weeks showed better regeneration response than those below the apex. In contrast, apical shoot tips reacted weaker than upper or middle axillary shoot tips when donor plants were subcultured after longer periods in a frame, every 5 or 7 weeks (Yoon et al. 2006). In contrast, Hirai and Sakai (1999) found little or no differences in cryopreservation results between axillary shoot tips of the first to third node counted from the apex. Next to the physiological state, also the explant size seems to have a significant effect. Halmagyi et al. (2005) observed the highest regeneration level after cryopreservation with shoot tips of 3–4 mm in length, whereas smaller (1–2 mm) or larger (5–6 mm) shoot tips showed weaker results. Schäfer-Menuhr et al. (1994) also reported that larger shoot tips (2–3 mm long, 0.5–1 mm thick) produced the best regeneration rates. Such explants include the meristem together with a few enveloping leaf bases. Optimum length of the shoot tip in S. tuberosum ‘Dejima’ was 1.5–2 mm and in S. stenotomum ‘STN13’ 1.–1.5 mm (Yoon et al. 2006), showing that the most favourable sizes of shoot tips can be genotype- or species-dependent. In addition, the shoot tips size within each genotype is dependent on the physiological age and environmental culture conditions (Keller et al. 2006).
Preculture of Shoot Tips
Most vitrification and encapsulation/dehydration protocols require a preculture step to increase shoot tip tolerance to dehydration and cooling stress conditions caused by submerging in LN. Sucrose is the most widely used compound to induce such tolerance (Bouafia et al. 1996; Fabre and Dereuddre 1990; Grospietsch et al. 1999; Hirai and Sakai 1999; Kim et al. 2006; Yoon et al. 2006). In addition, glucose, mannitol and sorbitol have been applied (Sarkar and Naik 1998; Halmagyi et al. 2005). Preculture of shoot tips can be made in one step or alternatively in several steps by increasing sugar concentration. Sucrose is most successfully applied in concentrations ranging from 0.3 to 0.75 M (Fabre and Dereuddre 1990; Bouafia et al. 1996; Hirai and Sakai 1999; Halmagyi et al. 2005; Kim et al. 2006). Comparing different sucrose concentrations ranging from 0.3 to 1.5 M for 24 h after shoot tips isolation, Halmagyi et al. (2005) discovered the optimum regeneration response in ‘Désirée’, ‘Ostara’ and ‘Santé’ through applying 0.5 M sucrose in preculture. By comparing sucrose, glucose, mannitol and sorbitol at their concentrations of 0.5 M they revealed the best results with sucrose (Halmagyi et al. 2005). Sarkar and Naik (1998) observed improved survival rates of shoot tips after preculture on medium supplemented with combination of 0.3 M sucrose and 0.2 M mannitol. In contrast, stepwise increase of sucrose concentration in the preculture medium, i.e. 0.3 M sucrose for 24 h followed by 0.7 M sucrose for 7 h caused the highest response in two tested accessions (Yoon et al. 2006). Preculture of donor plants with 2 M sucrose for 5 days followed by shoot tip incubation in 0.7 M sucrose for 1 day significantly increased internal soluble sugars and proline contents and thereby also finally the cryopreservation results (Grospietsch et al. 1999).
Loading and Cryoprotection
Loading or osmoprotection is needed in protocols which rely on vitrification. In the loading process, usually sucrose in combination with glycerol is used to increase osmolarity within shoot tips and to reduce the free water content. Glycerol being a cell membrane penetrating cryoprotectant reduces the concentration of salt by reducing the amount of water frozen on a simple colligative basis (Meryman et al. 1977). In contrast sucrose can only penetrate the cell wall (Tao and Li 1986). Sucrose acts as cryoprotectant by withdrawing water from the cell over plasmolysis and by protecting the plasmalemma against freeze-induced dehydration (Tao and Li 1986). Using sucrose alone, very low success in cryopreservation was obtained (Hirai and Sakai 1999). In contrast, using sucrose in combination with glycerol, a higher plant regeneration capacity was observed and the optimum results were found by applying 0.6 M sucrose with 2 M glycerol in the preculture medium (Hirai and Sakai 1999). The same solution was successfully used for loading of ‘Atlantic’ and ‘Superior’ in a vitrification protocol (Zhao et al. 2005). Supercool X1000 ice-blocking agent added to the PVS2 solution further improved the cryopreservation response (Zhao et al. 2005). In vitrification-based protocols, PVS2 is the most commonly applied cryoprotectant (Sarkar and Naik 1998; Hirai and Sakai 1999, 2000; Halmagyi et al. 2005; Yoon et al. 2006; Kim et al. 2006, Panta et al. 2009a, 2009b; Zhao et al. 2005). Shoot tips are either directly put into 100% PVS2 solution (Halmagyi et al. 2005), or the PVS2 concentration is stepwise increased (Sarkar and Naik 1998). Incubation time in PVS2 depends on working temperature (room temperature or 0 °C), shoot tip size and genotype (Halmagyi et al. 2005). A separate method of cryoprotection is performed in the DMSO droplet method. After isolation the shoot tips are incubated in MSTo medium overnight, and the next day they are cryoprotected in MSTo medium with 10% DMSO (Schäfer-Menuhr et al. 1994, Kaczmarczyk 2008).
Cooling
First applied potato cryopreservation protocols utilised the two-step cooling system (Table 2). Bajaj (1977) slowly cooled the explants by tying cryovials together with the explants to a cord and by gradually sinking them into LN, firstly by exposing the samples to the vapour of LN and then directly transferring them into LN. Towill (1981a) used cooling speed of 0.3 °C min−1 until −40 °C before plunging the explants directly into LN. Similar speed of cooling was used by Henshaw et al. (1985). Explants in cryovials were then submerged into a methanol bath at −5 °C for 15 min. Freezing of cryoprotectant solution in the cryovials was induced by touching the walls with pre-cooled forceps. After 15 min, the methanol bath was cooled down at speed of 0.3 °C min−1 until −30 °C. Explants were afterwards plunged directly in LN (Henshaw et al. 1985). Benson et al. (1989) used a programmable freezing unit, were shoot tips were cooled first at 1 °C min−1 to 0 °C then with 0.5 °C min−1 until −10 °C before transfer to LN. In comparison, two-step cooling had lower shoot regeneration than ultra-rapid cooling (Benson et al. 1989). Additional disadvantages of two-step cooling are difficult handling, longer processing time of the method (Schäfer-Menuhr et al. 1997) and need of special equipment (Kartha and Engelmann 1994). Recently developed methods, such as DMSO droplet method, vitrification, encapsulation/dehydration, encapsulation/vitrification and droplet vitrification apply rapid cooling by plunging precultured and cryoprotected shoot tips directly in LN. The fastest cooling rate (24,000 °C min−1; Pennycooke and Towill 2000) is implemented by using partially solidified nitrogen (LN slush, approximately −208 °C (Pennycooke and Towill 2000)), which was also applied in potato by Kim et al. (2006). The cooling rate of 360 °C min−1 is reached using PVS2 solution within cryovials (Towill and Bonnart 2003) and can be further increased by using aluminium foil strips (7,800 °C min−1, Towill and Bonnart 2003). Explants are exposed directly to LN on aluminium foils, whereas explants in cryovials are less fast cooled because of the air or cryoprotectant barrier to LN.
Rewarming
In potato shoot tip cryopreservation, either slow, medium or rapid rewarming has been applied. Slow rewarming was carried out by putting the explants from LN to room temperature (Fabre and Dereuddre 1990; Bouafia et al. 1996). Medium rewarming speed was achieved by plunging and shaking explants from LN into room temperatured medium with rewarming speeds of approximately 3,000 °C min−1 (Schäfer-Menuhr et al. 1996). In most cases, rapid rewarming was carried out by transferring explants from LN into a 35–40 °C water bath for 1–3 min (Hirai and Sakai 1999; Kryszczuk et al. 2006; Sarkar and Naik 1998; Zhao et al. 2005). Comparing different warming rates, Kim et al. (2006) found higher regeneration response in shoot tips warmed by dipping the foils with explants directly in preheated (40 °C) 0.8 M sucrose medium for 30 s than by warming the explants in 40 °C water bath for 5 s and then adding 0.8 M sucrose (40 °C). In the vitrification-based protocols, unloading with high sucrose concentrations is applied to dilute and remove PVS2 from explants, which could be toxic to cells and thereby reduce shoot regeneration capacity. In the successfully applied potato cryopreservation protocols unloading solutions with 0.8–1.2 M sucrose for 10–30 min have been introduced (Hirai and Sakai 1999; Sarkar and Naik 1998; Zhao et al. 2005; Kim et al. 2006; Yoon et al. 2006).
