
Abstract
Background
The impact of dose and simultaneous use of acid-reducing agents (ARAs) on the effectiveness of vemurafenib is unknown.
Objectives
To determine the association between progression of metastatic BRAF V600 mutated melanoma and (1) dose reductions of vemurafenib and (2) simultaneous use of vemurafenib and ARAs.
Patient and Methods
A retrospective cohort study of 112 first-line vemurafenib users for melanoma was conducted (March 2012–March 2016), using electronic patient records and pharmacy dispensing records of a Dutch academic hospital. Cox regression analysis was used to estimate the risk of progression with full-dose (n = 64) versus reduced-dose vemurafenib (n = 48) and with simultaneous use of vemurafenib and ARAs (n = 35) versus vemurafenib alone (n = 77). Analyses were adjusted for age and sex.
Results
In total, disease progression occurred in 55% of treated patients on vemurafenib, with a median progression-free survival of 6.0 (95% confidence interval [CI] 5.0–6.9) months. Compared to patients on vemurafenib alone, there was no increased risk of progression among patients requiring vemurafenib at a reduced dose or among patients receiving simultaneous therapy with vemurafenib and ARAs. In addition, there was no increased risk of progression among patients who used reduced-dose vemurafenib and ARAs versus those receiving full-dose vemurafenib as sole therapy. However, a tendency for progression was observed among patients who used full-dose vemurafenib and ARAs versus full-dose vemurafenib alone (adjusted hazard ratio [HRa] 2.37; 95% CI 0.97–5.76), which became statistically significant in a sensitivity analysis (HRa 4.56; 95% CI 1.51–13.75).
Conclusions
There was no association between the use of vemurafenib in a reduced dose or the simultaneous use of vemurafenib and ARAs and the risk of progression. In addition, there was no association between the simultaneous use of vemurafenib in a reduced dose and ARAs and the risk of progression. However, patients tolerating full-dose vemurafenib simultaneously with ARAs might have an increased risk of progression. This finding requires prospective validation.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Until recently, the prognosis for melanoma patients with distant metastases was poor, with 1-year survival rates ranging from 33 to 62% among patients with stage IV melanoma [1]. Mutations in the BRAF gene can be detected in approximately 40–60% of metastatic melanoma cases and lead to the constitutive activation of downstream MAP kinase/ERK-signaling, resulting in cell proliferation and survival [2]. Vemurafenib is a potent, selective inhibitor of the mutated BRAF V600 protein kinase [2]. In the BRIM-3 trial, which evaluated vemurafenib as first-line treatment of melanoma with BRAF V600 mutations, progression-free survival (PFS) was significantly longer for patients receiving vemurafenib than for those on dacarbazine, with a median PFS of 6.9 versus 1.6 months, respectively (hazard ratio [HR] 0.38; 95% confidence interval [CI] 0.32–0.46; p < 0.001) [2, 3]. Moreover, this improvement was also reflected in a significantly longer median overall survival (OS) of 13.6 months for vemurafenib as compared to 9.7 months for dacarbazine (HR 0.70; 95% CI 0.57–0.87; p = 0.0008) [3]. This trial resulted in the approval of vemurafenib as first-line treatment of metastatic BRAF V600 mutated melanoma by the U.S. Food and Drug Administration in August 2011, followed by the approval by the European Medicines Agency in December 2011.
Currently, vemurafenib is not dosed on an individual basis, and no dose increments are allowed. The standard starting dose is 960 mg orally twice daily, which may be reduced in case of toxicity or frailty. After oral intake, the bioavailability of vemurafenib is assumed to be modest [4]. Vemurafenib shows dose-proportional pharmacokinetics from a dose of 240 mg twice daily to 960 mg twice daily [5,6,7]. Due to the large intra- and interpatient pharmacokinetic variability of vemurafenib, the recommended fixed starting dose will lead to substantial differences in vemurafenib exposure and may thereby result in either under- or overexposure [7,8,9]. Both are likely to affect treatment outcome, since an exposure–response relationship has been shown for vemurafenib [5, 9,10,11]. Patients tolerating full-dose vemurafenib may be the subgroup underexposed to vemurafenib, while patients experiencing toxicities due1 to vemurafenib may reflect a subgroup of patients who are overexposed, and thus in need of dose reduction.
