
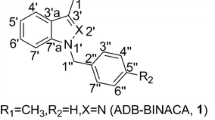
Abstract
Indazole-derived synthetic cannabinoids (SCs) featuring an alkyl substituent at the 1-position and l-valinamide at the 3-carboxamide position (e.g., AB-CHMINACA) have been identified by forensic chemists around the world, and are associated with serious adverse health effects. Regioisomerism is possible for indazole SCs, with the 2-alkyl-2H-indazole regioisomer of AB-CHMINACA recently identified in SC products in Japan. It is unknown whether this regiosiomer represents a manufacturing impurity arising as a synthetic byproduct, or was intentionally synthesized as a cannabimimetic agent. This study reports the synthesis, analytical characterization, and pharmacological evaluation of commonly encountered indazole SCs AB-CHMINACA, AB-FUBINACA, AB-PINACA, 5F-AB-PINACA and their corresponding 2-alkyl-2H-indazole regioisomers. Both regioisomers of each SC were prepared from a common precursor, and the physical properties, 1H and 13C nuclear magnetic resonance spectroscopy, gas chromatography–mass spectrometry, and ultraviolet–visible spectroscopy of all SC compounds are described. Additionally, AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA were found to act as high potency agonists at CB1 (EC50 = 2.1–11.6 nM) and CB2 (EC50 = 5.6–21.1 nM) receptors in fluorometric assays, while the corresponding 2-alkyl-2H-indazole regioisomers demonstrated low potency (micromolar) agonist activities at both receptors. Taken together, these data suggest that 2-alkyl-2H-indazole regioisomers of AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA are likely to be encountered by forensic chemists and toxicologists as the result of improper purification during the clandestine synthesis of 1-alkyl-1H-indazole regioisomers, and can be distinguished by differences in gas chromatography–mass spectrometry fragmentation pattern.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Synthetic cannabinoids (SCs) are the most rapidly growing class of novel psychoactive substances (NPSs) [1]. SC “designer drugs” are intended to mimic the psychoactive effects of Δ9-tetrahydrocannabinol (Δ9-THC, 1, Fig. 1), the principal bioactive component of cannabis. Unlike Δ9-THC—a partial agonist at both cannabinoid type 1 (CB1) and type 2 (CB2) receptors—most SCs possess high efficacy agonist activities at both CB receptor subtypes. Since the discovery of CB1/CB2 agonist JWH-018 (2) in consumer products in Germany, Austria, and Japan in 2008 [2, 3], more than 130 SCs have been reported in Europe, with 30 identified in 2014 alone [4].

Selected natural and synthetic cannabinoids (SCs)
The identification and subsequent prohibition of individual SCs have motivated clandestine chemists to produce analogues of increasing structural diversity, intended to evade legal restriction [5, 6]. Many newer SCs have no precedent in the scientific literature and little, if anything, is known of their activity or toxicity [7–14]. One particularly prevalent class of SCs, presumably inspired by recent Pfizer patents [15, 16], is comprised of an indazole core decorated at the 1-position with various aliphatic, alicyclic, or aromatic groups, and at the 3-position with valine- or tert-leucine-derived carboxamides. Unlike cannabis itself, these newer SCs are associated with exposures resulting in hospitalization or death in Europe, the USA, Japan, and Russia [17–25].
(S)-N-(1-Amino-3-methyl-1-oxobutan-2-yl)-1-(cyclohexylmethyl)-1H-indazole-3-carboxamide (AB-CHMINACA, 3) was formally notified to the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) in 2014 following identification in Latvia [4], and was also detected in Japan [26] and Germany [27]. An analogue featuring a 4-fluorobenzene group in place of the cyclohexane ring, (S)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(4-fluorobenzyl)-1H-indazole-3-carboxamide (AB-FUBINACA, 4), was found in consumer products by Japanese researchers at the National Institute of Health Sciences in 2013 [28], and was also identified in Belgium [29] and Germany [27].
(S)-N-(1-Amino-3-methyl-1-oxobutan-2-yl)-1-(pentyl)-1H-indazole-3-carboxamide (AB-PINACA, 5), featuring the n-pentyl substituent of JWH-018, was also found alongside AB-FUBINACA in Japan in 2013 [28]. Consistent with the trend for incorporation of fluorine into newer SCs [30], the terminally fluorinated analogue of AB-PINACA, (S)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(5-fluoropentyl)-1H-indazole-3-carboxamide (5F-AB-PINACA, 6), was discovered in products originating from Sweden in 2013 [29], Japan in 2014 [26], and Germany in 2015 [27].
Recently, AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA were shown to act as potent and efficacious agonists at human CB1 and CB2 receptors in vitro [31, 32]. Moreover, AB-CHMINACA, AB-FUBINACA, and AB-PINACA exert potent cannabimimetic effects on locomotion, body temperature, heart rate, and nociception in mice and rats, as well as substituting for Δ9-THC in drug discrimination assays [31–33]. Taken together, these data indicate that a range of alkyl substituents are tolerated at the 1-indazole position in this class of SCs.
These SCs appear more toxic than earlier examples, and multiple overdoses and fatalities in the USA have been attributed to AB-CHMINACA, AB-FUBINACA, and AB-PINACA [34–36]. Due to the abuse potential and toxicity of these newer SCs, the Federal Government of the USA has used emergency scheduling laws to temporarily place AB-FUBINACA, AB-PINACA, and AB-CHMINACA into Schedule I, the most restrictive category of the Controlled Substances Act. Similar legislation has been enacted in Germany, Singapore, and elsewhere [37, 38].
Forensic chemists and toxicologists have developed methods for the detection of AB-CHMINACA [39, 40], AB-FUBINACA [41–44], AB-PINACA [41, 45, 46], 5F-AB-PINACA [45, 46], and their metabolites in various biological matrices [47]. However, the changing legality of these substances, and the development of methods for their detection, increases the likelihood that newer analogues of this class will begin to appear on the drug market.
Isomerism may be used to generate new SC analogues [48–50], and the 2H-indazole regioisomer of AB-CHMINACA (AB-CHMINACA 2-isomer, 7) was recently detected in SC products in Japan [51]. It is currently unclear if this analogue is an impurity occurring as an unintended byproduct of AB-CHMINACA synthesis, or if the 2-isomer was willfully prepared as an intended cannabimimetic agent. Incidentally, the 2H-indazole analogue of APINACA was reported in China recently [52]. One study of bulk powders used as the active ingredients for SC products found purity to range from 78 to 96 %, consistent with the potential presence of manufacturing impurities in the raw materials [53].
We recently reported the synthesis, structural characterization, in vitro cannabinoid activity, and in vivo biotelemetry of several indazole synthetic cannabinoid designer drugs [31]. During the preparation of 5F-AB-PINACA, it was noted that alkylation of methyl indazole-3-carboxylate predominantly produced the desired 1-alkylated intermediate, with the 2-alkyl regiosiomer formed as a minor product. Failure to remove the unwanted 2-alkyl intermediate before the remaining synthetic steps would be expected to form 5F-AB-PINACA 2-isomer as an impurity. However, the cannabinoid activity of the 2H-indazole regioisomers of this class of SCs has never been reported, and AB-CHMINACA 2-isomer was detected in consumer products at a concentration similar to those reported for other SC products.
In this work, we synthesized AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA (3–6) and the corresponding 2H-indazole regiosiomers 7–10 from a common precursor. The characterization of these regioisomers was achieved using melting point range determinations, nuclear magnetic resonance (NMR) spectroscopy, ultraviolet (UV)-visible (Vis) spectroscopy, infrared (IR) spectroscopy, and gas chromatography–mass spectrometry (GC–MS). Additionally, the in vitro cannabinoid activities of 3–10 at human CB1 and CB2 receptors were evaluated using fluorometric imaging plate reader (FLIPR) assays.