Further Culture and Viability Assessment
The loss of regenerating plantlets does often occur in cryopreservation because of not surviving or damaged shoot tips (Grout and Henshaw 1980; Schäfer-Menuhr et al. 1994; Benson et al. 1996; Kaczmarczyk et al. 2008a). After recovery, MS culture media with 3% sucrose and plant hormones have generally been used. Absence of phytohormones resulted in reduced numbers, sometime even no regenerated plants (Marion Grübe, IPK, Gatersleben, personal communication). In the DMSO droplet method, a combination of 0.5 mg/l-1 zeatin riboside, 0.2 mg/l-1 GA3, and 0.5 mg/l-1 IAA (Towill 1983) was used. In certain protocols higher phytohormone concentrations were applied in the first weeks of the regeneration phase. Subsequently during the culture, lower levels or no phytohormones at all were supplied (Bouafia et al. 1996; Sarkar and Naik 1998; Yoon et al. 2006). Bouafia et al. (1996) employed medium supplemented with 0.1 mg/l-1 BAP, 0.01 mg/l-1 NAA and 5 mg/l-1 GA3 for a period of 1 week. In the same protocol, the shoot tips were later transferred to a medium with only 0.001 mg/l-1 GA3 to avoid callus formation. Sarkar and Naik (1998) used a similar procedure, first by regenerating shoot tips on medium with 2 mg/l-1 GA3 and 0.23 mg/l-1 BAP for 1 week and then on medium with 1 mg/l-1 GA3. Yoon et al. (2006) used medium with 0.05 mg/l-1 IAA, 0.3 mg/l-1 zeatin and 0.05 mg/l-1 GA3 for 1 week followed by a culture on medium without phytohormones. Increase in plant regeneration was obtained by addition of polyoxy ethylene (POE)-polyoxypropylene (trade name PluronicF-68) at concentrations of 0.0005% to the regeneration media (Zhao et al. 2005). The consistency of the regeneration medium can also have an influence on cryopreservation results. Kryszczuk et al. (2006) observed the highest regeneration rates in four cultivated potato varieties using a vitrification protocol and solid medium, whereas the highest recovery using the DMSO droplet method was attained with liquid regeneration medium. In contrast, Kaczmarczyk (2008) also used the DMSO droplet method in two cultivated potato varieties to compare liquid and solid regeneration media obtaining better response on the solid medium in terms of plant regeneration and plantlet structure. Light regime also plays an important role in shoot tip regeneration. As proven by Benson et al. (1996) ‘Désirée’ showed only high survival rate, if the light intensity was low (15 μmol m−2 s−1) during the first week of recovery. Afterwards it was necessary to cultivate plants in higher light conditions (45 μmol m−2 s−1) to ensure regular morphological development (Benson et al. 1989).
Ultrastructure
Ultrastructural studies on potato applying transmission electron microscopy have revealed structural damage on both surviving and killed explants resulting from different cryopreservation methods (Golmirzaie et al. 2000; Grout and Henshaw 1980; Kaczmarczyk et al. 2008a). First ultrastructural studies on potato shoot tips were conducted by Grout and Henshaw (1980) using Solanum goniocalyx and ultra-rapid freezing. After rewarming of shoot tips it was not possible visually to discriminate between killed and surviving explants during the first 24 h. After rewarming and culture for 24 h, shoot tips showed areas of massive damage often associated with rupture of the epidermis that was also visible under light microscope (Grout and Henshaw 1980). Ultrastructural analyses revealed considerable cellular damage in certain areas with total breakdown of protoplasts and sometimes cell wall rupture (Grout and Henshaw 1980). The influence of the cell wall in freezing tolerance of cultured potato cells was analysed by Tao et al. (1983). In this study, the cell wall of cultured potato cells accounted for freezing injury because of mechanical stress between cell wall and plasma membrane during a freeze-thaw cycle. Surviving explants were characterised by single cells or small groups of cells with electron-dense cytoplasm which were located in primordia and dome regions of the explants (Grout and Henshaw 1980). Survival involved either regrowth of the shoot apex or callus proliferation, although it was sometimes difficult to be certain whether the original apical meristem resumed growth or whether it was replaced by another meristem rapidly regenerated from the callus (Grout and Henshaw 1980). Some studies indicate that the original meristem does not survive cryopreservation and that only small groups of cells are enough for regeneration of complete shoots (Fukai and Oe 1990; Grout and Henshaw 1980; Haskins and Kartha 1980). Insufficient numbers of undamaged cells could lead to callus formation instead of shoot development (Grout and Henshaw 1980).
In a later study, four accessions of S. stenotomum were studied, which had been cryopreserved by the vitrification method (Golmirzaie et al. 2000). After cryopreservation, abnormal cytoplasm, cell plasmolysis at different stages and a large number of small vesicles were found. Further to a lesser extent cell wall rupture, protoplast outflow and anomalous nucleus shape were also discovered. Golmirzaie et al. (2000) could observe a negative correlation between the percentage of survival and cellular damage meaning the genotype with the highest survival capacity had the least ultrastructural damage.
Suzuki et al. (1997) analysed the influence of sugar preculture on ultrastructure of shoot tips of Asparagus officinalis. They found surviving cells after sucrose preculture with many developed plastids that contained starch grains and extensive rough endoplasmic reticulum (rER) in the cytosol (Suzuki et al. 1997). Jitsuyama et al. (2002) obtained similar results. They observed drastic changes in cell ultrastructure with the appearance of rER after sugar incubation (Jitsuyama et al. 2002). These ultrastructural changes resemble those occurring in cold-acclimated cells, thus, being a characteristic feature of freezing tolerance (Ishikawa 1996, Stefanowska et al. 2002). Sucrose preculture could, therefore, substitute cold acclimation and increase freezing tolerance in tissue subjected to cryopreservation.
Ultrastructural studies on S. tuberosum ‘Désirée’ using the DMSO droplet method, showed changes, when shoot tips had been incubated in the cryoprotectant solution for 2 h (10% DMSO in MSTo medium, Kaczmarczyk et al. 2008a). After this time period, cells contained numerous small vesicles. Mitochondria and chloroplasts were increased in size and vacuoles assumed an irregular shape. Directly after cooling in LN and rewarming in MS medium, cells had been unchanged in comparison to the conditions prior to freezing. However, first visible changes were found 2 days after rewarming, when parts of the meristematic dome and epidermis showed signs of extensive damage. Rupture of plasmalemma, plasmolysis and destruction of cell organelles as well as strong heterochromatisation of nuclei were observed. Survival and regeneration of cells were mainly found in leaf primordial regions, where cells were physiologically very active and contained many mitochondria and intact or regenerating chloroplasts (Kaczmarczyk et al. 2008a).
Genetic and Epigenetic Factors
Genomic DNA Stability
In theory, all metabolic activities at temperatures of LN are reduced to zero, so that after rewarming from cryopreservation, true-to-type plants are expected (Panis et al. 2001). Since cryopreservation protocols do not only involve cooling in and rewarming from LN but also in vitro culture and regeneration processes, phenotypic and genomic changes can occur due to somaclonal variation. Therefore, the verification of true-to-type plants after cryopreservation is necessary (Harding 2004).