Vemurafenib absorption can be decreased with simultaneous use of acid-reducing agents (ARAs), since the solubility of vemurafenib drops when gastric pH is > 2.8 [5, 12]. Data from two American healthcare databases showed that the prevalence of ARA use ranged from 14 to 29% among melanoma patients [12]. Simultaneous use of proton pump inhibitors (PPIs) was shown to reduce the area under the concentration–time curve of other tyrosine kinase inhibitors by 46 to 61% [13,14,15]. Patients using vemurafenib simultaneously with ARAs may therefore be at risk of underexposure to vemurafenib due to the potential interaction between ARAs and vemurafenib [12, 16].
It is currently unknown whether the use of reduced-dose vemurafenib positively affects treatment outcomes of vemurafenib and whether the use of ARAs negatively affects treatment outcomes of vemurafenib. The objectives of this study were, therefore, to determine the association between the progression of metastatic BRAF V600 mutated melanoma and (1) dose reductions of vemurafenib and (2) simultaneous use of ARAs.
2 Materials and Methods
2.1 Data Source
The treatment of patients with metastatic melanoma in the Netherlands is centralized in 14 designated medical centers. Requirements of the national reimbursement system are such that vemurafenib prescriptions can only be dispensed at the pharmacy that is affiliated to the respective melanoma medical center, which ensures a perfect capture of vemurafenib exposure. Data for this study were obtained from both the pharmacy dispensing data and the electronic patient records of an academic hospital in the Netherlands, which were linked at an individual patient level. The pharmacy dispensing data included information on (co-)medication, such as ARAs, cobimetinib, potent inductors or inhibitors of cytochrome P450 enzyme 3A4 (CYP3A4) and P-glycoprotein (P-gp), the prescribed dose of vemurafenib, and the starting date of vemurafenib treatment. The electronic patient records contained information on the date and reason for vemurafenib discontinuation (disease progression, toxicity, hospital admission, death, switch to different lines of treatment, and/or surgical interventions). All data entries were checked by independent researchers.
2.2 Study Population
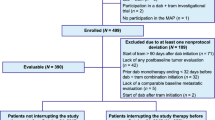
With the approval of the Research Ethics Board (Arnhem/Nijmegen), we conducted a retrospective cohort study and reviewed pharmacy dispensing data to identify patients who had started vemurafenib as first-line treatment for metastatic BRAF V600 mutated melanoma between 17 March 2012 and 17 March 2016. The first vemurafenib dispensing by the pharmacy was defined as the start of follow-up (index date). A total of 115 patients were selected based on the pharmacy dispensing data, and we included all patients aged 18+ years at the start of follow-up who had least one vemurafenib prescription dispensed. Two patients were excluded because their melanoma was not BRAF V600 mutated after revision by our molecular biology laboratory. One patient was excluded because the electronic patient record was lacking information on the safety and effectiveness of vemurafenib (Fig. 1). No other exclusion criteria were defined in order to reflect a ‘real-life’ retrospective cohort.

Flow chart of the study population according to entry criteria
2.3 Exposure
The follow-up was divided into 7-day intervals. Patients were followed up to death, censoring, or disease progression (the outcome of interest) as recorded in the electronic patient files, whichever came first. Reasons for censoring included switching to different lines of therapy before disease progression occurred, toxicity, surgical interventions, loss to follow-up, and the end of the study period.
At the start of each interval, the most recently dispensed dose of vemurafenib was determined using the pharmacy dispensing data. These data were then compared to the dispensed dose of the penultimate vemurafenib dispensed to assess whether a dose change had taken place. person-time was divided into three categories (‘before dose reduction;’ ‘after dose reduction;’ ‘no dose reduction’) since it was hypothesized that patients experiencing toxicities while using full-dose vemurafenib could be overexposed and therefore in need of a dose reduction. When a dose reduction did occur, time was separated into two different exposure categories: ‘before dose reduction’ and ‘after dose reduction’. Patients using full-dose vemurafenib during their entire treatment were classified as ‘no dose reduction’ (960 mg twice daily).
Furthermore, prior to the start of each interval, exposure to ARAs (PPIs, H2 receptor antagonists, or antacids) was determined using the pharmacy dispensing data. ARAs were identified by the following Anatomical Therapeutic Chemical (ATC) codes: A02BC, A02BA, and A02A [17]. A current ARA user was defined as someone who had his most recent ARA dispensed within the 30 days prior to the start of an interval, while past ARA users had received their most recent ARA dispensed > 30 days previously.