Materials and methods
General chemical synthesis details
The synthesis of 3–10 is shown in Fig. 2. All reactions were performed under an atmosphere of nitrogen or argon unless otherwise specified. Anhydrous tetrahydrofuran (THF), methanol, acetonitrile, and dimethyl sulfoxide (DMSO) (Sigma Aldrich, St. Louis, MO, USA) were used as purchased. Commercially available chemicals (Sigma-Aldrich) were used as purchased. Analytical thin-layer chromatography was performed using Merck aluminum-backed silica gel 60 F254 (0.2 mm) plates (Merck, Darmstadt, Germany), which were visualized using shortwave (254 nm) UV fluorescence. Flash chromatography was performed using Merck Kieselgel 60 (230–400 mesh) silica gel. Melting point (m.p.) ranges were measured in open capillaries using a Stuart SMP10 m.p. apparatus (Bibby Scientific, Staffordshire, UK) and were uncorrected. NMR spectra were recorded at 300 K using either a Bruker AVANCE DRX400 (400.1 MHz) or AVANCE III 500 Ascend (500.1 MHz) spectrometer (Bruker, Bremen, Germany). The data are reported as chemical shift (δ ppm) relative to the residual protonated solvent resonance, relative integral, multiplicity (s, singlet; br s, broad singlet; d, doublet; t, triplet; quin., quintet; sept., septet; dt, doublet of triplets; m, multiplet), coupling constants (J Hz) and assignment (for 13C, quat., quaternary). Assignment of signals was assisted by correlation spectroscopy (COSY), distortionless enhancement by polarization transfer (DEPT), heteronuclear single quantum coherence (HSQC), and heteronuclear multiple-bond correlation (HMBC) experiments where necessary. Low-resolution mass spectra (LRMS) were recorded using electrospray ionization (ESI) recorded on a Finnigan LCQ ion trap mass spectrometer (ThermoFisher Scientific, Waltham, MA, USA). High-resolution mass spectra (HRMS) were run on a Bruker 7T Apex Qe Fourier Transform Ion Cyclotron resonance mass spectrometer equipped with an Apollo II ESI/APCI/MALDI Dual source by the Mass Spectrometry Facility of the School of Chemistry at The University of Sydney. IR absorption spectra were recorded on a Bruker ALPHA FT-IR spectrometer as solid or thin film from ethanol, and the data are reported as vibrational frequencies (cm−1). Please see the supplementary material for 1H and 13C NMR spectra and Fourier-transform infrared (FTIR) spectra of all final compounds.

Synthesis of indazole SC regioisomers 3–10. Reagents and conditions: a conc. sulfuric acid, methanol, reflux, 24 h, 95 %; b bromoalkane, potassium tert-butoxide, tetrahydrofuran, 0 °C–rt, 48 h, 65–84 %; c bromoalkane, potassium carbonate, potassium iodide, acetonitrile, reflux, 24 h, 20–32 %; d sodium hydroxide, methanol, rt, 24 h, 76–91 %; e l-valinamide, (benzotriazol-1-yl-oxy)tripyrrolidinophosphonium hexafluorophosphate, N,N-diisopropylethylamine, dimethyl sulfoxide, rt, 24 h, 55–87 %
Methyl 1H-indazole-3-carboxylate (12)
To a cooled (0 °C) solution of indazole-3-carboxylic acid (11, 3.0 g, 18.5 mmol) in methanol (45 mL), sulfuric acid (3 mL) was slowly added and the solution heated at reflux for 24 h. The solvent was evaporated under reduced pressure and the residue partitioned between ethyl acetate (100 mL) and water (100 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (2 × 100 mL). The combined organic phases were washed with sat. aq. sodium bicarbonate (3 × 100 mL), brine (100 mL), dried (MgSO4), and the solvent evaporated under reduced pressure. The crude material was purified using flash chromatography (hexane/ethyl acetate 50:50, R f 0.31) to give 12 (3.1 g, 95 %) as a white solid. m.p. 165–169 °C; 1H NMR (300 MHz, CDCl3): δ 8.24 (1H, d, J = 8.2 Hz), 7.69 (1H, d, J = 8.4 Hz), 7.48 (1H, t, J = 7.0 Hz), 7.35 (1H, t, J = 7.2 Hz), 4.07 (3H, s); 13C NMR (75 MHz, CDCl3): δ 163.6 (CO), 141.5 (quat.), 136.3 (quat.), 127.6 (CH), 123.5 (CH), 122.5 (quat.), 121.8 (CH), 111.5 (CH), 52.3 (CH3); LRMS (+ESI) m/z 198.95 ([M+Na]+, 54 %), 374.99 ([2M + Na]+, 100 %).
General procedure A: 1-alkylation of methyl 1H-indazole-3-carboxylate using potassium tert-butoxide
To a cooled (0 °C) solution of 1H-indazole-3-carboxylate (500 mg, 2.84 mmol, 1.0 equiv.) in THF (15 mL), potassium tert-butoxide (350 mg, 3.12 mmol, 1.1 equiv.) was added portionwise, and the mixture stirred at ambient temperature for 1 h. The cooled (0 °C) mixture was treated dropwise with appropriate bromoalkane (2.98 mmol, 1.05 equiv.) and stirred at ambient temperature for 48 h. The mixture was poured onto H2O (100 mL), extracted with ethyl acetate (3 × 100 mL), dried (MgSO4), and the solvent evaporated under reduced pressure. Following purification by flash chromatography (hexane/ethyl acetate 80:20 unless otherwise stated), methyl 1-alkyl-1H-indazole-3-carboxylate was obtained as the major product, and methyl 2-alkyl-2H-indazole-3-carboxylate as the minor product.
Methyl 1-(cyclohexylmethyl)-1H-indazole-3-carboxylate (13)
Subjecting (bromomethyl)cyclohexane (415 µL, 2.98 mmol, 1.05 equiv.) to general procedure A gave, after purification by flash chromatography (hexane/ethyl acetate 85:15), 13 (505 mg, 65 %) as a pale yellow oil. R f 0.60 (hexane/ethyl acetate 80:20); 1H NMR (300 MHz, CDCl3): δ 8.22 (1H, dt, J = 8.1, 0.9 Hz), 7.48–7.39 (2H, m), 7.33–7.28 (1H, m), 4.28 (2H, d, J = 7.5 Hz), 4.03 (3H, s), 2.08 (1H, m), 1.74–1.52 (5H, m), 1.32–0.98 (5H, m); 13C NMR (75 MHz, CDCl3): δ 163.3 (CO), 141.3 (quat.), 134.6 (quat.), 126.8 (CH), 123.7 (quat.), 123.1 (CH), 122.3 (CH), 110.0 (CH), 56.1 (CH2), 52.1 (CH3), 38.9 (CH2), 31.7 (CH), 26.3 (CH2), 25.7 (CH2); LRMS (+ESI) m/z 295.05 ([M+Na]+, 100 %); IR (diamond cell, thin film): 2925 (s), 2851 (m), 1710 (s), 1477 (s), 1441 (m), 1224 (s), 1161 (s), 1121 (s), 751 (s).
Methyl 1-(4-fluorobenzyl)-1H-indazole-3-carboxylate (14)
Subjecting 4-fluorobenzyl bromide (371 µL, 2.98 mmol, 1.05 equiv.) to general procedure A gave 14 (570 mg, 71 %) as a colorless oil. R f 0.35 (hexane/ethyl acetate 80:20); 1H NMR (300 MHz, CDCl3): δ 8.25 (1H, d, J = 7.8 Hz), 7.42–7.29 (3H, m), 7.21 (2H, t, J = 6.9 Hz), 6.99 (2H, t, J = 8.1 Hz), 5.67 (2H, s), 4.06 (3H, s); 13C NMR (75 MHz, CDCl3): δ 163.1 (CO), 162.6 (d, 1 J C–F = 245.3 Hz, quat.), 140.6 (quat.), 135.3 (quat.), 131.6 (d, 4 J C–F = 3.8 Hz, quat.), 129.2 (d, 3 J C–F = 8.3 Hz, CH), 127.3 (CH), 124.3 (quat.), 123.5 (CH), 122.5 (CH), 116.0 (d, 2 J C–F = 21.8 Hz, CH), 53.5 (CH2), 52.2 (CH3); 19F NMR (282 MHz, CDCl3): δ −113.8 (s); LRMS (+ESI): m/z 307.00 ([M+Na]+, 100 %). IR (diamond cell, thin film): 3071 (w), 2952 (w), 1712 (s), 1510 (s), 1479 (s), 1268 (s), 1157 (s), 749 (s).