In potato, many studies on genetic integrity after cryopreservation have been performed. Morphological and phenotypic (Barandalla et al. 2003; Benson et al. 1996; Harding and Benson 1994; Hirai and Sakai 2000; Mix-Wagner 1999; Perazzo et al. 1999–2000; Schäfer-Menuhr 1996a, 1996b, 1997; Zarghami et al. 2008), cytological (Barandalla et al. 2003; Benson et al. 1996; Mix-Wagner 1999; Schäfer-Menuhr et al. 1996a, 1996b, 1997; Ward et al. 1993; Zarghami et al. 2008) and molecular (Harding 1991; Harding and Benson 2000; Harding and Benson 2001; Hirai and Sakai 1999; Mix-Wagner 1999; Schäfer-Menuhr et al. 1996a, 1996b, 1997; Zarghami et al. 2008) comparisons were conducted revealing that plant material was genetically stable as a result of cryopreservation.
Shoot tips of diverse potato species with different ploidy levels (diploid, tetraploid and hexaploid) exhibited normal developmental patterns (flowering, berry set and tuber formation) after regeneration from cryopreservation which had been carried out through the encapsulation/dehydration method (Benson et al. 1996). The ploidy status of the regenerated plants was maintained and chromosomal abnormalities were not observed based on cytological studies (Benson et al. 1996). Harding (1991) and Harding and Benson (2000) also verified the stability at the molecular level through the analyses of the ribosomal RNA genes (rDNA) as well as the nuclear-chloroplast DNA in the chloroplast genome in potato plants regenerated from cryopreserved shoot apices.
Using microsatellite fragments, regenerated plants from cryopreserved shoot tips of ‘Brodick’ and ‘Golden Wonder’ were compared to the control materials. Microsatellite profiles were identical in the regenerated material to those of the parental plants and their progeny. This demonstrates the stable somatic inheritance of genomic regions containing the measured simple sequences (Harding and Benson 2001).
Applying the DMSO droplet method, genetic stability was also confirmed in a random sample of the collection using morphological parameters, flow cytometric measurements and restriction fragment length polymorphism (RFLP) analyses by Schäfer-Menuhr et al. (1997). The comparison of morphological parameters displayed only one abnormal and a few weak plants in a set of approximately 1,000 regenerated plants from 98 varieties tested. The vigour was not increasingly reduced compared to the regular variation occuring in the control plants, which had not been cryopreserved (Schäfer-Menuhr et al. 1997). RFLP analysis and flow cytometry did not show genetic abnormalities within 161 samples of regrown plants (Schäfer-Menuhr et al. 1996a, 1996b, 1997). Storage in LN for up to 10 years was found to have no adverse effect on the regeneration rates (Keller et al. 2006; Mix-Wagner et al. 2003). Concluding from these results, it seems likely that the cryopreservation technique does not induce somaclonal variation (Schäfer-Menuhr et al. 1997). However, certain wild species, such as Solanum microdentum and S. pinnatisectum and dihaploid (2n = 2x = 24) S. tuberosum lines ‘PDH 40’, ‘PDH 135’, ‘PDH 634’ and ‘PDH 638’ produced more than one shoot after rewarming from cryopreservation (Ward et al. 1993). The authors pointed out that a certain portion of these shoots may have been originating from callus and could, therefore, have shown evidence of somaclonal variation. Plants recovered from cryopreservation were tested concerning their ploidy levels, but only one tetraploid plant was found, which could be due to spontaneous chromosome doubling. Since the number of regenerated shoots revealing abnormality was low, it was concluded that cryopreservation did not induce ploidy changes (Ward et al. 1993). The capacity to generate tubers was not affected by the cryopreservation process in the cryopreserved plant materials, but the development of flowers was impaired in comparison to tuber-derived control plants (Harding and Benson 1994). Perazzo et al. (1999–2000) performed phenotypical comparisons between material cryopreserved for 1 year and the original clones maintained in vitro. A number of 18 accessions were analysed belonging to the following potato species and interspecific hybrids: S. stenotomum, S. tuberosum ssp. andigena, S. goniocalyx, Solanum chaucha, S. tuberosum ssp. andigena x S. tuberosum, S. phureja, and Solanum curtilobum. Even 23 different morphological descriptors were compared. Differences were found in six of the 18 accessions. Of these, there were two misidentifications. The remaining four accessions displayed small morphological differences, which were attributable to pathological and environmental factors (Perazzo et al. 1999–2000). Phenotypic integrity between cryopreserved and control stock plants of S. tuberosum ‘Agria’ and ‘Marfona’ was found by Zarghami et al. (2008), when the encapsulation/dehydration method was applied. In the same study, genetic stability was found using AFLP analyses and by using seven primer pairs. In addition, ploidy comparison did not reveal any differences between control and cryopreserved plants (Zarghami et al. 2008). Applying the encapsulation/vitrification method, Hirai and Sakai (1999) compared control plants with cryopreserved materials using RAPD analyses with 17 primers. Genetic integrity was also found for each analysed plant that had been cryopreserved (Hirai and Sakai 1999).
Epigenetic Factors
Genetic changes are characterised through various alterations in the DNA nucleotide sequences (single copy sequences, low copy and repetitive sequences). In contrast, epigenetic modifications do not change the original DNA sequence (Boyko and Kovalchuk 2008), but may, nevertheless, result in heritable changes of gene expression potential. Typical features associated with epigenetic processes such as regulation of gene expression, activity of transposable elements, defence against foreign DNA and inheritance of specific gene expression patterns are DNA methylation, particular histone modifications and alterations in chromatin structure (Boyko and Kovalchuk 2008). DNA methylation is required for regular development of plants and may have an impact on vigour and morphogenesis (Chan et al. 2005). Some studies have discussed putative effects of changes in DNA methylation on genomic DNA stability and rearrangements in micropropagated and cryopreserved stocks (Harding 2004; Harding et al. 2009; Peredo et al. 2008). Modification of epigenetic features due to the stress during in vitro culture could possibly result in altered phenotypes in the recovered plants. First comparative studies of epigenetic marks in plants before and after cryopreservation have already been done. Modifications in DNA methylation have been determined in almond (Channuntapipat et al. 2003), papaya (Kaity et al. 2008), chrysanthemum (Martín and González-Benito 2006), Ribes (Johnston et al. 2009), strawberry (Hao et al. 2002a) and citrus (Hao et al. 2002b). In potato, no studies on epigenetic modifications in connection with cryopreservation have been published so far, but first analyses at IPK Gatersleben pointed to methylation changes, although it is not clear yet, whether these changes are due to cryopreservation or long-term in vitro storage (A Kaczmarczyk, unpublished results). Analysis of epigenetic changes induced by ex situ storage systems like in vitro culture has been carried out in potato so far in only one study (Harding 1994). DNA methylation changes have been found in mature greenhouse plants of S. tuberosum ‘Désirée’ and ‘Golden Wonder’ after they had grown on MS with 6% (w/v) mannitol for 6 months. The growth on mannitol-enriched medium seems to be correlated with subsequent DNA hypermethylation, which may be an adaptive reaction to abnormal environmental conditions related to high osmotic stress. This result has implications for vegetatively multiplied potato plants, because the methylation status could be inherited in the tuber progeny, and changed phenotypes may subsequently be expressed in following generations (Harding 1994). Overall it is suggested that the methylation changes may not be induced by cryopreservation per se but may be the results of the whole process of in vitro culture and regeneration phases (Channuntapipat et al. 2003; Harding 2004).
Thermal Analysis
The state of water, meaning ice or glass formation, during cryopreservation is important for the success of the method. Whereas ice crystallisation within cells during cooling and warming leads to membrane damage and cell death, the glass formation, which is a solid amorphous state of water in solutions, can alternatively preserve the cells.
DSC is a thermal analysis method that can be applied for measurement and determination of glass transitions and ice crystallisation during cryopreservation (Bilavčík et al. 2007). It measures heat flow and water transitions in samples as a function of temperature and/or time (Benson et al. 2006; Höhne et al. 2003). Transitions between liquid, amorphous glassy, and ice states can be detected by heat flow data through endo- and exothermic peaks or sigmoidal changes in the record diagram. The point of change from the liquid to the glass state is the glass transition temperature. Thermal profiles provide important information about the cryoprotective treatments required to obviate lethal ice formation and to stabilise glasses (Benson et al. 2006).