2.4 Outcomes
The outcome of interest was PFS in patients with metastatic BRAF V600 mutated melanoma on first-line vemurafenib. Progression was either assessed radiologically or clinically by the treating physician. PFS was defined as the time from dispensing of first vemurafenib prescription until the occurrence of new lesions or the progression of existing lesions.
2.5 Potential Confounders
Age and sex were considered to be potential confounders and were incorporated as covariates into the analyses. Sex was determined at baseline, and age was determined time dependently.
2.6 Co-Medication
The influence of concomitant administration of potent inductors or inhibitors of CYP3A4 and P-gp was assessed because vemurafenib is mainly metabolized by CYP3A4 and is a substrate of P-gp.
2.7 Statistical Analysis
Regression analysis with the Cox proportional hazards model (PHREG procedure, SAS version 9.4; SAS Institute, Cary, NC, USA) was used to estimate disease progression among (1) full-dose vemurafenib users (n = 64) versus reduced-dose vemurafenib users (n = 48); (2) simultaneous ARA use (n = 35) versus no ARA use (n = 77); (3) the combination of both exposure groups. Multivariate analysis was used to adjust for age and sex. PFS was estimated using the Kaplan–Meier method.
2.8 Sensitivity Analysis
A sensitivity analysis was performed using only pharmacy dispensing data to determine the discontinuation date of vemurafenib exposure. This discontinuation date was added to the previously defined censoring events. Discontinuation of vemurafenib was defined as the estimated end date of the dispensing based on the total number of dispensed tablets and the number of prescribed tablets per day. In the case of no new dispensing during the 28 days after the estimated end data, patients were censored at the estimated end date of the dispensing. This maximum tolerated gap period of vemurafenib was similar to the maximum discontinuation period in clinical trials. In another sensitivity analysis, the time-window of the time since the most recent ARA prescription (i.e., current use) was varied to 15 or 60 days, respectively, prior to the start of an interval. ARAs are likely to be dispensed for either a short period of time or for long period of time, but they may also be used by patients on an ‘as needed basis’.
3 Results
3.1 Study Population at Baseline
Among the 112 patients fulfilling the inclusion criteria (Fig. 1), 45 were female (40.2%) and the median age at index date was 60 (range 26–81) years. In total, 64 patients were using full-dose vemurafenib during the entire study period, and 35 patients were using ARAs. The ARAs used by the patients in this study were the PPIs esomeprazole, omeprazole, and pantoprazole, the H2-receptor antagonist ranitidine, and the antacids magnesium hydroxide and ordinary salt combinations. Further, four patients were treated with vemurafenib in combination with cobimetinib. Of those four patients, two were using full-dose vemurafenib during the entire study period and two were using ARAs. None of the patients treated with vemurafenib concomitantly used strong CYP3A4 or P-gp inducers or inhibitors.
3.2 Study Population at Outcome
The patient outcome characteristics are shown in Table 1. In total, 62 patients (55.4%) showed disease progression on first-line vemurafenib. Censoring occurred in 50 patients (44.6%), mainly due to switching to immunotherapy before disease progression occurred (20.5%), toxicity (12.5%), surgical resection (3.6%), or loss to follow-up (2.7%). Six patients (5.4%) were still on vemurafenib at the end of the follow-up period. The median PFS was 6.0 (95% CI 5.0–6.9) months.
3.3 Dose Reductions of Vemurafenib and/or Simultaneous Use of ARAs and Risk of Disease Progression
Patients using vemurafenib in a reduced dose had no increased risk of disease progression as compared to full-dose vemurafenib users (adjusted HR [HRa] 1.12; 95% CI 0.64–1.61) (Table 2). Vemurafenib users who had used ARAs within the past 30 days had no increased risk of disease progression (HRa 1.23; 95% CI 0.53–2.85) as compared to vemurafenib users not using ARAs. There was no increased risk of progression among patients who used reduced-dose vemurafenib and ARAs versus those on full-dose vemurafenib alone (HRa 1.00; 95% CI 0.45–2.20). However, a tendency for disease progression was observed among patients who had used full-dose vemurafenib and ARAs at the same time versus those who used full-dose vemurafenib alone (HRa 2.37; 95% CI 0.97–5.76).