Methyl 1-pentyl-1H-indazole-3-carboxylate (15)
Subjecting 1-bromopentane (370 µL, 2.98 mmol, 1.05 equiv.) to general procedure A gave 15 (585 mg, 84 %) as a colorless oil. R f 0.55 (hexane/ethyl acetate 80:20); 1H NMR (300 MHz, CDCl3): δ 8.24 (1H, d, J = 8.0 Hz), 7.50–7.44 (2H, m), 7.35–7.27 (1H, m), 4.47 (2H, t, J = 7.4 Hz), 4.04 (3H, s), 1.97 (2H, quin., J = 7.0 Hz), 1.32 (4H, m), 0.87 (3H, t, J = 6.6 Hz); 13C NMR (75 MHz, CDCl3): δ 163.3 (CO), 140.6 (quat.), 134.6 (quat.), 126.8 (CH), 123.9 (quat.), 123.1 (CH), 122.3 (CH), 109.7 (CH), 52.1 (CH2), 50.1 (CH3), 29.7 (CH2), 29.0 (CH2), 22.4 (CH2), 14.0 (CH3); LRMS (+ESI): m/z 269.03 ([M+Na]+, 60 %), 515.16 ([2 M + Na]+, 100 %); IR (diamond cell, thin film): 2954 (m), 2932 (m), 2860 (w), 1709 (s), 1477 (s), 1215 (s), 1159 (s), 1117 (s), 751 (s).
Methyl 1-(5-fluoropentyl)-1H-indazole-3-carboxylate (16)
Subjecting 1-bromo-5-fluoropentane (370 µL, 2.98 mmol, 1.05 equiv.) to general procedure A gave 16 (560 mg, 75 %) as a colorless oil. R f 0.30 (hexane/ethyl acetate 80:20); 1H NMR (300 MHz, CDCl3): δ 8.24 (1H, d, J = 8.1 Hz), 7.49–7.45 (2H, m), 7.32 (1H, t, J = 6.3 Hz), 4.52–4.47 (3H, m), 4.33 (1H, t, J = 5.7 Hz), 4.04 (3H, s), 2.03 (2H, quin., J = 7.2 Hz), 2.00–1.63 (2H, m), 1.46 (2H, quin., J = 12.0 Hz); 13C NMR (75 MHz, CDCl3): δ 163.2 (CO), 140.6 (quat.), 134.8 (quat.), 127.0 (CH), 123.9 (quat.), 123.2 (CH), 122.4 (CH), 109.6 (CH), 83.8 (d, 1 J C–F = 163.5 Hz, CH2F), 52.1 (CH2), 49.8 (CH3), 30.0 (d, 2 J C–F = 19.5 Hz, CH2), 29.6 (CH2), 22.8 (d, 3 J C–F = 4.5 Hz, CH2); 19F NMR (282 MHz, CD3OD): δ −218.6 (m); LRMS (+ESI): m/z 287.03 ([M+Na]+, 100 %); IR (diamond cell, thin film): 2950 (m), 2867 (w), 1729 (s), 1710 (s), 1478 (s), 1163 (s), 1118 (s), 752 (s).
General procedure B: 2-alkylation of methyl 1H-indazole-3-carboxylate using potassium carbonate
To a stirred mixture of methyl 1H-indazole-3-carboxylate (500 mg, 2.84 mmol, 1.0 equiv.), potassium carbonate (785 mg, 5.68 mmol, 2.0 equiv.) and potassium iodide (24 mg, 0.14 mmol, 0.05 equiv.) in acetonitrile (5 mL), the appropriate bromoalkane (2.98 mmol, 1.05 equiv.) was added dropwise. The suspension was heated at reflux for 24 h and the cooled reaction was poured onto H2O (30 mL) and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were dried (MgSO4) and the solvent removed under reduced pressure. Following purification by flash chromatography (hexane/ethyl acetate 80:20 unless otherwise stated), methyl 1-alkyl-1H-indazole-3-carboxylate was obtained as the major product, and methyl 2-alkyl-2H-indazole-3-carboxylate as the minor product.
Methyl 2-(cyclohexylmethyl)-2H-indazole-3-carboxylate (17)
Subjecting (bromomethyl)cyclohexane (415 µL, 2.98 mmol, 1.05 equiv.) to general procedure B gave, after purification by flash chromatography (hexane/ethyl acetate 93:7), 17 (235 mg, 30 %) as a colorless solid. m.p. 83–85 °C; R f 0.72 (hexane/ethyl acetate 80:20); 1H NMR (300 MHz, CDCl3): δ 8.02 (1H, d, J = 8.4 Hz), 7.79 (1H, d, J = 8.7 Hz), 7.35 (1H, t, J = 7.2 Hz), 7.30 (1H, t, J = 7.8 Hz), 4.78 (2H, d, J = 7.2 Hz), 4.03 (3H, s), 2.12–2.01 (1H, m), 1.70–1.56 (5H, m), 1.20–1.05 (5H, m); 13C NMR (75 MHz, CDCl3): δ 160.9 (CO), 147.4 (quat.), 126.3 (CH), 125.0 (CH), 123.9 (quat.), 123.6 (quat.), 121.5 (CH), 118.4 (CH), 59.3 (CH2), 52.0 (CH3), 39.6 (CH), 30.7 (CH2), 26.4 (CH), 25.8 (CH2); LRMS (+ESI): m/z 295.08 ([M+Na]+, 100 %). IR (diamond cell, thin film): 2923 (m), 2850 (m), 1711 (s), 1462 (s), 1250 (s), 1153 (s), 997 (s), 758 (s).
Methyl 2-(4-fluorobenzyl)-2H-indazole-3-carboxylate (18)
Subjecting 4-fluorobenzyl bromide (370 µL, 2.98 mmol, 1.05 equiv.) to general procedure B gave, after purification by flash chromatography (hexane/ethyl acetate 85:15), 18 (225 mg, 28 %) as a white solid. m.p. 116.5–118.5 °C; R f 0.57 (hexane/ethyl acetate 80:20); 1H NMR (300 MHz, CDCl3): δ 8.01 (1H, d, J = 8.1 Hz), 7.81 (1H, d, J = 8.4 Hz), 7.35–7.28 (4H, m), 6.98 (2H, t, J = 7.8 Hz), 6.07 (2H, s), 4.01 (3H, s); 13C NMR (75 MHz, CDCl3): δ 162.6 (d, quat., 1 J C–F = 246.6 Hz), 160.8 (CO), 147.8 (quat.), 132.2 (quat.), 130.0 (d, 3 J C–F = 8.3 Hz), 126.7 (CH), 125.4 (CH), 123.8 (quat.), 123.5 (quat.), 121.6 (CH), 118.5 (CH), 115.6 (d, 2 J C–F = 21.8 Hz, CH), 56.0 (CH2), 52.2 (CH3); 19F NMR (282 MHz, CDCl3): δ −114.2 (s); LRMS (+ESI): m/z 307.02 ([M+Na]+, 100 %); IR (diamond cell, thin film): 3054 (w), 2954 (w), 1710 (s), 1554 (s), 1510 (s), 1222 (s), 1210 (s), 1083 (s), 758 (s).
Methyl 2-pentyl-2H-indazole-3-carboxylate (19)
Subjecting 1-bromopentane (370 µL, 2.98 mmol, 1.05 equiv.) to general procedure B gave, after purification by flash chromatography (hexane/ethyl acetate 93:7), 19 (175 mg, 25 %) as a colorless oil. R f 0.69 (hexane/ethyl acetate 80:20); 1H NMR (500 MHz, CD3OD): δ 8.01 (1H, d, J = 8.5 Hz), 7.70 (1H, d, J = 8.5 Hz), 7.36 (1H, t, J = 7.0 Hz), 7.28 (1H, t, J = 7.5 Hz), 4.86 (2H, t, J = 7.0 Hz), 4.02 (3H, s), 1.94 (2H, quin., J = 7.0 Hz), 1.39–1.31 (4H, m), 0.90 (3H, t, J = 6.5 Hz); 13C NMR (125 MHz, CD3OD): δ 161.8 (CO), 148.5 (quat.), 127.7 (CH), 126.0 (CH), 124.8 (quat.), 124.6 (quat.), 122.5 (CH), 118.6 (CH), 54.4 (CH2), 52.5 (CH3), 31.6 (CH2), 29.8 (CH2), 23.2 (CH2), 14.2 (CH3); LRMS (+ESI): m/z 269.03 ([M+Na]+, 60 %), 515.16 ([2M + Na]+, 100 %); IR (diamond cell, thin film): 2955 (m), 2930 (w), 2860 (w), 1710 (s), 1463 (s), 1278 (s), 1206 (s), 1078 (s), 758 (s).