Using the DMSO droplet method in S. tuberosum ‘Désirée’ and S. demissum DSC analyses were conducted with freshly isolated shoot tips and following incubation in cryoprotectant solution (10% DMSO in MS medium with 3% sucrose, Kaczmarczyk 2008). The explants were thermally characterised by an ice crystallisation peak during cooling, and melting peak of ice during rewarming. In addition the cryoprotected shoot tips of S. tuberosum ‘Désirée’ exhibited a glass transition at −117 °C. The cryoprotected accessions showed crystallisation and melting of ice, but the heat flow was much lower compared to freshly isolated shoot tips (Kaczmarczyk 2008). This means that the cryoprotectant withdraws water from shoot tips. Also melting and crystallisation points were shifted to lower temperatures, which means that the cryoprotectant decreased the ice crystallisation temperature. It was concluded that the DMSO droplet method relies on ultra-rapid cooling and rewarming with marginal ice formation, which does not detain shoot tips from regeneration after rewarming. Survival and regeneration of shoot tips with marginal intracellular ice crystallisation was also reported by other authors (Gonzalez-Arnao et al. 2008; Mazur 1977; Santos and Stushnoff 2003). In contrast to the DMSO droplet method, the vitrification method uses PVS2 as cryoprotectant. Depending on incubation time, the cryoprotected potato shoot tips showed either crystallisation of ice together with glass transition or only glass transition at −108 °C without ice crystallisation at all. The longer the incubation time, the less ice crystallisation was found and as a result of 60 min incubation in PVS2, only glass transition was detected (Kaczmarczyk 2008). Therefore, DSC analysis can be used to find optimum cryoprotectant incubation times, although it should additionally be considered that DSC instruments cannot manage the speed of cooling and rewarming used in cryopreservation experiments and there are differences between DSC analyses and actual cryopreservation experiments.
Biochemical Analysis and Proteome Research
Biochemical characterization of explants can be assessed through determination of the plant protein expression. Proteomics approaches are used to analyse the complex functions of plants at different levels and are supported through the progress obtained in sensitive and rapid protein identification by mass spectrometry (Canovas et al. 2004).
In potato, proteome studies have been carried out mainly on tuber tissue (Bauw et al. 2006; Delaplace et al. 2005; Lehesranta et al. 2005, 2006) and in connection with investigation on the action of phytopathogens such as Pectobacterium atrosepticum (Coulthurst et al. 2006, Mattinen et al. 2007) and the aphid Macrosiphum euphorbiae (Nguyen et al. 2007). The application of protein expression studies for cryopreservation is a new research area. Carpentier et al. (2007) used banana as a model to study protein expression associated with sucrose-mediated osmotic stress in the meristem. A protocol for two-dimensional gels was developed for small amounts of tissue. Polypeptide databases obtained by mass spectrometry were used for discriminative identification of proteins from closely related plant species. From those results, the conclusion was drawn that the maintenance of an osmoprotective intracellular sucrose concentration, the enhanced expression of particular genes of glycolysis and the conservation of the cell wall integrity are all essential to maintain homeostasis, and to acclimate to and to survive from dehydration influencing the explant prior to cryopreservation (Carpentier et al. 2007). Results were, however, not further evaluated and no identification of proteins was presented (Carpentier et al. 2005). First proteome analyses in connection with cryopreservation have been made for potato shoot tips by Criel et al. (2008) and Kaczmarczyk (2008). Criel et al. (2008) analysed proteins of S. tuberosum ‘Désirée’ shoot tips and leaves after preculture of donor plants for 21 days on regular MS medium and MS complemented with 0.055, 0.11 and 0.22 M sorbitol. In their study, the regeneration rate after cryopreservation was increased from 50% to 80% with increased sorbitol concentration in the medium. Differences in 2D-difference-gel-electrophoresis patterns were observed for samples precultured on various sorbitol-enriched media. In total, 63 and 15 differently expressed proteins were determined in the shoot tips and leaves, respectively. Of them 43 and 3, respectively, were identified by matrix-assisted laser desorption/ionisation mass spectrometry. The results indicated strong alteration of the primary metabolism and in carbon fixation. These results were additionally confirmed by parallel biochemical analyses. Increase in carbohydrates was measured in plants treated with up to 0.11 M sorbitol, whereas the concentration of 0.22 M sorbitol showed decreased levels of carbohydrates. In conclusion, better cryopreservation response could be obtained due to high internal sugar concentration and alterations in primary metabolism as a result of the cryoprotectant function of sugars (Criel et al. 2008). Kaczmarczyk (2008) analysed S. tuberosum ‘Désirée’ and S. demissum after two different preculture conditions followed by cryopreservation; 1 week culture at 22/8 °C day/night temperature in comparison to 1 week culture at constant temperature of 22 °C. The alternating temperature preculture of donor plants revealed higher survival and regeneration rates after cryopreservation. In protein expression, only minor changes were found between the preculture treatments in the two analysed species, meaning that 2.7% or 0.4% of the proteins were changed in S. demissum and S. tuberosum ‘Désireé’, respectively. Proteins were mostly down-regulated in their expression level after preculture with alternating temperature. The identified proteins belonged to the functional groups of metabolism, signal transduction, defence, transcription, energy and secondary metabolism (Kaczmarczyk 2008).
Cryotherapy
Besides long-term storage, cryopreservation can also be used for virus elimination, a method named cryotherapy (Benson 2008b; Wang and Valkonen 2009a, 2009b; Wang et al. 2008a, 2008b, 2009). Cooling and freezing of explants down to LN temperatures facilitates viral elimination in larger meristems, because many viruses are unable to infect the meristem proper, and virus-free plants can be grown from excised apices of small size (Martin and Postman 1999). Cryotherapy works as a micro-scalpel, where larger, more hydrated cells outside the meristem die because of freezing, and only small cells within and close to the meristem survive cryopreservation (Benson 2008b; Helliot et al. 2002). Cryotherapy has been successfully applied to eliminate or reduce viruses in banana (Helliot et al. 2002), grapevine (Wang et al. 2003), Prunus (Brison et al. 1997), raspberry (Wang and Valkonen 2009b; Wang et al. 2009), citrus (Ding et al. 2008) and sweet potato (Wang and Valkonen 2008). Also potato shoot tips were freed from viruses by cryotherapy. Wang et al. (2006) applied encapsulation/dehydration, encapsulation/vitrification and DMSO droplet method to eliminate Potato leafroll virus (PLRV) and Potato virus Y (PVY) from virus-infected potato shoot tips. Each method produced similar rates of virus-free plants, between 83% and 86% for PLRV, and 91% and 95% for PVY. Frequencies of virus-free plantlets produced by cryotherapy were actually higher than those derived from meristem culture (56% for PLRV and 62% for PVY) and from thermotherapy (50% for PLRV and 65% for PVY), and comparable to those by thermotherapy followed by meristem culture (90% for PLRV and 93% for PVY). Regrowth rates of cryopreserved shoot tips were 85–89% and, thus, higher than regrowth ability found by using meristem culture (55%) alone, and thermotherapy followed by meristem culture (50%), but comparable to the results obtained by thermotherapy (87%). In addition, the required working time for cryotherapy was much less (about 55 days) in comparison to thermotherapy or thermotherapy followed by shoot tip culture (87 days). In total, cryotherapy has shown its advantages being easier in handling and simpler in procedures for plant regeneration. It has also been verified that cryotherapy produced higher rates of regrowth and virus eradication and shorter time for production of virus-free plants was needed than in using other virus elimination methods (Wang et al. 2006). Since virus-free donor material is required for successful cryopreservation and conservation of accessions, Wang et al. (2006) proposed the use of cryopreservation simultaneously for long-term storage of potato accessions and the production of virus-free plants.
