
Abstract
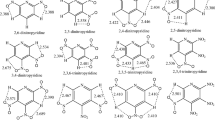
Density functional theory (DFT) method has been employed to study the effect of nitroamino group as a substituent in cyclopentane and cyclohexane, which usually construct the polycyclic or caged nitramines. Molecular structures were investigated at the B3LYP/6-31G** level, and isodesmic reactions were designed for calculating the group interactions. The results show that the group interactions accord with the group additivity, increasing with the increasing number of nitroamino groups. The distance between substituents influences the interactions. Detonation performances were evaluated by the Kamlet-Jacobs equations based on the predicted densities and heats of formation, while thermal stability and pyrolysis mechanism were studied by the computations of bond dissociation energy (BDE). It is found that the contributions of nitroamino groups to the detonation heat, detonation velocity, detonation pressure, and stability all deviate from the group additivity. Only 3a, 3b, and 9a–9c may be novel potential candidates of high energy density materials (HEDMs) according to the quantitative criteria of HEDM (ρ ≈ 1.9 g/cm3, D ≈ 9.0 km/s, P ≈ 40.0 GPa). Stability decreases with the increasing number of N-NO2 groups, and homolysis of N-NO2 bond is the initial step in the thermolysis of the title compounds. Coupled with the demand of thermal stability (BDE > 20 kcal/mol), only 1,2,4-trinitrotriazacyclohexane and 1,2,4,5-tetranitrotetraazacyclohexane are suggested as feasible energetic materials. These results may provide basic information for the molecular design of HEDMs.
Similar content being viewed by others

References
Choi C S, Boutin H P. A Study of the crystal structure of β-cyclotetramethylenetetranitramine by neutron diffraction. Acta Crystal B, 1970, 26: 1235–1240
Choi C S, Prince E. The crystal structure of cyclotrimethylene-trinitramine. Acta Crystal B, 1972, 28: 2857–2862
Karpowicz R J, Bill T B. Comparison of the molecular structure of hexahydro-1,3,5-trinitro-s-triazine in the vapor, solution, and solid phases. J Phys Chem, 1984, 88, 348–352
Kohno Y, Ueda K, Imamura A. Molecular dynamics simulations of initial decomposition processes on the unique N-N bond in nitramines in the crystalline state. J Phys Chem, 1996, 100: 4701–4712
Sorescu D C, Rice B M, Thompson D L. Intermolecular packing for the hexahydro-1,3,5-trinitro-1,3,5-s-triazine crystal (RDX): A crystal packing, Monte Carlo and molecular dynamics study. J Phys Chem B, 1997, 101: 798–808
Thomas D S. Monte Carlo calculations of the hydrostatic compression of hexahydro-1,3,5-trinitro-1,3,5-triazine and β-octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine. J Appl Phys, 1998, 83: 4142–4145
Sorescu D C, Rice B M, Thompson D L. Theoretical studies of the hydrostatic compression of RDX, HMX, HNIW, and PETN crystals. J Phys Chem B, 1999, 103: 6783–6790
Dmitry B, Grant D S, Thomas D S. Temperature-dependent shear viscosity coefficient of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX): A molecular dynamics simulation study. J Chem Phys, 2000, 112: 7203–7208
James P L, Thomas D S, Richard B E, Gregory A V. Electronic structure calculation of the structures and energies of the three pure polymorphic forms of crystalline HMX. J Phys Chem B, 2000, 104: 1009–1013
Xiao J J, Zhang J, Yang D, Xiao H M. DFT Comparative studies on the structures and properties of heterocyclic nitramines. Acta Chim Sin, 2002, 60(12): 2110–2114
Ji G F, Xiao H M, Dong H S. High level calculations on structure and properties of crystalline β-HMX. Acta Chim Sin, 2002, 60(2): 194–199
Manaa M R, Fried L E, Melius C F, Elstner M, Frauenheim T. Decomposition of HMX at extreme conditions: A molecular dynamics simulation. J Phys Chem A, 2002, 106: 9024–9029
Sewell T D, Menikoff R, Bedrov D, Smith G S. A molecular dynamics simulation study of elastic properties of HMX. J Chem Phys, 2003, 119: 7417–7426