Methyl 2-(5-fluoropentyl)-2H-indazole-3-carboxylate (20)
Subjecting 1-bromo-5-fluoropentane (370 µL, 2.98 mmol, 1.05 equiv.) to general procedure B gave 20 (152 mg, 20 %) as a colorless oil. R f 0.50 (hexane/ethyl acetate 80:20); 1H NMR (300 MHz, CDCl3): δ 8.02 (1H, d, J = 8.4 Hz), 7.78 (1H, d, J = 8.6 Hz), 7.36 (1H, t, J = 7.9 Hz), 7.29 (1H, t, J = 7.7 Hz), 4.93 (2H, t, J = 7.4 Hz), 4.43 (2H, dt, J = 47.5 Hz, J = 6.2 Hz), 4.04 (3H, s), 2.03 (2H, quin., J = 7.5 Hz), 1.76 (2H, m), 1.50 (2H, quin., J = 7.6 Hz); 13C NMR (75 MHz, CDCl3): δ 160.8 (CO), 147.5 (quat.), 126.4 (CH), 125.1 (CH), 123.6 (quat.), 123.5 (quat.), 121.5 (CH), 118.3 (CH), 83.9 (d, 1 J C–F = 163.5 Hz, CH2F), 53.5 (CH3), 52.1 (CH2), 30.6 (CH2), 30.1 (d, 2 J C–F = 20.3 Hz, CH2), 22.6 (d, 3 J C–F = 5.3 Hz, CH2); 19F NMR (282 MHz, CDCl3): δ −218.7 (m); LRMS (+ESI): m/z 287.05 ([M+Na]+, 100 %); IR (diamond cell, thin film): 2954 (w), 2865 (w), 1708 (s), 1463 (s), 1278 (s), 1205 (s), 1042 (s), 758 (s).
General procedure C: ester hydrolysis of methyl 1-alkyl-1H-indazole-3-carboxylates and methyl 2-alkyl-2H-indazole-3-carboxylates
To a solution of the appropriately substituted indazole-3-carboxylic acid methyl ester (0.43 mmol) in methanol (5 mL), a solution of 1 M aq. sodium hydroxide (650 µL, 0.65 mmol, 1.5 equiv.) was added dropwise, and the solution stirred at ambient temperature for 18 h. Solvent was evaporated in vacuo, and sat. aq. sodium hydrogen carbonate (50 mL) was added to the residue. The aqueous phase was washed with diethyl ether (10 mL), adjusted to pH ~2 using 1 M aq. hydrochloric acid solution, and extracted with diethyl ether (3 × 50 mL). The combined organic phases were washed with brine (50 mL), dried (MgSO4), and the solvent evaporated under reduced pressure.
1-(Cyclohexylmethyl)-1H-indazole-3-carboxylic acid (21)
Subjecting 13 (475 mg, 1.74 mmol) to general procedure C gave 21 (388 mg, 86 %) as a white solid. m.p. 124–126 °C; 1H NMR (300 MHz, CD3OD): δ 8.16 (1H, d, J = 8.1 Hz), 7.66 (1H, d, J = 8.4 Hz), 7.47 (1H, t, J = 7.2 Hz), 7.31 (1H, t, J = 7.8 Hz), 4.34 (2H, d, J = 7.2 Hz), 2.05 (1H, m), 1.72–1.66 (3H, m), 1.56 (2H, d, J = 12.9 Hz), 1.26–1.02 (5H, m); 13C NMR (75 MHz, CD3OD): δ 165.5 (CO), 142.6 (quat.), 135.9 (quat.), 128.0 (CH), 124.5 (quat.), 124.1 (CH), 123.0 (CH), 111.4 (CH), 56.5 (CH2), 40.0 (CH), 31.7 (CH2), 27.7 (CH), 26.7 (CH2); LRMS (–ESI): m/z 257.16 ([M−H]−, 100 %); IR (diamond cell, thin film): 3060 (bs), 2926 (s), 2851 (m), 1707 (s), 1479 (s), 1230 (s), 1174 (s), 752 (s).
1-(4-Fluorobenzyl)-1H-indazole-3-carboxylic acid (22)
Subjecting 14 (560 mg, 1.97 mmol) to general procedure C gave 22 (480 mg, 91 %) as a white solid. m.p. 198.5–199.5 °C; 1H NMR (300 MHz, CD3OD): δ 8.17 (1H, d, J = 8.1 Hz), 7.62 (1H, d, J = 8.7 Hz), 7.44 (1H, t, J = 7.5 Hz), 7.31 (3H, m), 7.04 (2H, t, J = 8.4 Hz), 5.71 (2H, s, CH2).; 13C NMR (75 MHz, CD3OD): δ 165.5 (CO), 163.9 (d, 1 J C–F = 244.5 Hz, quat.), 142.1 (quat.), 136.7 (quat.), 133.7 (d, 4 J C–F = 3.0 Hz, quat.), 130.6 (d, 3 J C–F = 8.3 Hz, quat.), 128.3 (CH), 125.0 (quat.), 124.3 (CH), 123.1 (CH), 116.5 (d, 2 J C–F = 21.8 Hz, quat.), 111.4 (CH), 53.6 (CH2); 19F NMR (285 MHz, CD3OD): δ −118.0 (s); LRMS (–ESI): m/z 269.07 ([M−H]−, 100 %); IR (diamond cell, thin film): 3058 (bs), 2926 (w), 1696 (s), 1510 (s), 1481 (s), 1224 (s), 1170 (s), 1157 (s), 749 (s).
1-Pentyl-1H-indazole-3-carboxylic acid (23)
Subjecting 15 (600 mg, 2.58 mmol) to general procedure C gave 23 (510 mg, 85 %) as a white solid. m.p. 76.5–78.0 °C; 1H NMR (300 MHz, CD3OD): δ 8.26 (1H, d, J = 8.1 Hz), 7.52–7.44 (2H, m), 7.35 (1H, t, J = 7.8 Hz), 4.48 (2H, t, J = 7.2 Hz), 1.99 (2H, quin., J = 7.2 Hz), 1.34 (4H, m), 0.89 (3H, t, J = 6.0 Hz); 13C NMR (75 MHz, CD3OD): δ 165.4 (CO), 142.0 (quat.), 135.9 (quat.), 127.9 (CH), 124.5 (quat.), 124.0 (CH), 122.9 (CH), 111.0 (CH), 49.7 (CH2), 30.4 (CH2), 29.8 (CH2), 23.1 (CH2), 14.1 (CH3); LRMS (–ESI): m/z 231.12 ([M−H]−, 100 %); IR (diamond cell, thin film): 3053 (bs), 2956 (m), 2931 (m), 2860 (w), 1687 (s), 1503 (s), 1218 (s), 1176 (s), 1121 (s), 752 (s).
1-(5-Fluoropentyl)-1H-indazole-3-carboxylic acid (24)
Subjecting 16 (750 mg, 2.84 mmol) to general procedure C gave 24 (580 mg, 81 %) as a white solid. m.p. 199–200 °C; 1H NMR (300 MHz, CD3OD): δ 8.36 (1H, d, J = 7.8 Hz), 7.52 (2H, m), 7.33 (1H, t, J = 7.5 Hz), 4.53–4.49 (3H, m), 4.34 (1H, t, J = 5.7 Hz), 2.03 (2H, quin., J = 7.2 Hz), 1.82–1.65 (2H, m), 1.50 (2H, quin., J = 6.9 Hz); 13C NMR (75 MHz, CD3OD): δ 165.5 (CO), 142.2 (quat.), 136.1 (quat.), 128.1, 124.7 (quat.), 124.2 (CH), 123.0 (CH), 111.2 (CH), 84.6 (d, 1 J C–F = 162.8 Hz, CH2), 50.3 (CH2), 31.0 (d, 2 J C–F = 20.3 Hz, CH2), 30.4 (CH2), 23.6 (d, 3 J C–F = 5.3 Hz, CH2); 19F NMR (282 MHz, CD3OD): δ −221.8 (m); LRMS (–ESI): m/z 249.12 ([M−H]−, 100 %); IR (diamond cell, thin film): 3052 (bs), 2941 (m), 2866 (w), 1685 (s), 1480 (s), 1167 (s), 1120 (s), 751 (s).
2-(Cyclohexylmethyl)-2H-indazole-3-carboxylic acid (25)
Subjecting 17 (100 mg, 0.37 mmol) to general procedure C gave 25 (72 mg, 76 %) as a white solid. m.p. 178–179.5 °C; 1H NMR (300 MHz, CD3OD): δ 8.07 (1H, d, J = 8.1 Hz), 7.69 (1H, d, J = 8.4 Hz), 7.35 (1H, t, J = 7.2 Hz), 7.25 (1H, t, J = 6.9 Hz), 4.75 (2H, d, J = 7.2 Hz), 2.03 (1H, m), 1.72–1.63 (3H, m), 1.53 (2H, m), 1.22–1.05 (5H, m); 13C NMR (75 MHz, CD3OD): δ 162.6 (CO), 148.4 (quat.), 127.6 (CH), 126.1 (quat.), 125.7 (CH), 124.9 (quat.), 122.9 (CH), 118.4 (CH), 59.7 (CH2), 40.8 (CH), 31.6 (CH2), 27.4 (CH2), 26.8 (CH2); LRMS (–ESI): m/z 257.17 ([M−H]−, 100 %); IR (diamond cell, thin film): 2923 (m), 2851 (m), 2567 (bs), 1706 (s), 1471 (s), 1271 (s), 1208 (s), 1197 (s), 1053 (s), 757 (s).