3.4 Sensitivity Analysis
In a sensitivity analysis in which we used pharmacy dispensing data to determine the discontinuation date of vemurafenib exposure, only 41 patients showed disease progression (Table 3). In this sensitivity analysis, there was no association between full-dose vemurafenib use versus reduced-dose vemurafenib use and disease progression. In addition, there was no increased risk of progression among patients who used reduced-dose vemurafenib and ARAs versus those on full-dose vemurafenib alone (HRa 0.99; 95% CI 0.35–2.75). However, a statistically significant 4.6-fold increased risk of disease progression was found among patients who had used full-dose vemurafenib and ARAs at the same time versus those who had used full-dose vemurafenib alone (HRa 4.56; 95% CI 1.51–13.75). In a second sensitivity analysis, current ARA use was determined by varying the time since the most recent ARA prescription to 15 or 60 days, respectively (Table 4). For both time-windows, we found no associations for current and past ARA use with disease progression as compared to no ARA use.
4 Discussion
In the present study we found no increased risk of the progression of metastatic BRAF V600 mutated melanoma after dose reductions of vemurafenib, or with simultaneous use of vemurafenib and ARAs, versus vemurafenib alone. In addition, there was no increased risk of progression among patients who used reduced-dose vemurafenib and ARAs versus those who used full-dose vemurafenib alone. However, a tendency for disease progression was observed among patients who used full-dose vemurafenib and ARAs at the same time versus those on full-dose vemurafenib alone when determining exposure to vemurafenib using electronic patient records. The risk for disease progression became significantly elevated to almost fivefold in a sensitivity analysis in which a discontinuation date of vemurafenib (based on pharmacy dispensing records) was added as a censoring event.
To the best of our knowledge, this is the first study to investigate the influence of vemurafenib dose reductions, simultaneous use of vemurafenib and ARA, and full-dose vemurafenib use simultaneously with ARAs on the risk of disease progression in melanoma patients [12, 16]. It is presumed that co-treatment with ARAs will reduce vemurafenib plasma concentrations. Vemurafenib has only limited solubility at pH 1 (e.g., the physiologic pH of the stomach), but has even less solubility at pH > 2.8 (i.e., the pKa value of vemurafenib) [5]. Since the use of ARAs may increase the gastric pH to ≥ 4, it is possible that an ARA–vemurafenib interaction occurs that affects vemurafenib uptake [5, 12].
Several previous studies have shown results in line with our findings. Our median PFS was 6.0 (95% CI 5.1–6.9) months, which is comparable to the PFS reported in the study of McArthur et al. (6.9 months) and Kramkimel et al. (5.0 months) [3, 11]. Furthermore, we found dose reductions in 43% of the patients using vemurafenib, which is comparable to 38–50% of the patients requiring a dose reduction in other studies [2, 9, 18]. In addition, we identified a complete discontinuation of vemurafenib due to toxicity in 13% of our patients, which is comparable to the 12% identified in the study by Kramkimel et al. [11]. These results suggest that our population of ‘real-life’ melanoma patients was comparable to earlier studied melanoma patient populations.
Other studies have reported a relationship between vemurafenib plasma concentrations and treatment outcome (i.e., toxicity and effectiveness), identifying a minimal target steady state concentration of 42 μg/mL [9, 11]. High inter- and intrapatient variability at steady-state pharmacokinetics was observed, also when focusing only on the patients taking full-dose vemurafenib [9, 18]. In addition, patients treated with a reduced dose of vemurafenib due to adverse events had nearly similar plasma concentrations as patients who tolerated full-dose vemurafenib [9]. These observations indicate that patients requiring dose reductions might be overexposed when taking full-dose vemurafenib [9]. The effect of simultaneous ARA use on treatment outcome will likely be most pronounced in patients with plasma concentrations close to the target concentration of 42 μg/mL during the entire treatment period, i.e., the group of patients tolerating full-dose vemurafenib [9]. Therefore, patients tolerating full-dose vemurafenib while simultaneously using ARAs may be of particular risk of early progression due to vemurafenib plasma concentrations below the target level, a possibility which is supported by our results. Modifying the vemurafenib dose based on plasma concentrations could be an elegant and patient friendly way to optimize vemurafenib therapy for individual patients [18]. Unfortunately, data on vemurafenib plasma concentrations were not collected for the patients included in our cohort; such data could be the starting point for future studies.