Conclusions
Improvement in the development and successful application of potato shoot tip cryopreservation protocols have been made over the last 30 years. Many studies have been conducted on the influence of various factors affecting cryopreservation such as the physiological state of donor plants and shoot tips, as well as specific cryogenic factors, e.g., type of cryoprotectant(s) and speed of cooling and rewarming. So far, all known cryopreservation methods of plants already applied for shoot tip tissue, like two-step cooling, ultra-rapid cooling, DMSO droplet method (droplet freezing), encapsulation/dehydration, encapsulation/vitrification, vitrification and droplet vitrification, have been successfully employed for a wide range of species. It remains to be seen whether one particular technique can become the ‘standard’ for plant cryogenic storage (e.g. droplet vitrification) or whether, as it already exists, many protocols will remain in common use as long as they work effectively. Although many successful protocols have been established, none of the cryopreservation techniques can guarantee the conservation of all genotypes. A critical point in potato cryopreservation is the diverse response between different genotypes in terms of their regeneration capacities as a result of cryopreservation. Therefore, weakly reacting genotypes need to be maintained using tissue or field culture. In addition, certain genotypes may not even have tissue culture ability and need to be preserved as tubers. Finally, none of the conservation strategies (cryopreservation, tissue culture, field culture) can be completely safe, because materials may always be lost regardless of the conservation technique.
Studies on the genomic DNA stability conducted on morphological and molecular level have revealed no changes and thereby confirmed the successful conservation of cultivar characteristics (true-to-type status) as a result of cryopreservation. However, no studies on the epigenetic level followed by cryopreservation have been published so far. In addition to long-term conservation, the cryopreservation methods have also effectively been applied for virus elimination in potato (i.e. cryotherapy). Therefore, it can be seen as an alternative method to chemo- or thermotherapy with the advantage that the cryotreated shoot tips can also be simultaneously stored for long-term conservation. Additional fundamental studies of freeze tolerance and influence of LN on plant tissues need to be carried out to further understand the processes occurring after various steps in the cryopreservation protocols in order to develop more practical cryogenic methods.
Abbreviations
- AFLP:
-
Amplified fragment length polymorphism
- BAP:
-
6-benzylaminopurine
- DSC:
-
Differential scanning calorimetry
- DMSO:
-
Dimethyl sulfoxide
- GA3 :
-
Gibberellic acid
- IAA:
-
Indole-3-acetic acid
- LN:
-
Liquid nitrogen
- MS:
-
Murashige and Skoog (1962)
- MSTo:
-
Medium composition after Towill (Towill 1983)
- NAA:
-
1-naphthaleneacetic acid
- PVS2:
-
Plant vitrification solution 2 (Sakai et al. 1990)
- RAPD:
-
Random amplified polymorphic DNA
- RFLP:
-
Restriction fragment length polymorphism
References
Bajaj YPS (1977) Initiation of shoots and callus from potato-tuber sprouts and axillary buds frozen at −196 °C. Crop Improv 4:48–53
Bajaj YPS (1995) Cryopreservation of germplasm of potato (Solanum tuberosum L.) and cassava (Manihot esculenta Crantz). In: Bajaj YPS (ed) Biotechnology in agriculture and forestry Vol. 32 cryopreservation of plant germplasm I. Springer-Verlag, Berlin, pp 398–416
Barandalla L, Sánchez I, Ritter E, Ruiz de Galarreta JI (2003) Conservation of potato (Solanum tuberosum L.) cultivars by cryopreservation. Span J Agric Res 1:9–13
Bauw G, Nielsen HV, Emmersen J, Nielsen KL, Jorgensen M, Welinder KG (2006) Patatins, Kunitz protease inhibitors and other major proteins in tuber of potato cv. Kuras. FEBS J 273:3569–3584
Benson EE (2008a) Cryopreservation theory. In: Reed BM (ed) Plant cryopreservation: a practical guide. Springer, New York, pp 15–32
Benson EE (2008b) Cryopreservation of phytodiversity: a critical appraisal of theory practice. Crit Rev Plant Sci 27:141–219
Benson EE, Harding K, Smith H (1989) Variation in recovery of cryopreserved shoot-tips of Solanum tuberosum exposed to different pre- and post-freeze light regimes. Cryo-Letters 10:323–344
Benson EE, Chabrillange N, Engelmann F (1992) Mise au point de méthodes de cryoconservation de méristemes pour la conservation à long terme des ressources génétiques du manioc (Manihot spp.). Rapport de fin d’etude, Laboratoire de Ressources Génétiques et Amélioration des Plantes Tropicale. ORSTOM, Montpellier
Benson EE, Wilkinson M, Todd A, Ekuere U, Lyon J (1996) Developmental competence and ploidy stability in plants regenerated from cryopreserved potato shoot-tips. Cryo-Letters 17:119–128
Benson EE, Johnston J, Muthusamy JA, Harding K (2006) Physical and engineering perspectives of in vitro plant cryopreservation. In: Gupta SD, Ibaraki Y (eds) Plant tissue culture engineering. Springer, Dordrecht, pp 441–476
Bilavčík A, Zámečník J, Faltus M (2007) The use of differential scanning calorimetric analysis of plant shoot tips in cryopreservation. COST 871 Action, Oviedo, Spain, 13–14 April 26–27. pp 26–27
Bouafia S, Jelti N, Lairy G, Blanc A, Bonnel E, Dereuddre J (1996) Cryopreservation of potato shoot tips by encapsulation-dehydration. Potato Res 39:69–78
Boyko A, Kovalchuk I (2008) Epigenetic control of plant stress response. Environ Mol Mutagen 49:61–72
Brison M, De Boucaud MT, Pierronnet A, Dosba F (1997) Effect of cryopreservation on the sanitary state of a cv of Prunus rootstock experimentally contaminated with Plum Pox Potyvirus. Plant Sci 123:189–196
Canovas FM, Dumas-Gaudot E, Recorbet G, Jorrin J, Mock HP, Rossignol M (2004) Plant proteome analysis. Proteomics 4:285–298
Carpentier SC, Witters E, Laukens K, Deckers P, Swennen R, Panis B (2005) Preparation of protein extracts from recalcitrant plant tissues: an evaluation of different methods for two-dimensional gel electrophoresis analysis. Proteomics 5:2497–2507
Carpentier SC, Witters E, Laukens K, Van Onckelen H, Swennen R, Panis B (2007) Banana (Musa spp.) as a model to study the meristem proteome: acclimation to osmotic stress. Proteomics 7:92–105
Chan SWL, Henderson IR, Jacobsen SE (2005) Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat Rev Genet 6:351–360
Chang Y, Reed BM (2000) Extended alternating-temperature cold acclimation and culture duration improve pear shoot cryopreservation. Cryobiology 40:311–322
Chang Y, Barker RE, Reed BM (2000) Cold acclimation improves recovery of cryopreserved grass (Zoysia and Lolium sp.). Cryo-Letters 21:107–116
Channuntapipat C, Sedgley M, Collins G (2003) Changes in methylation and structure of DNA from almond tissues during in vitro culture and cryopreservation. J Amer Soc Hort Sci 128:890–897
Coulthurst SJ, Lilley KS, Salmond GPC (2006) Genetic and proteomic analysis of the role of luxS in the enteric phytopathogen, Erwinia carotovora. Mol Plant Pathol 7:31–45
Criel B, Panis B, Oufir M, Swennen R, Renault D, Hausman JF (2008) Protein and carbohydrate analyses of abiotic stress underlying cryopreservation in potato. Agrifood Res Working Pap 153:37–38