2-(4-Fluorobenzyl)-2H-indazole-3-carboxylic acid (26)
Subjecting 18 (100 mg, 0.35 mmol) to general procedure C gave 26 (75 mg, 79 %) as a white solid. m.p. 207.5–210 °C; 1H NMR (500 MHz, CD3OD): δ 8.07 (1H, d, J = 8.5 Hz), 7.72 (1H, d, J = 8.5 Hz), 7.37 (1H, t, J = 7.0 Hz), 7.33 (2H, t, J = 7.0 Hz), 7.27 (1H, t, J = 7.5 Hz), 7.02 (2H, t, J = 8.0 Hz), 6.09 (2H, s); 13C NMR (125 MHz, CD3OD): δ 163.8 (d, 1 J C–F = 242.5 Hz), 162.5 (CO), 148.9 (quat.), 134.3 (quat.), 130.8 (d, 3 J C–F = 8.8 Hz, CH), 127.9 (CH), 125.9 (CH), 125.8 (quat.), 125.0 (quat.), 122.9 (CH), 118.6 (CH), 116.3 (d, 2 J C–F = 21.3 Hz, CH), 56.3 (CH2); 19F NMR (470 MHz, CD3OD): δ −116.6 (s); LRMS (–ESI): m/z 269.12 ([M−H]−, 100 %); IR (diamond cell, thin film): 2922 (w), 2525 (bs), 1712 (s), 1605 (m), 1511 (s), 1214 (m), 1152 (s), 1018 (s), 755 (s).
2-Pentyl-2H-indazole-3-carboxylic acid (27)
Subjecting 19 (100 mg, 0.43 mmol) to general procedure C gave 27 (84 mg, 90 %) as a white solid. m.p. 141.5–142.5 °C; 1H NMR (300 MHz, CD3OD): δ 8.26 (1H, d, J = 8.1 Hz), 7.52–7.44 (2H, m), 7.35 (1H, t, J = 7.8 Hz), 4.48 (2H, t, J = 7.2 Hz), 1.99 (2H, quin., J = 7.2 Hz), 1.34 (4H, m), 0.89 (3H, t, J = 6.0 Hz); 13C NMR (75 MHz, CD3OD): δ 161.0 (CO), 147.0 (CH), 134.6 (quat.), 126.1 (CH), 124.1 (CH), 123.4 (quat.), 121.3 (CH), 116.8 (CH), 52.7 (CH2), 30.2 (CH2), 28.3 (CH2), 21.8 (CH2), 12.7 (CH3); LRMS (–ESI): m/z 231.13 ([M−H]−, 100 %); IR (diamond cell, thin film): 2953 (m), 2927 (m), 2869 (m), 2474 (bs), 1701 (s), 1477 (m), 1428 (m), 1274 (s), 1209 (s), 1083 (s), 753 (s).
2-(5-Fluoropentyl)-2H-indazole-3-carboxylic acid (28)
Subjecting 20 (100 mg, 0.38 mmol) to general procedure C gave 28 (78 mg, 82 %) as a white solid. m.p. 137–138 °C; 1H NMR (300 MHz, CD3OD): δ 8.08 (1H, d, J = 8.1 Hz), 7.70 (1H, d, J = 8.7 Hz), 7.37 (1H, t, J = 7.2 Hz), 7.27 (1H, t, J = 7.2 Hz), 4.93 (2H, t, J = 7.2 Hz), 4.48 (1H, t, J = 6.0 Hz), 4.32 (1H, t, J = 5.7 Hz), 2.01 (2H, quin., J = 7.2 Hz), 1.80–1.66 (2H, m), 1.45 (2H, quin., J = 7.5 Hz); 13C NMR (75 MHz, CD3OD): δ 162.5 (CO), 148.6 (quat.), 127.7 (CH), 125.75 (CH), 125.74 (quat.), 124.9 (quat.), 122.8 (CH), 118.4 (CH), 84.6 (d, 1 J C–F = 162.8 Hz, CH2F), 54.0 (CH2), 31.6 (CH2), 31.0 (d, 2 J C–F = 19.5 Hz, CH2), 23.4 (d, 3 J C–F = 5.3 Hz, CH2); 19F NMR (282 MHz, CD3OD): δ −222.0 (m); LRMS (–ESI): m/z 249.14 ([M−H]−, 100 %); IR (diamond cell, thin film): 2927 (w), 2863 (w), 2548 (bs), 1692 (s), 1428 (s), 1390 (m), 1202 (s), 1079 (s).
General procedure D: preparation of indazole-3-carboxamides
A cooled (0 °C) solution of the appropriate 1-alkyl-1H- or 2-alkyl-2H-indazole-3-carboxylic acid (0.19 mmol, 1.0 equiv.), l-valinamide (31 mg, 0.20 mmol, 1.05 equiv.), and (benzotriazol-1-yl-oxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP®) (106 mg, 0.20 mmol, 1.05 equiv.) in DMSO (3 mL) was treated dropwise with N,N-diisopropylethylamine (DIPEA, 68 µL, 0.39 mmol, 2.0 equiv.) and stirred at ambient temperature for 2 h. The reaction was poured onto sat. aq. sodium hydrogen carbonate and extracted with diethyl ether (3 × 50 mL). The combined organic layers were dried (MgSO4) and the solvent evaporated under reduced pressure. The crude products were purified using flash chromatography.
(S)-N-(1-Amino-3-methyl-1-oxobutan-2-yl)-1-(cyclohexylmethyl)-1H-indazole-3-carboxamide (AB-CHMINACA, 3)
Subjecting 21 (150 mg, 0.58 mmol) to general procedure D gave, following purification by flash chromatography (hexane/ethyl acetate 60:40), 3 (135 mg, 65 %) as a white solid. R f 0.25 (hexane/ethyl acetate 50:50); m.p. 88.5–92.5 °C; 1H NMR (400 MHz, DMSO-d 6): δ 8.16 (1H, dt, J = 8.0, 0.8 Hz), 7.77 (1H, d, J = 8.4 Hz), 7.68 (1H, d, J = 8.4 Hz), 7.67 (1H, br s), 7.45 (1H, ddd, J = 8.4, 6.8, 1.2 Hz), 7.27 (1H, ddd, J = 8.2, 7.2, 0.8 Hz), 7.22 (1H, br s), 4.42 (1H, dd, J = 9.0, 6.4 Hz), 4.34 (2H, d, J = 7.2 Hz), 2.14–2.05 (1H, m), 2.00–1.88 (1H, m), 1.68–1.55 (3H, m), 1.54–1.43 (2H, m), 1.22–0.99 (5H, m), 0.94 (3H, d, J = 6.8 Hz), 0.90 (3H, d, J = 6.8 Hz); 13C NMR (100 MHz, DMSO-d 6): δ; 172.6 (CO), 161.4 (CO), 141.2 (quat.), 136.4 (quat.), 126.6 (CH), 122.4 (CH), 121.8 (quat.), 121.6 (CH), 110.7 (CH), 56.8 (CH), 54.6 (CH2), 38.4 (CH), 31.3 (CH), 30.02 (CH2), 29.99 (CH2), 25.8 (CH2), 25.11 (CH2), 25.08 (CH2), 19.4 (CH3), 18.0 (CH3); LRMS (+ESI): m/z 379.16 ([M+Na]+, 100 %); HRMS (+ESI): m/z [M+H]+ calculated 357.2291, found 357.2286; IR (diamond cell, thin film): 3388 (bs), 3190 (bs), 2926 (m), 2851 (w), 1650 (s), 1528 (s), 1491 (m), 1176 (m), 778 (w), 749 (m).