Xiao J J, Fang G Y, Ji G F, Xiao H M. Simulation investigation in the binding energy and mechanical properties of HMX-based polymer-bonded explosives. Chin Sci Bull, 2005, 50: 21–26
Bell J A, Dunstan I. Chemistry of nitramines. Part III. Cyclic nitramines derived from trimethylenedinitramine. J Chem Soc C, 1966, 870–872
Gilbert P S, Jack A. Research towards novel energetic materials. J Energ Mater, 1986, 45: 5–28
Archibald T G, Gilardi R, Baum K, George C. Synthesis and X-ray crystal structure of 1,3,3-trinitroazetidine. J Org Chem, 1990, 55: 2920–2924
Jalovy Z, Zeman S, Sucesks M. 1,3,3-trinitroazetidine (TNAZ) Part I: Synthesis and properties. J Energ Mater, 2001, 19: 219–239
Xiao J J, Gong X D, Xiao H M. The DFT predictions of the structure and property of cyclopentamethylenepentanitramine. Chin J Phys Chem, 2002, 15(6): 433–437
Fried L E, Manaa M R, Pagoria P F, Simpson R L. Design and synthesis of energetic materials. Annu Rev Mater Res, 2001, 31: 291–321
Xiao H M, Xu X J, Qiu L. Theoretical Design of High Energy Density Materials. Beijing: Science Press, 2008
Eck G, Piteau M. Preparation of 2,4,6,8-tetranitro-2,4,6,8-tetrazabicyclo[3.3.0]octane. Brit. UK Pat. Appl. GB 2303849 A1, 5 Mar 1997
Willer R L. Trans-1,4,5,8-tetranitro-1,4,5,8-tetraazadecalin. US4443602, Apr.17, 1984
Nielsen A T, Chafin A P, Christian S L, Moore D W, Nadler M P, Nissan R A, Vanderah D J, Gilardi R D, George C F, Flippen-Anderson J L. Synthesis of polyazapolycyclic caged polynitramines. Tetrahedron. 1998, 54: 11793–11812
Koppes W M, Chaykovsky M, Adolph H G, Gilardi R, George C. Synthesis and structure of some peri-substituted 2,4,6,8-tetraazabicyclo[3.3.0]octanes. J Org Chem, 1987, 52: 1113–1119
Brill T B, Oyumi Y. Thermal decomposition of energetic materials. 18. Relationship of molecular composition to nitrous acid formation: bicyclo and spiro tetranitramines. J Phys Chem, 1986, 90(26): 6848–6853
Nielsen A T, Nissan R A, Chafin A P, Gilardi R D, George C F. Polyazapolycyclics by condensation of aldehydes with amines. 3. Formation of 2,4,6,8-tetrabenzyl-2,4,6,8-tetraazabicyclo[3.3.0] octanes from formaldehyde, glyoxal, and benzylamines. J Org Chem, 1992, 57: 6756–6759
Prabhakaran K V, Bhide N M, Kurian E M. Spectroscopic and thermal studies on 1,4,5,8-tetranitrotetraazadecalin (TNAD). Thermochim Acta, 1995, 249: 249–258
Gilardi R, Flippen-Anderson J L, Evans R. cis-2,4,6,8-Tetranitro-1H, 5H-2,4,6,8-tetraazabicyclo[3.3.0]octane, the energetic compound ‘bicyclo-HMX’. Acta Crystal E, 2002, 58(9): 972–974
Liu M H, Chen C, Hong Y S. Theoretical study on the detonation properties of energetic TNAD molecular derivatives. J Mol Struct (THEOCHEM), 2004, 710: 207–214
Qiu L, Xiao H M, Ju X H, Gong X D. Theoretical study on the structures and properties of cyclic nitramines: Tetranitrotetraazadecalin (TNAD) and its isomers. Int J Quant Chem, 2005, 105: 48–56
Qiu L, Xiao H M, Zhu W H, Xiao J J, Zhu W. Ab initio and molecular dynamics studies of crystalline TNAD (trans-1,4,5,8-tetranitro-1,4,5,8-tetraazadecalin). J Phys Chem B, 2006, 110, 10651–10661
Xu X J, Xiao J J, Zhu W, Xiao H M, Huang H, Li J S. Molecular dynamics simulations for pure ɛ-CL-20 and ɛ-CL-20-based PBXs. J Phys Chem B, 2006, 110: 7203–7207
Qiu L, Zhu W H, Xiao J J, Zhu W, Xiao H M, Huang H, Li J S. Molecular dynamics simulations of TNAD (trans-1,4,5,8-tetranitro-1, 4,5,8-tetraazadecalin)-based PBXs. J Phys Chem B, 2007, 111: 1559–1566
Xu X J, Zhu W H, Xiao H M. DFT Studies on the four polymorphs of crystalline CL-20 and the influences of hydrostatic pressure on E-CL-20 crystal. J Phys Chem B, 2007, 111: 2090–2097
Xu X J, Xiao J J, Huang H, Li J S, Xiao H M. Molecular dynamics simulations on the structures and properties of ɛ-CL-20-based PBXs—Primary theoretical studies on HEDM formulation design. Sci Chin Ser B-Chem, 2007, 37(6): 556–563
Qiu L, Zhu W H, Xiao J J, Xiao H M. Theoretical studies of solid bicyclo-HMX: Effects of hydrostatic pressure and temperature. J Phys Chem B, 2008, 112: 3882–3893
Hrovat D A, Borden W T, Eaton P E, Kahr B. A computational study of the interactions among the nitro groups in octanitrocubane. J Am Chem Soc, 2001, 123: 1289–1293