Information regarding the exposure–response relationship of vemurafenib used in combination with cobimetinib is lacking. The target might be equal or below the earlier identified target for monotherapy with vemurafenib of 42 μg/mL, since efficacy was demonstrated with the same dose as used in monotherapy and no interaction between both drugs was identified [19]. It is therefore yet unknown if the increased risk of progression is still present in a population of metastatic BRAF V600 patients who are treated with vemurafenib and cobimetinib and simultaneously using ARAs.
It is important to note the shortcomings of this retrospective database study. True causality cannot be provided with this study. Further, the simultaneous use of vemurafenib with ARAs was difficult to assess. ARAs could be obtained at multiple pharmacies and are available over the counter (without dispensing), making an accurate estimation of co-use challenging. The electronic network linkage of pharmacies allowed us to use drug dispensing data from other healthcare providers; however, completeness of the pharmacy dispensing data could not be guaranteed. This potential misclassification of exposure may have resulted in an underestimation of the effect. Additionally, it is likely that our observed associations are not without residual confounding. Factors that could not be retrieved from the medical records of the patients, and thus were not adjusted for, were food intake (e.g., use of vemurafenib with or without food), the use of food supplements (e.g., grapefruit juice), and herbal supplements (e.g., St. John’s wort) [4, 16, 20]. Lastly, confounding by disease severity might be present. Patients with brain metastases are often treated with the CYP3A4 inductor dexamethasone, which might result in a lower exposure of vemurafenib. We recommend future prospective studies to correct for factors such as food and/or herbal intake, disease severity, and simultaneous use of dexamethasone.
Notwithstanding, our study has a number of important strengths. First of all, we were able to include a relatively large group of BRAF V600 mutated metastatic melanoma patients using vemurafenib as first-line treatment. Moreover, we applied no other exclusion criteria, thereby providing a representative sample of ‘real-life’ melanoma patients who can therefore be extrapolated to the general population of melanoma patients. Second, we were able to obtain information from both the pharmacy dispensing data and the electronic patient record. The data collected is assumed to be reliable, since all data entries were checked independently by researchers. Moreover, due to different data sources we were able to investigate the presence of potential misclassification of vemurafenib exposure. In the sensitivity analyses using only pharmacy dispensing data to determine a discontinuation date of vemurafenib, we found a significant risk of progression in patients using full-dose vemurafenib and ARAs at the same time versus those using full-dose vemurafenib alone. The presence of non-differential misclassification of exposure results in an effect towards the null [21], which might explain the difference in the results between using the pharmacy data and the electronic patient records data (HRa 4.56; 95% CI 1.51–13.75 and HRa 2.37; 95% CI 0.97–5.76, respectively). Third, in order to control for confounding by indication, we only included patients using first-line vemurafenib. It has been found that pretreated patients have an impaired PFS, translating into a trend towards an unfavorable OS [22].
In conclusion, there was no association between the use of vemurafenib in a reduced dose or the simultaneous use of vemurafenib and ARAs and the risk of progression. In addition, there was no increased risk of progression among patients who used reduced-dose vemurafenib and ARAs. However, patients using full-dose vemurafenib simultaneously with ARAs might have an increased risk of progression. This finding requires prospective validation. Moreover, future studies are necessary to investigate the added value of measuring vemurafenib concentrations in order to optimize therapy.
References
Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–206.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. BRIM-3 study group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–32.
Zelboraf. Summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002409/WC500124317.pdf. 2012. Accessed 9 Jan 2017.
CDER (Center for Drug Evaluation and Research) (2011) Application number: 202429Orig1s000. Clinical pharmacology and biopharmaceutics review(s). Center for Drug Evaluation and Research, Food and Drug Administration, U.S. Department of Health and Human Services, Silver Spring, MD. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202429Orig1s000ClinPharmR.pdf Accessed January 9, 2017.
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19.
Grippo JF, Zhang W, Heinzmann D, et al. A phase I, randomized, open-label study of the multiple-dose pharmacokinetics of vemurafenib in patients with BRAF V600E mutation-positive metastatic melanoma. Cancer Chemother Pharmacol. 2014;73(1):103–11.
Alvarez JC, Funck-Brentano E, Abe E, Etting I, Saiag P, Knapp A. A LC/MS/MS micro-method for human plasma quantification of vemurafenib. Application to treated melanoma patients. J Pharm Biomed Anal. 2014;97:29–32.