(S)-N-(1-Amino-3-methyl-1-oxobutan-2-yl)-1-(4-fluorobenzyl)-1H-indazole-3-carboxamide (AB-FUBINACA, 4)
Subjecting 22 (75 mg, 0.28 mmol) to general procedure D gave, following purification by flash chromatography (hexane/ethyl acetate 50:50), 4 (84 mg, 81 %) as a white solid. R f 0.15 (hexane/ethyl acetate 50:50); m.p. 163.0–165.5 °C; 1H NMR (400 MHz, DMSO-d 6): δ 8.18 (1H, d, J = 8.4 Hz), 7.77 (1H, t, J = 9.4 Hz), 7.66 (1H, br s), 7.46 (1H, ddd, J = 8.4, 6.8, 0.8 Hz), 7.34–7.27 (3H, m), 7.22 (1H, br s), 7.18–7.15 (2H, m), 5.78 (2H, s), 4.42 (1H, dd, J = 9.2, 6.4 Hz), 2.15–2.06 (1H, m), 0.95 (3H, d, J = 6.8 Hz), 0.90 (3H, d, J = 6.8 Hz); 13C NMR (100 MHz, DMSO-d 6): δ 172.6 (CO), 161.6 (d, CF, J = 243.8 Hz), 161.3 (CO), 140.6 (quat.), 137.1 (quat.), 133.0 (d, quat., J = 3.0 Hz), 129.5 (d, CH, J = 8.4 Hz), 127.0 (CH), 122.7 (CH), 122.3 (quat.), 121.8 (CH), 115.5 (d, CH, J = 21.6 Hz), 110.6 (quat.), 56.9 (CH), 51.6 (CH2), 31.2 (CH), 19.4 (CH3), 18.0 (CH3); LRMS (+ESI): m/z 391.10 ([M+Na]+, 100 %); HRMS (+ESI): m/z calculated [M+Na]+ 391.1546, found 391.1541; IR (diamond cell, thin film): 3337 (bs), 3231 (bs), 2966 (w), 1683 (m), 1651 (s), 1510 (s), 1491 (s), 1470 (m), 1225 (m), 748 (m).
(S)-N-(1-Amino-3-methyl-1-oxobutan-2-yl)-1-pentyl-1H-indazole-3-carboxamide (AB-PINACA, 5)
Subjecting 23 (50 mg, 0.22 mmol) to general procedure D gave, following purification by flash chromatography (hexane/ethyl acetate 70:30), 5 (63 mg, 87 %) as a white solid. R f 0.55 (hexane/ethyl acetate 40:60); m.p. 100–103 °C; 1H NMR (400 MHz, DMSO-d 6): δ 8.16 (1H, d, J = 8.4 Hz), 7.77 (1H, d, J = 8.4 Hz), 7.69 (1H, d, J = 9.2 Hz), 7.66 (1H, br s), 7.45 (1H, ddd, J = 8.4, 7.2, 1.2 Hz), 7.27 (1H, t, J = 7.4 Hz), 7.22 (1H, br s), 4.49 (2H, t, J = 7.0 Hz), 4.39 (1H, dd, J = 9.2, 6.0 Hz), 2.14–2.06 (1H, m), 1.86 (2H, quin., J = 7.2 Hz), 1.35–1.19 (4H, m), 0.95 (3H, d, J = 6.8 Hz), 0.90 (3H, d, J = 6.8 Hz), 0.82 (3H, t, J = 7.2 Hz); 13C NMR (100 MHz, DMSO-d 6): δ 172.6 (CO), 161.3 (CO), 140.6 (quat.), 136.4 (quat.), 126.6 (CH), 122.4 (CH), 122.0 (quat.), 121.6 (CH), 110.4 (CH), 56.7 (CH), 48.7 (CH2), 31.3 (CH), 29.1 (CH2), 28.3 (CH2), 21.6 (CH2), 19.4 (CH3), 17.9 (CH3), 13.8 (CH3); LRMS (+ESI): m/z 353.13 ([M+Na]+, 100 %); HRMS (+ESI): m/z calculated [M+Na]+ 353.1954, found 353.1948; IR (diamond cell, thin film): 3390 (bs), 3198 (m), 2960 (m), 2931 (m), 1651 (s), 1530 (s), 1468 (m), 1182 (m), 750 (m).
(S)-N-(1-Amino-3-methyl-1-oxobutan-2-yl)-1-(5-fluoropentyl)-1H-indazole-3-carboxamide (5F-AB-PINACA, 6)
Subjecting 24 (75 mg, 0.30 mmol) to general procedure D gave, following purification by flash chromatography (hexane/ethyl acetate 60:40), 6 (70 mg, 67 %) as a white solid. R f 0.45 (hexane/ethyl acetate 40:60); m.p. 121–124 °C; 1H NMR (400 MHz, DMSO-d 6): δ 8.16 (1H, d, J = 8.4 Hz), 7.79 (1H, dt, J = 8.4, 0.9 Hz), 7.69 (1H, d, J = 9.2 Hz), 7.66 (1H, br s), 7.46 (1H, ddd, J = 8.4, 6.8, 1.2 Hz), 7.28 (1H, ddd, J = 8.2, 7.0, 0.8 Hz), 7.22 (1H, br s), 4.52 (2H, t, J = 7.0 Hz), 4.42 (1H, dd, J = 9.2, 6.0 Hz), 4.39 (1H, dt, J = 47.6, 6.0 Hz), 2.14–2.06 (1H, m), 1.91 (2H, quin., J = 7.6 Hz), 1.74–1.60 (2H, m), 1.39–1.31 (2H, m), 0.95 (3H, d, J = 6.8 Hz), 0.90 (3H, d, J = 6.8 Hz); 13C NMR (100 MHz, DMSO-d 6): δ 172.7 (CO), 161.4 (CO), 140.6 (quat.), 136.4 (quat.), 126.7 (CH), 122.5 (CH), 122.0 (quat.), 121.7 (CH), 110.5 (quat.), 83.6 (d, CH2F, J = 161.8 Hz), 56.8 (CH), 48.6 (CH2), 31.3 (CH), 29.3 (d, CH2, J = 19.3 Hz), 29.0 (CH2), 22.0 (d, CH2, J = 5.3 Hz), 19.4 (CH3), 17.9 (CH3); 19F NMR (470 MHz, DMSO-d 6): δ −217.0 (1F, s, CH2F); LRMS (+ESI): m/z 371.12 ([M+Na]+, 100 %); HRMS (+ESI): m/z calculated [M+Na]+ 371.1859, found 371.1855; IR (diamond cell, thin film): 3393 (bs), 3195 (bs), 2963 (m), 2873 (w), 1650 (s), 1529 (s), 1491 (m), 1172 (m), 751 (s).
(S)-N-(1-Amino-3-methyl-1-oxobutan-2-yl)-2-(cyclohexylmethyl)-2H-indazole-3-carboxamide (AB-CHMINACA 2-regioisomer, 7)
Subjecting 25 (50 mg, 0.19 mmol) to general procedure D gave, following purification by flash chromatography (hexane/ethyl acetate 70:30), 7 (50 mg, 74 %) as a white solid. R f 0.60 (hexane/ethyl acetate 40:60); m.p. 203–204 °C; 1H NMR (400 MHz, DMSO-d 6): δ 8.36 (1H, d, J = 8.8 Hz), 7.81 (1H, d, J = 8.8 Hz), 7.69 (1H, d, J = 8.8 Hz), 7.51 (1H, br s), 7.34–7.30 (1H, m), 7.23–7.20 (1H, m), 7.18 (1H, br s), 4.59 (1H, dd, J = 12.8, 7.2 Hz), 4.49 (1H, dd, J = 12.8, 7.2 Hz), 4.39 (1H, dd, J = 8.8, 6.8 Hz), 2.19–2.10 (1H, m), 1.96–1.85 (1H, m), 1.66–1.54 (3H, m), 1.49–1.45 (2H, m), 1.18–1.07 (3H, m), 1.03–0.92 (2H, overlapping), 0.99 (3H, d, J = 6.8 Hz), 0.97 (3H, d, J = 6.8 Hz); 13C NMR (100 MHz, DMSO-d 6): δ 172.6 (CO), 159.8 (CO), 146.4 (quat.), 129.1 (quat.), 125.8 (CH), 122.8 (CH), 120.4 (quat.), 120.2 (CH), 117.4 (CH), 58.4 (CH), 57.3 (CH2), 30.2 (CH), 30.01 (CH), 29.99 (CH2), 25.8 (CH2), 25.2 (CH2), 19.5 (CH3), 18.3 (CH3); LRMS (+ESI): m/z 379.16 ([M+Na]+, 100 %); HRMS (+ESI): m/z calculated [M+Na]+ 379.2110, found 379.2105; IR (diamond cell, thin film): 3345 (bs), 3275 (bs), 3159 (bs), 2956 (w), 2925 (m), 2854 (w), 1679 (s), 1649 (s), 1624 (s), 532 (m).