Delaplace P, Dierick JF, Fauconnier ML, Van Der Wal F, Cordewener JG, America TA, du Jardin P (2005) Characterization of potato (Solanum tuberosum L.) tuber ageing using physiological and proteomic markers (2D-PAGE). FEBS J 272:71–71
Ding F, Jin SX, Hong N, Zhong Y, Cao Q, Yi GJ, Wang GP (2008) Vitrification-cryopreservation, an efficient method for eliminating Candidatus Liberobactus asiaticus, the citrus Huanglongbing pathogen, from in vitro adult shoot tips. Plant Cell Rep 27:241–250
Dodds JH, Huaman Z, Lizarraga R (1991) Potato germplasm conservation. In: Dodds JH (ed) In vitro methods for conservation of plant genetic resources. Chapman and Hall, London, pp 93–109
Dumet D, Chang Y, Reed BM, Benson EE (2000) Replacement of cold acclimation with high sucrose pretreatment in black currant cryopreservation. In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm. Current research progress and application. Japan International Research Center for Agricultural Sciences, Tsukuba, pp 385–387
Edesi J, Kotkas K, Mattila AM, Häggman H (2009) Preservation of potato (Solanum tuberosum) germplasm by the mean of cryopreservation. Meeting Workgroup 2 COST Action 871, Integration of cryopreservation in genebank srategies, Gatersleben, Germany, 9–11 September 2009. pp 34
Fabre J, Dereuddre J (1990) Encapsulation-dehydration: a new approach to cryopreservation of Solanum shoot-tips. Cryo-Letters 11:423–426
Faltus M, Bilavčík A, Zámečník J, Svoboda P, Domkarova J (2008) Establishment of cryobank of potato and hop apices in the Czech Republic. Agrifood Res Working Pap 153:65–66
FAO (2007). Available at: http://www.potato2008.org/en/potato/. Accessed 8 May 2008
Fukai S, Oe M (1990) Morphological observations of chrysanthemum shoot tips cultured after cryoprotection and freezing. J Jpn Soc Hortic Sci 59:383–387
Gavrilenko T (2008) Ex situ conservation of plant genetic resources in Russia: history, current status and persectives. Agrifood Res Working Pap 153:65–66
Gnanapragasam S, Vasil IK (1992) Ultrastructural changes in suspension culture cells of Panicum maximum during cryopreservation. Plant Cell Rep 11:169–174
Golmirzaie AM, Panta A (1997) Advances in potato cryopreservation by vitrification. CIP Program Report. pp 71–76
Golmirzaie AM, Panta A (2000) Advances in potato cryopreservation at CIP. In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm. Current research progress and application. Japan International Research Center for Agricultural Sciences, Tsukuba, pp 250–254
Golmirzaie A, Toledo J (1997) In vitro conservation of potato and sweetpotato germplasm. CIP Program Report. pp 351–356
Golmirzaie AM, Panta A, Delgado C (2000) Structural observations on potato shoot-tips after thawing from liquid nitrogen. In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm. Current research progress and application. Japan International Research Center for Agricultural Sciences, Tsukuba, pp 388–389
Gonzalez-Arnao MT, Panta A, Roca WM, Escobar RH, Engelmann F (2008) Development and large scale application of cryopreservation techniques for shoot and somatic embryo cultures of tropical crops. Plant Cell Tissue Organ Cult 92:1–13
Grospietsch M, Stodůlková E, Zámečník J (1999) Effect of osmotic stress on the dehydration tolerance and cryopreservation of Solanum tuberosum shoot tips. Cryo-Letters 20:339–346
Grout BWW, Henshaw GG (1978) Freeze preservation of potato shoot-tip cultures. Ann Bot 42:1227–1229
Grout BWW, Henshaw GG (1980) Structural observations on the growth of potato shoot-tip cultures after thawing from liquid nitrogen. Ann Bot 46:243–248
Halmagyi A, Deliu C, Coste A (2005) Plant regrowth from potato shoot tips cryopreserved by a combined vitrification-droplet method. Cryo-Letters 26:313–322
Hao YJ, You CX, Deng XX (2002a) Analysis of ploidy and the patterns of amplified fragment length polymorphism and methylation sensitive amplified polymorphism in strawberry plants recovered from cryopreservation. Cryo-Letters 23:37–46
Hao YJ, You CX, Deng XX (2002b) Effects of cryopreservation on developmental competency, cytological and molecular stability of citrus callus. Cryo-Letters 23:27–35
Harding K (1991) Molecular stability of the ribosomal RNA genes in Solanum tuberosum plants recovered from slow growth and cryopreservation. Euphytica 55:141–146
Harding K (1994) The methylation status of DNA derived from potato plants recovered from slow growth. Plant Cell Tissue Organ 37:31–38
Harding K (2004) Genetic integrity of cryopreserved plant cells: a review. Cryo-Letters 25:3–22
Harding K, Benson EE (1994) A study of growth, flowering, and tuberisation in plants derived from cryopreserved potato shoot-tips: implications for in vitro germplasm collections. Cryo-Letters 15:59–66
Harding K, Benson EE (2000) Analysis of nuclear and chloroplast DNA in plants regenerated from cryopreserved shoot-tips of potato. Cryo-Letters 21:279–288
Harding K, Benson EE (2001) The use of microsatellite analysis in Solanum tuberosum L. in vitro plantlets derived from cryopreserved germplasm. Cryo-Letters 22:199–208
Harding K, Johnston JW, Benson EE (2009) Exploring the physiological basis of cryopreservation success and failure in clonally propagated in vitro crop plant germplasm. Agr Food Sci 18:3–16
Haskins RH, Kartha KK (1980) Freeze preservation of pea meristems: cell survival. Can J Bot 58:833–840
Hawkes JG (1978) Biosystematics of the potato. In: Harris PM (ed) The potato crop. Chapman & Hall, London, pp 15–69
Hawkes JG (1990) The potato: evolution, biodiversity and genetic resources. Belhaven, London
Heller R (1953) Recherches sur la nutrition minérale des tissus gétaux cultivés in vitro. Ann Sc Nat Bot Biol Veg 14:1–22
Helliot B, Panis B, Poumay Y, Swennen R, Lepoivre P, Frison E (2002) Cryopreservation for the elimination of cucumber mosaic and banana streak viruses from banana (Musa spp.). Plant Cell Rep 20:1117–1122
Henshaw GG, Keefe PD, O’Hara JF (1985) Cryopreservation of potato meristems. In: Schäfer-Menuhr A (ed) In vitro techniques: propagation and long term storage. Martinus Nijhoff/Dr. W. Junk Publishers, Brussels, pp 155–160
Hils U, Pieterse L (2009) World catalogue of potato varieties 2009/10. AGRIMEDIA, Clenze
Hirai D, Sakai A (1999) Cryopreservation of in vitro-grown meristems of potato (Solanum tuberosum L.) by encapsulation-vitrification. Potato Res 42:153–160
Hirai D, Sakai A (2000) Cryopreservation of in vitro-grown meristems of potato (Solanum tuberosum L.) by encapsulation-vitrification. In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm. Current research progress and application. Japan International Research Center for Agricultural Sciences, Tsukuba
Höhne GWH, Hemminger WF, Flammersheim H-J (2003) Differential scanning Calorimetry. Springer-Verlag, Berlin
IPK 2007. Annual Report. Available at: http://www.ipk-gatersleben.de/Internet/Infrastruktur/Oeffentlichkeitsarbeit/Forschungsberichte. Accessed 26 October 2010
Ishikawa HA (1996) Ultrastructural features of chilling injury: injured cells and the early events during chilling of suspension-cultured mung bean cells. Am J Bot 83:825–835
Jitsuyama Y, Suzuki T, Harada T, Fujikawa S (2002) Sucrose incubation increases freezing tolerance of Asparagus (Asparagus officinalis L.) embryogenic cell suspensions. Cryo-Letters 23:103–112