Kamlet M J, Jacobs S J. Chemistry of detonations. I. Simple method for calculating detonation properties of C-H-N-O explosives. J Chem Phys, 1968, 48: 23–35
Blanksby S J, Ellison G B. Bond dissociation energies of organic molecules. Acc Chem Res, 2003, 36: 255–263
Chung G, Schmidt M W, Gordon M S. An ab initio study of potential energy surfaces for N8 isomers. J Phys Chem A, 2000, 104: 5647–5650
Becke A D. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys, 1993, 98: 5648–5652
Lee C, Yang W, Parr R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B, 1988, 37: 785–789
Hariharan P C, Pople J A. Self-consistent-field molecular orbital methods. XII. Further extension of Gaussian-type basis sets. Theor Chim Acta, 1973, 28: 213–222
Qiu L, Xiao H M, Gong X D, Ju X H, Zhu W H. Crystal density predictions for nitramines based on quantum chemistry. J Hazard Mater, 2007, 141: 280–288
Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Montgomery J A, Jr., Vreven T, Kudin K N, Burant J C, Millam J M, Iyengar S S, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson G A, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox J E, Hratchian H P, Cross J B, Adamo C, Jaramillo J, Gomperts R, Stratmann R E, Yazyev O, Austin A J, Cammi R, Pomelli C, Ochterski J W, Ayala P Y, Morokuma K, Voth G A, Salvador P, Dannenberg J J, Zakrzewski V G, Dapprich S, Daniels A D, Strain M C, Farkas O, Malick D K, Rabuck A D, Raghavachari K, Foresman J B, Ortiz J V, Cui Q, Baboul A G, Clifford S, Cioslowski J, Stefanov B B, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin R L, Fox D J, Keith T, Al-Laham M A, Peng C Y, Nanayakkara A, Challacombe M, Gill P M W, Johnson B, Chen W, Wong M W, Gonzalez C, Pople J A. Gaussian 03, Revision C.02. Wallingford CT: Gaussian, Inc., 2004
Oxley J C, Kooh A B, Szekeres R, Zheng W. Mechanisms of nitramine thermolysis. J Phys Chem, 1994, 98: 7004–7008
Mulliken R S. Electronic population analysis on LCAO-MO molecular wave functions. I J Chem Phys, 1955, 23: 1833–1840
Xiao H M. Molecular Orbital Theory for Nitro Compounds. Beijing: National Defence industry Press, 1993
Dong H S, Zhou F F. High Energy Explosives and Correlative Physical Properties. Beijing: Science Press, 1989
Roth J. Encyclopedia of explosives and related items; Large Caliber Weapon System Laboratory, Armament Research and Development Command, U.S. Army, Dover, NJ, 1978, 8: 57
Kamlet M J, Adolph H G. The relationship of impact sensitivity with structure of organic high explosives. II. Polynitroaromatic explosives. Prop Explos Pyrotech, 1979, 4(2): 30–34
Politzer P, Lane P. Comparison of density functional calculations of C-NO2, N-NO2 and C-NF2 dissociation energies. J Mol Struct (Theochem), 1996, 388: 51–55
Harris N J, Lammertsma K. Ab initio density functional computations of conformations and bond dissociation energies for hexahydro-1,3,5-trinitro-1,3,5-triazine. J Am Chem Soc, 1997, 119: 6583–6589
Rice B M, Sahu S, Owens F J. Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules. J Mol Struct (Theochem), 2002, 583: 69–72
Owens F J. Molecular orbital calculation of decomposition pathways of nitrocubanes and nitroazacubanes. J Mol Struct (Theochem), 1999, 460: 137–140
Curtiss L A, Raghavachari K, Trucks G W, Pople J A. Gaussian-2 theory for molecular energies of first-and second-row compounds. J Chem Phys, 1991, 94: 7221–7230
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by the National Natural Science Foundation of China (Grant Nos. 10576030 and 10576016) and National 973 Project (Grant No. 61337)
Rights and permissions
About this article
Cite this article
Qiu, L., Gong, X., Ju, X. et al. Substituent effect on the molecular stability, group interaction, detonation performance, and thermolysis mechanism of nitroamino-substituted cyclopentanes and cyclohexanes. Sci. China Ser. B-Chem. 51, 1231–1245 (2008). https://doi.org/10.1007/s11426-008-0141-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11426-008-0141-1