(S)-N-(1-Amino-3-methyl-1-oxobutan-2-yl)-2-(4-fluorobenzyl)-2H-indazole-3-carboxamide (AB-FUBINACA 2-regioisomer, 8)
Subjecting 26 (50 mg, 0.19 mmol) to general procedure D gave, following purification by flash chromatography (hexane/ethyl acetate 70:30), 8 (55 mg, 79 %) as a white solid. R f 0.55 (hexane/ethyl acetate 40:60); m.p. 264–266 °C; 1H NMR (500 MHz, DMSO-d 6): δ 8.38 (1H, d, J = 8.5 Hz), 7.81 (1H, d, J = 8.5 Hz), 7.70 (1H, d, J = 8.5 Hz), 7.56 (1H, br s), 7.35–7.32 (3H, m), 7.23 (1H, ddd, J = 8.3, 6.8, 0.5 Hz), 7.19 (1H, br s), 7.14–7.09 (2H, m), 5.90–5.82 (2H, m), 4.37 (1H, dd, J = 9.0, 7.0 Hz), 2.15–2.08 (1H, m), 0.95 (3H, d, J = 6.8 Hz), 0.91 (3H, d, J = 6.8 Hz); 13C NMR (125 MHz, DMSO-d 6): δ 172.9 (CO), 161.9 (d, CF, J = 244.0 Hz, quat.), 159.8 (CO), 147.0 (quat.), 133.2 (d, J = 3.0 Hz, quat.), 130.3 (d, CH, J = 8.4 Hz, CH), 128.8 (quat.), 126.4 (CH), 123.4 (CH), 120.7 (quat.), 120.5 (CH), 117.7 (CH), 115.4 (d, CH, J = 21.5 Hz, CH), 58.7 (CH), 54.4 (CH2), 30.3 (CH), 19.6 (CH3), 18.4 (CH3); 19F NMR (470 MHz, DMSO-d 6): δ −114.5 (s); LRMS (+ESI): m/z 391.09 ([M+Na]+, 100 %); HRMS (+ESI): m/z calculated [M+Na]+ 391.1546, found 391.1541; IR (diamond cell, thin film): 3350 (bs), 3257 (bs), 2960 (w), 2926 (w), 2854 (w), 1673 (s), 1636 (s), 1618 (s), 1536 (m), 1110 (w), 755 (w).
(S)-N-(1-Amino-3-methyl-1-oxobutan-2-yl)-2-pentyl-2H-indazole-3-carboxamide (AB-PINACA 2-regioisomer, 9)
Subjecting 27 (50 mg, 0.22 mmol) to general procedure D gave, following purification by flash chromatography (hexane/ethyl acetate 70:30), 9 (40 mg, 55 %) as a white solid. R f 0.30 (hexane/ethyl acetate 50:50); m.p. 224–226 °C; 1H NMR (400 MHz, DMSO-d 6): δ 8.30 (1H, d, J = 8.8 Hz), 7.80 (1H, dt, J = 8.4, 1.0 Hz), 7.69 (1H, dt, J = 8.4, 1.0 Hz), 7.56 (1H, br s), 7.32 (1H, ddd, J = 8.4, 6.8, 1.2 Hz), 7.22 (1H, ddd, J = 8.4, 6.8, 1.2 Hz), 7.17 (1H, br s), 4.72–4.57 (2H, m), 4.39 (1H, dd, J = 8.8, 6.9 Hz), 2.18–2.10 (1H, m), 1.84 (2H, quin., J = 7.4 Hz), 1.29–1.16 (4H, m), 0.98 (3H, d, J = 6.8 Hz), 0.96 (3H, d, J = 6.8 Hz), 0.81 (3H, t, J = 7.4 Hz); 13C NMR (100 MHz, DMSO-d 6): δ 172.9 (CO), 159.9 (CO), 146.6 (quat.), 128.8 (quat.), 126.0 (CH), 123.1 (CH), 120.5 (quat.), 120.3 (CH), 117.6 (CH), 58.6 (CH), 51.9 (CH2), 30.4 (CH), 30.1 (CH2), 28.3 (CH2), 21.8 (CH2), 19.6 (CH3), 18.4 (CH3), 13.9 (CH3); LRMS (+ESI): m/z 353.15 ([M+Na]+, 100 %); HRMS (+ESI): m/z calculated [M+Na]+ 353.1954, found 353.1948; IR (diamond cell, thin film): 3369 (bs), 3269 (bs), 3205 (bs), 2958 (w), 2932 (w), 2859 (w), 1672 (s), 1619 (s), 1528 (m), 756 (w).
(S)-N-(1-Amino-3-methyl-1-oxobutan-2-yl)-2-(5-fluoropentyl)-2H-indazole-3-carboxamide (5F-AB-PINACA 2-regioisomer, 10)
Subjecting 28 (50 mg, 0.20 mmol) to general procedure D gave, following purification by flash chromatography (hexane/ethyl acetate 65:35), 10 (57 mg, 82 %) as a white solid. R f 0.50 (hexane/ethyl acetate 40:60); m.p. 213–214 °C; 1H NMR (500 MHz, DMSO-d 6): δ 8.32 (1H, d, J = 8.8 Hz), 7.82 (1H, d, J = 8.5 Hz), 7.70 (1H, d, J = 8.8 Hz), 7.55 (1H, br s), 7.34–7.31 (1H, m), 7.24–7.21 (1H, m), 7.18 (1H, br s), 4.74–4.62 (2H, m), 4.40 (1H, dd, J = 8.8, 6.9 Hz), 4.39 (2H, dt, J = 47.5, 6.1 Hz), 2.14 (1H, sept., J = 6.8 Hz), 1.90 (2H, quin., J = 7.6 Hz), 1.68–1.58 (2H, m), 1.33–1.27 (2H, m), 1.00 (3H, d, J = 6.8 Hz), 0.97 (3H, d, J = 6.8 Hz); 13C NMR (125 MHz, CDCl3): δ 172.6 (CO), 159.7 (CO), 146.5 (quat.), 128.7 (quat.), 125.8 (CH), 122.9 (CH), 120.4 (quat.), 120.2 (CH), 117.5 (quat.), 83.5 (d, J = 162.2 Hz, CH2F), 58.4 (CH), 51.6 (CH2), 30.3 (CH), 29.9 (CH2), 29.3 (d, J = 18.9 Hz, CH2), 21.8 (d, J = 5.0 Hz, CH2), 19.5 (CH3), 18.3 (CH3); 19F NMR (470 MHz, DMSO-d 6): δ −216.9 (s); LRMS (+ESI): m/z 371.14 ([M+Na], 100 %); HRMS (+ESI): m/z calculated [M+Na]+ 371.1859, found 371.1854; IR (diamond cell, thin film): 3368 (bs), 3267 (bs), 3204 (bs), 2959 (w), 2869 (w), 1672 (s), 1631 (s), 1530 (m), 758 (w).
Ultraviolet absorption spectroscopy
UV absorption spectroscopy was performed using a Cary 60 UV–Vis Spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). UV absorbances of solutions of 3–10 in methanol (0.001 % w/v) in quartz cuvettes were recorded over 200–800 nm.
Gas chromatography–mass spectrometry
All SCs were analyzed by gas chromatography–mass spectrometry (GC–MS) using a ThermoQuest Trace gas chromatograph with Finnigan Polaris Q ion trap mass spectrometer (ThermoQuest, Madison, CT, USA) in positive ion electron ionization (EI) mode and a Phenomenex ZB Wax column (30 m × 0.25 mm internal diameter, 0.25 μm film thickness; Phenomenex, Torrance, CA, USA) with helium gas as carrier at 1.3 mL/min. The conditions were: electron energy, 70 eV; injector temperature, 200 °C; injection, splitless mode; injection volume, 1 μL; oven temperature program, 80 °C (1.2 min hold), increase at a rate of 50 °C/min to 190 °C (15 min-hold) and then increase rate of 10 °C/min to 310 °C (10 min-hold); transfer line temperature, 280 °C; scan range, m/z 46–650 (3 min solvent delay).