Funck-Brentano E, Alvarez JC, Longvert C, Abe E, Beauchet A, Funck-Brentano C, et al. Plasma vemurafenib concentrations in advanced BRAFV600mut melanoma patients: impact on tumour response and tolerance. Ann Oncol. 2015;26(7):1470–5.
Goldwirt L, Chami I, Feugeas JP, Pages C, Brunet-Possenti F, Allayous C, et al. Reply to ‘plasma vemurafenib concentrations in advanced BRAFV600mut melanoma patients: impact on tumour response and tolerance’ by Funck-Brentano et al. Ann Oncol. 2016;27(2):363–4.
Kramkimel N, Thomas-Schoemann A, Sakji L, Golmard J, Noe G, Regnier-Rosencher E, et al. Vemurafenib pharmacokinetics and its correlation with efficacy and safety in outpatients with advanced BRAF-mutated melanoma. Target Oncol. 2016;11(1):59–69.
Smelick GS, Heffron TP, Chu L, Dean B, West DA, Duvall SL, et al. Prevalence of acid-reducing agents (ARA) in cancer populations and ARA drug-drug interaction potential for molecular targeted agents in clinical development. Mol Pharm. 2013;10(11):4055–62.
Sprycel. Summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000709/WC500056998.pdf 2009. Accessed 17 Jan 2017.
Iressa. Summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001016/WC500036358.pdf 2009. Accessed 17 Jan 2017.
Tarceva. Summary of product characteristics. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000618/human_med_001077.jsp&mid=WC0b01ac058001d124 2009. Accessed 17 Jan 2017.
Willemsen AE, Lubbermans FJ, Tol J, Gerritsen WR, van Herpen CM, van Erp NP. Effect of food and acid-reducing agents on the absorption of oral targeted therapies in solid tumors. Drug Discov Today. 2016;21(6):962–76.
World Health Organization Collaborating Centre for Drug Statistics Methodology. https://www.whocc.no/atc_ddd_index/ Accessed 9 Jan 2017.
Nijenhuis CM, Huitema AD, Blank C, Haanen JB, van Thienen JV, Rosing H, et al. Clinical pharmacokinetics of Vemurafenib in BRAF-mutated melanoma patients. J Clin Pharmacol. 2017;57(1):125–8.
Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76.
Ribas A, Zhang W, Chang I, Shirai K, Ernstoff MS, Daud A, et al. The effects of a high-fat meal on single-dose vemurafenib pharmacokinetics. J Clin Pharmacol. 2014;54(4):368–74.
Rothman KJ, Greenland S, Lash TL. Modern Epidemiology. 3rd ed. Philadelphia: Lippincott, Williams & Wilkins; 2008.
Ugurel S, Loquai C, Kähler K, Hassel J, Berking C, Zimmer L, et al. A multicenter DeCOG study on predictors of vemurafenib therapy outcome in melanoma: pretreatment impacts survival. Ann Oncol. 2015;26(3):573–82.
Acknowledgements
We thank Guillemette E. Benoit, Emilie M. Gieling, and Floor J.E. Lubberman for checking the data recorded in the database.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript. This article was published open access under a Springer Compact agreement with Dutch universities and Academy institutes.
Conflict of Interest
LK, JD, BvV, SC, SS, and FdV declare that they have no conflicts of interest that might be relevant to the contents of this manuscript. RK received consultancy fees from MSD, BMS, and Novartis and research funding from BMS and Roche. AC received research funding from BMS, Merck, Viiv, and Janssen. WG received speaker fees from Bayer and MSD, consultancy fees from Bristol-Myers Squibb, Astellas, Janssen-Cilag, Bayer, Sanofi, Amgen, Morphosys, CureVac, and Aglaia Biomedical Ventures, and research funding from Bayer, Astellas, and Janssen-Cilag. DB received research funding from BMS, Viiv, and Janssen. NvE received research funding from Novartis, Janssen-Cilag BV, Astellas, Gilead, Boehringer-Ingelheim, Pfizer, AstraZeneca, and Sanofi. All fees and funding were received outside the context of the submitted work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Knapen, L.M., Koornstra, R.H.T., Driessen, J.H.M. et al. The Impact of Dose and Simultaneous Use of Acid-Reducing Agents on the Effectiveness of Vemurafenib in Metastatic BRAF V600 Mutated Melanoma: a Retrospective Cohort Study. Targ Oncol 13, 363–370 (2018). https://doi.org/10.1007/s11523-018-0564-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-018-0564-3