Johnston JW, Benson EE, Harding K (2009) Cryopreservation induces temporal DNA methylation epigenetic changes and differential transcriptional activity in Ribes germplasm. Plant Physiol Biochem 47:123–131
Kaczmarczyk A (2008) Physiological, biochemical, histological and ultrastructural aspects of cryopreservation in meristematic tissue of potato shoot tips. Dissertation. Verlag Dr. Köster Berlin, Germany
Kaczmarczyk A, Rutten T, Melzer M, Keller ERJ (2008a) Ultrastructural changes associated with cryopreservation of potato (Solanum tuberosum L.) shoot tips. Cryo-Letters 29:145–156
Kaczmarczyk A, Shvachko N, Lupysheva Y, Hajirezaei MR, Keller ERJ (2008b) Influence of alternating temperature preculture on cryopreservation results for potato shoot tips. Plant Cell Rep 27:1551–1558
Kaczmarczyk A, Grübe M, Keller ERJ (2009) History and development of the potato cryopreservation method and the cryopreserved collection at the IPK Gatersleben, Meeting of the WG2, COST Action 871 ‘Cryopreservation of Crop Species in Europe’, Gatersleben, Germany, 9.–11.09.2009. pp 37
Kaity A, Ashmore SE, Drew RA, Dulloo ME (2008) Assessment of genetic and epigenetic changes following cryopreservation in papaya. Plant Cell Rep 27:1529–1539
Kartha KK, Engelmann F (1994) Cryopreservation and germplasm storage. In: Vasil IK, Thorpe TA (eds) Plant cell and tissue culture. Kluwer Acad. Publ, Dordrecht, pp 165–230
Kartha KK, Leung NL, Mroginski LA (1982) In vitro growth responses and plant regeneration from cryopreserved meristems of Cassava (Manihot esculenta Crantz). Z Pflanzenphysiol 107:133–140
Keller ERJ (2005) Improvement of cryopreservation results in garlic using low temperature preculture and high-quality in vitro plantlets. Cryo-Letters 26:357–366
Keller ERJ, Dreiling M (2003) Potato cryopreservation in Germany - using the droplet method for the establishment of a new large collection. Acta Hortic 623(ISHS):193–200
Keller ERJ, Senula A, Leunufna S, Grübe M (2006) Slow growth storage and cryopreservation–tools to facilitate germplasm maintenance of vegetatively propagated crops in living plant collections. Int J Refrig 29:411–417
Keller ERJ, Senula A, Kaczmarczyk A (2008) Cryopreservation of Herbaceous Dicots. In: Reed BM (ed) Plant cryopreservation: a pratical guide. Springer, New York, pp 281–332
Kim HH, Yoon JW, Park YE, Cho EG, Sohn JK, Kim TS, Engelmann F (2006) Cryopreservation of potato cultivated varieties and wild species: critical factors in droplet vitrification. Cryo-Letters 27:223–234
Kryszczuk A, Keller J, Grübe M, Zimnoch-Guzowska E (2006) Cryopreservation of potato (Solanum tuberosum L.) shoot tips using vitrification and droplet method. Food. Agric Environ 4:196–200
Lehesranta SJ, Davies HV, Shepherd LVT, Nunan N, McNicol JW, Auriola S, Koistinen KM, Suomalainen S, Kokko HI, Karenlampi SO (2005) Comparison of tuber proteomes of potato varieties, landraces, and genetically modified lines. Plant Physiol 138:1690–1699
Lehesranta SJ, Davies HV, Shepherd LVT, Koistinen KM, Massat N, Nunan N, McNicol JW, Karenlampi SO (2006) Proteomic analysis of the potato tuber life cycle. Proteomics 6:6042–6052
Leunufna S, Keller ERJ (2005) Cryopreservation of yams using vitrification modified by including droplet method: effects of cold acclimation and sucrose. Cryo-Letters 26:93–102
Martín MC, González-Benito ME (2006) Sequence comparison in somaclonal variant of cryopreserved Dendranthema grandiflora shoot apices. Cryobiology 53:424
Martin RR, Postman JD (1999) Phytosanitary aspects of plant germplasm conservation. In: Benson EE (ed) Plant conservation biotechnology. Taylor and Francis Ltd., London, pp 63–82
Mattinen L, Nissinen R, Riipi T, Kalkkinen N, Pirhonen M (2007) Host-extract induced changes in the secretome of the plant pathogenic bacterium Pectobacterium atrosepticum. Proteomics 7:3527–3537
Mazur P (1977) The role of intracellular freezing in the death of cells cooled at supraoptimal rates. Cryobiology 14:251–272
Meryman HT, Williams RJ, Douglas MSJ (1977) Freezing injury from solution effects and its prevention by natural artificial cryoprotection. Cryobiology 14:287–302
Messer E (2000) II.B.3.–Potatoes. In: Kiple KF, Ornelas KC (eds) The Cambridge world history of food. Cambridge University Press, Cambridge, UK
Mikula A, Tykarska T, Kuras M (2005) Ultrastructure of Gentiana tibetica proembryogenic cells before and after cooling treatments. Cryo-Letters 26:367–378
Mix-Wagner G (1999) The conservation of potato cultivars. Potato Res 42:427–436
Mix-Wagner G, Schumacher HM, Cross RJ (2003) Recovery of potato apices after several years of storage in liquid nitrogen. Cryo-Letters 24:33–41
Morel G, Muller JF (1964) La Culture in Vitro Du Meristeme Apical De La Pomme De Terre. Compt Rend Acad Sci Paris 258:5250–5252
Morel G, Wetmore RH (1951) Fern callus tissue culture. Am J Bot 38:141–143
Mroginski L, Rey H (2007) Cryopreservation of plant germplasm in Argentina. Adv Hortic Sci 21:270–273
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497
Nguyen TTA, Michaud D, Cloutier C (2007) Proteomic profiling of aphid Macrosiphum euphorbiae responses to host-plant-mediated stress induced by defoliation and water deficit. J Insect Physiol 53:601–611
Niino T, Sakai A (1992) Cryopreservation of alginate-coated in vitro-grown shoot tips of apple, pear, and mulberry. Plant Sci 87:199–206
O’Hara JF, Keefe PD, Henshaw GG (1985) Cryopreservation of potato meristems. In: Schäfer-Menuhr A (ed) In vitro techniques: propagation and long term storage. Martinus Nijhoff/Dr. W. Junk Publishers, Brussels, pp 155–165
Panis B, Swennen R, Engelmann F (2001) Cryopreservation of plant germplasm. Acta Hortic 560:79–86
Panis B, Piette B, Swennen R (2005) Droplet vitrification of apical meristems: a cryopreservation protocol applicable to all Musaceae. Plant Sci 168:45–55
Panta A, Panis B, Ynouye C, Criel B, Swennen R, Roca W (2006) Improvement of potato cryopreservation for the long-term conservation of Andean landraces at the International Potato Center (CIP). Cryobiology 53:401
Panta A, Panis B, Sanchez D, Canepa P, Ynouye C, Geuns J, Swennen R, Roca W, Tay D (2009a) Improved potato cryopreservation based on the identification of biochemical cell compounds linked to response to abiotic stress. 1st International Symposium on Cryopreservation in Horticultural Species, Leuven, Belgium, 5–8 April. pp 16
Panta A, Panis B, Sanchez D, Ynouye C, Geuns J, Swennen R, Tay D, Roca W (2009b) Andean food tuber crops: cryopreservation and links with their response to abiotic stress. Cryo-Letters 30:88–88
Pennycooke JC, Towill LE (2000) Cryopreservation of shoot tips from in vitro plants of sweet potato [Ipomoea batatas (L.) Lam.] by vitrification. Plant Cell Rep 19:733–737
Perazzo G, Panta A, Rodriguez F, Gomez R, Toledo J, Huamán Z, Ghislain M, Golmirzaie AM, Roca. W (1999–2000) Clonal true-to-type verification of potato accessions retrieved from in vitro conservation and cryopreservation. CIP Program Report. pp 175–183
Peredo EL, Arroyo-Garcia R, Reed BM, Revilla MA (2008) Genetic and epigenetic stability of cryopreserved and cold-stored hops (Humulus lupulus L.). Cryobiology 57:234–241
Reed BM, Okut N, D’Achino J, Narver L, DeNoma J (2003) Cold storage and cryopreservation of hops (Humulus L.) shoot cultures through application of standard protocols. Cryo-Letters 24:389–396
Sakai A, Engelmann F (2007) Vitrification, encapsulation-vitrification and droplet-vitrification: a review. Cryo-Letters 28:151–172
Sakai A, Kobayashi S, Oiyama I (1990) Cryopreservation of nucellar cells of navel orange (Citrus sinensis Osb. var. brasiliensis Tanaka) by vitrification. Plant Cell Rep 9:30–33
Sant R, Panis B, Taylor M, Tyagi A (2008) Cryopreservation of shoot-tips by droplet vitrification applicable to all taro (Colocasia esculenta var. esculenta) accessions. Plant Cell Tissue Organ 92:107–111
Santos IRI, Stushnoff C (2003) Desiccation and freezing tolerance of embryonic axes from Citrus sinensis L. osb. pretreated with sucrose. Cryo-Letters 24:281–292
Sarkar D, Naik PS (1998) Cryopreservation of shoot tips of tetraploid potato (Solanum tuberosum L.) clones by vitrification. Ann Bot 82:455–461
Schäfer-Menuhr A (1996) Refinement of cryopreservation techniques for potato. Final report for the period September 1, 1991–August 31, 1996. International Plant Genetic Resources Institute, Rome, Italy. pp 1–41
Schäfer-Menuhr A, Schumacher HM, Mix-Wagner G (1994) Langzeitlagerung alter Kartoffelsorten durch Kryokonservierung der Meristeme in flüssigem Stickstoff. Landbauforsch Völkenrode 44:301–313
Schäfer-Menuhr A, Müller E, Mix-Wagner G (1996a) Cryopreservation: an alternative for the long-term storage of old potato varieties. Potato Res 39:507–513
Schäfer-Menuhr A, Müller E, Mix-Wagner G (1996b) Einsatz der Kryokonservierung als Routinemethode für die Erhaltung alter Kartoffelsorten. Landbauforsch Völkenrode Heft 2:65–75
Schäfer-Menuhr A, Schumacher HM, Mix-Wagner G (1997) Long-term storage of old potato varieties by cryopreservation of shoot-tips in liquid nitrogen. Plant Genet Resour Newsl 111:19–24
Schäfer-Menuhr A, Mix-Wagner G, Kelller ERJ, Schüler K (1998) Liquid nitrogen–a medium to store germplasm. Proceedings International Symposium Breeding Research on Potatoes. 23–26 June, Groß Lüsewitz (Rostock), Germany. pp 157–162
Seibert M, Wetherbee PJ (1977) Increased survival and differentiation of frozen herbaceous plant organ cultures through cold treatment. Plant Physiol 59:1043–1046
Senula A, Keller ERJ, Sanduijav T, Yohannes T (2007) Cryopreservation of cold-acclimated mint (Mentha spp.) shoot tips using a simple vitrification protocol. Cryo-Letters 28:1–12
Spooner DM, Salas A (2006) Structure, biosystematics and genetic resources. In: Gopal J, Khurana SM (eds) Handbook of potato production, improvement and post-harvest management, New York. pp 1–39
Stefanowska M, Kuraś M, Kacperska A (2002) Low temperature-induced modifications in cell ultrastructure and localization of phenolics in winter oilseed rape (Brassica napus L. var. oleifera L.) leaves. Ann Bot 90:637–645
Steponkus PL, Langis R, Fujikawa S (1992) Cryopreservation of plant tissues by vitrification. Adv Low Temp Biol 1:1–16
Suzuki T, Kaneko M, Harada T (1997) Increase in freezing resistance of excised shoot tips of Asparagus officinalis L. by preculture on sugar-rich media. Cryobiology 34:264–275
Tao DL, Li PH (1986) Classification of plant-cell cryoprotectants. J Theor Biol 123:305–310
Tao D, Li PH, Carter JV (1983) Role of cell-wall in freezing tolerance of cultured potato cells and their protoplasts. Physiol Plant 58:527–532
Taylor MJ, Song YC, Brockbank KGM (2004) Vitrification in tissue preservation: new developments. In: Fuller B, Lane N, Benson EE (eds) Life in the frozen state. London, New York, pp 603–644
Towill LE (1981a) Solanum etuberosum—a model for studying the cryobiology of shoot-tips in the tuber-bearing Solanum species. Plant Sci Lett 20:315–324
Towill LE (1981b) Survival at low temperatures of shoot-tips from cultivars of Solanum tuberosum group Tuberosum. Cryo-Letters 2:373–382
Towill LE (1983) Improved survival after cryogenic exposure of shoot tips derived from in vitro plantlet cultures of potato. Cryobiology 20:567–573
Towill LE (1984) Survival at ultra-low temperatures of shoot tips from Solanum tuberosum groups Andigena, Phureja, Stenotomum, Tuberosum, and other tuber-bearing Solanum species. Cryo-Letters 5:319–326
Towill LE, Bonnart R (2003) Cracking in a vitrification solution during cooling or warming does not affect growth of cryopreserved mint shoot tips. Cryo-Letters 24:341–346
Wang QC, Valkonen JPT (2008) Elimination of two viruses which interact synergistically from sweetpotato by shoot tip culture and cryotherapy. J Virol Methods 154:135–145
Wang Q, Valkonen JPT (2009a) Cryotherapy of shoot tips: novel pathogen eradication method. Trends Plant Sci 14:119–122
Wang Q, Valkonen JPT (2009b) Improved recovery of cryotherapy-treated shoot tips following thermotherapy of in vitro-grown stock shoots of raspberry (Rubus idaeus L.). Cryo-Letters 30:171–182
Wang QC, Mawassi M, Li P, Gafny R, Sela I, Tanne E (2003) Elimination of grapevine virus A (GVA) by cryopreservation of in vitro-grown shoot tips of Vitis vinifera L. Plant Sci 165:321–327
Wang Q, Liu Y, Xie Y, You M (2006) Cryotherapy of potato shoot tips for efficient elimination of potato leafroll virus (PLRV) and potato virus Y (PVY). Potato Res 49:119–129
Wang B, Yin Z, Feng C, Shi Xiao, Li Y, Wang Q (2008a) Cryopreservation of potato shoot tips. Fruit, Vegetable and Cereal Sci Biotechnol 2(Special Issue 1):46–53
Wang QC, Cuellar WJ, Rajamaki ML, Hirata Y, Valkonen JPT (2008b) Combined thermotherapy and cryotherapy for efficient virus eradication: relation of virus distribution, subcellular changes, cell survival and viral RNA degradation in shoot tips. Mol Plant Pathol 9:237–250
Wang QC, Panis B, Engelmann F, Lambardi M, Valkonen JPT (2009) Cryotherapy of shoot tips: a technique for pathogen eradication to produce healthy planting materials and prepare healthy plant genetic resources for cryopreservation. Ann Appl Biol 154:351–363
Ward ACW, Benson EE, Blackhall NW, Cooperbland S, Powell W, Power JB, Davey MR (1993) Flow-cytometric assessments of ploidy stability in cryopreserved dihaploid Solanum tuberosum and wild Solanum species. Cryo-Letters 14:145–152
Yoon J-W, Kim H-H, Ko H-C, Hwang H-S, Hong E-S, Cho E-G, Engelmann F (2006) Cryopreservation of cultivated and wild potato varieties by droplet vitrification: effect of subculture of mother-plants and of preculture of shoot tips. Cryo-Letters 27:211–222
Zámečník J, Faltus M, Bilavčík A (2007) Cryoprotocols used for cryopreservation of vegetatively propagated plants in the Czech cryobank. Adv Hortic Sci 21:247–250
Zarghami R, Pirseyedi M, Hasrak S, Sardrood BP (2008) Evaluation of genetic stability in cryopreserved Solanum tuberosum. Afr J Biotechnol 7:2798–2802
Zhao MA, Xhu YZ, Dhital SP, Khu DM, Song YS, Wang MY, Lim HT (2005) An efficient cryopreservation procedure for potato (Solanum tuberosum L.) utilizing the new ice blocking agent, Supercool X1000. Plant Cell Rep 24:477–481
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kaczmarczyk, A., Rokka, VM. & Keller, E.R.J. Potato Shoot Tip Cryopreservation. A Review. Potato Res. 54, 45–79 (2011). https://doi.org/10.1007/s11540-010-9169-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11540-010-9169-7