In vitro pharmacological assessment of 3–10
Mouse AtT-20 neuroblastoma cells stably transfected with human CB1 or human CB2 have been previously described [54] and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10 % fetal bovine serum (FBS), 100 U penicillin/streptomycin, and 300 μg/mL G418. Cells were passaged at 80 % confluency as required. Cells for assays were grown in 75 cm2 flasks and used at 90 % confluence. The day before, the assay cells were detached from the flask with trypsin/EDTA (Sigma-Aldrich) and resuspended in 10 mL of Leibovitz’s L-15 media supplemented with 1 % FBS, 100 U penicillin/streptomycin and 15 mM glucose (membrane potential assay and Ca5 calcium assay). The cells were plated in volume of 90 μL in black-walled, clear-bottomed 96-well microplates (Corning, Oneonta, NY, USA) which had been precoated with poly-l-lysine (Sigma-Aldrich). Cells were incubated overnight at 37 °C in ambient CO2.
Membrane potential was measured using a FLIPR membrane potential assay kit (blue) from Molecular Devices (Sunnyvale, CA, USA), as described previously [55]. The dye was reconstituted with assay buffer of composition (mM): NaCl 145, HEPES 22, Na2HPO4 0.338, NaHCO3 4.17, KH2PO4 0.441, MgSO4 0.407, MgCl2 0.493, CaCl2 1.26, glucose 5.56, and bovine serum albumin (0.1 mg/mL, pH 7.4, osmolarity 315 ± 5). Prior to the assay, cells were loaded with 90 μL/well of the dye solution without removal of the L-15, giving an initial assay volume of 180 μL/well. Plates were then incubated at 37 °C at ambient CO2 for 60 min. Fluorescence was measured using a FlexStation 3 (Molecular Devices) microplate reader with cells excited at a wavelength of 530 nm and emission measured at 565 nm. Baseline readings were taken every 2 s for at least 2 min, at which time either drug or vehicle was added in a volume of 20 μL. The background fluorescence of cells without dye or dye without cells was negligible. Changes in fluorescence were expressed as a percentage of baseline fluorescence after subtraction of the changes produced by vehicle addition, which was less than 2 % for drugs dissolved in assay buffer or DMSO. The final concentration of DMSO was always 0.1 %.
Data were analyzed with PRISM (GraphPad Software Inc., San Diego, CA), using four-parameter nonlinear regression to fit concentration–response curves. In all plates, a maximally effective concentration (1 µM) of CP 55,940 (Cayman Chemical, Ann Arbor, MI, USA) was added to allow for normalization between assays.
Results and discussion
The synthesis of 3–10 started with indazole-3-carboxylic acid (11), which was subjected to Fischer esterification to give the corresponding methyl ester (12). Alkylation of 12 with the appropriate bromoalkane, in the presence of potassium tert-butoxide in THF at ambient temperature or potassium carbonate in acetonitrile at reflux, produced a mixture of major 1-alkyl (13–16) and minor 2-alkyl (17–20) regioisomeric intermediates. Although 1-alkylated products 13–16 always predominated, regardless of choice of base, solvent, and temperature, the overall yield of minor 2-alkylated intermediates 17–20 was greater using potassium carbonate than potassium tert-butoxide. Saponification of esters 13–20 gave the corresponding acids 21–28, which were coupled to l-valinamide using PyBOP and DIPEA to furnish 1H-indazoles 3–6 and their 2H-regioisomers 7–10 (Fig. 2).
The alkylation of methyl 1H-indazole-3-carboxylate (12) proceeded regioselectively to favour the formation of 1-substituted 1H-indazole intermediates as major products, entirely consistent with the known reactivity of indazoles towards nucleophilic substitution [56–58]. To quantify the regioselectivity of each alkylation method, the isolated yields of the regioisomeric products obtained following alkylation of 12 under each set of conditions for two representative bromoalkanes were determined are shown in Table 1. All reactions were performed in parallel, on the same scale, using identical reagents. Alkylation of 12 with 4-fluorobenzyl bromide or 1-bromopentane using potassium tert-butoxide as the base produced 14 and 15, respectively, as the major products with excellent regioselectivity for the 1-position (49:1 and 12.3:1, respectively). The corresponding 2-alkylated regiosiomers (18 and 19, respectively) were obtained as minor products in <10 % yield. While use of potassium carbonate as a base also furnished 14 and 15 as the major products, regioselectivity was reduced and appreciable quantities of 18 and 19 were obtained. The latter set of conditions (general procedure B) required flash chromatography to separate regiosiomeric intermediates of similar polarity and retention time; so it is unlikely that this method is used on an industrial scale to produce this class of SCs. In contrast, general procedure A proceeded with high regioselectivity, and it is feasible that incomplete purification of the crude product from this reaction allows 2-alkyl-2H-indazole intermediate to be carried through the synthesis, resulting in the production of regioisomeric SCs.
All SCs were analyzed using GC–MS, and total ion current chromatograms (TICs) and EI mass spectra for (S)-N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-alkyl-1H-indazole-3-carboxamides (3–6) and the corresponding 2-alkyl-2H-indazole regioisomers (7–10) are shown in Figs. 3 and 4, respectively.

Gas chromatography–mass spectrometry (GC–MS) analyses showing total ion current chromatograms (TICs) (left) and electron ionization (EI)-mass spectra (right) for a 3, b 4, c 5, and d 6. Proposed fragmentation ions are superimposed

GC–MS analyses showing TICs (left) and EI-mass spectra (right) for a 7, b 8, c 9, and d 10. Proposed fragmentation ions are superimposed
All 1-alkyl regioisomers failed to show molecular ion peaks, but similar fragmentation patterns were evident in each case. For AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA, the most prominent fragment ions could be attributed to cleavage of the terminal carboxamide subunit of the pendant l-valinamide group (m/z 312, 324, 286, and 304 respectively), as well as C–N scission of the indazole-3-carboxamide bond (m/z 241, 253, 215, and 233). 5F-AB-PINACA also showed a peak at m/z 213, consistent with a protonated species arising from defluorination of fragment m/z 233.
In the cases of AB-CHMINACA, AB-PINACA, and 5F-AB-PINACA, a common fragment of 145 mass units was observed, likely due to the protonated species arising from indazole-3-carboxamide bond scission and loss of the 1-alkyl group. AB-FUBINACA was devoid of peak at m/z 145, but did feature the 4-fluorobenzyl fragmentation ion (m/z 109).
The fragmentation patterns for the corresponding 2-alkyl-2H-indazoles 7–10 were distinct from their 1-alkyl regioisomers. Only in the case of AB-FUBINACA 2-isomer, the molecular ion (m/z 368) and fragment corresponding to cleavage of the terminal carboxamide group (m/z 324) were observed. Again, common fragment m/z 145 arising from dealkylated indazole-3-acylium ion could be seen in the spectra of 7, 9, and 10. A fragment corresponding to scission of the entire 3-substituent of indazoles 7, 9, and 10 (m/z 213, 187, and 205 respectively) was present as the base peak in these spectra. Fragmentation of the neighboring amide bond proximal to the indazole core was seen for 8, 9, and 10 (m/z 253, 215, and 233 respectively), but not for 7.
The UV–Vis absorption spectra of 3–10 are shown in Fig. 5, and peak absorbances are similar for each 1-alkyl-1H-indazoles and their regioisomers, indicating that ultraviolet spectroscopy is not a suitable method for differentiation of regiosiomeric indazole cannabinoids of this class.

Ultraviolet absorption spectra of a 3, b 4, c 5, d 6, e 7, f 8, g 9, and h 10
The cannabinoid activities of indazole SCs 3–10 at CB1 and CB2 receptors are shown in Table 2. Murine AtT-20 neuroblastoma cells were stably transfected with human CB1 or CB2 receptors, and activities of CP 55,940 and 3–10 were evaluated using FLIPR membrane potential assays whereby endogenously expressed G protein-gated inwardly rectifying K+ channels (GIRKs) are activated by agonists at the coexpressed CB1 or CB2 receptors [55, 59]. The maximum effects of 3–10 were compared to the high efficacy CB1/CB2 receptor agonist CP 55,940, which produced a maximal decrease in fluorescence, corresponding to cellular hyperpolarization, of 14 ± 2 % in the AtT-20-CB1 cells and 33 ± 2 % in the AtT-20-CB2 cells.
AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA, and their 2-alkylated regiosiomers displayed distinct profiles at CB1 and CB2 receptors. The 1-alkyl isomers AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA showed potent activity at CB1 receptors, with nanomolar EC50 values (EC50 = 2.1–7.8 nM) exceeding that of CP 55,940 itself. Moreover, 3–6 were highly efficacious CB1 agonists, eliciting 132–151 % of the response achieved by 1 µM CP 55,940. The 3–6 showed slightly lower potency at CB2 receptors (1.5–3.2 times less), but were still potent, efficacious agonists.
In contrast, the 2-regioisomers 7–10 were not potent at stimulating CB1 and CB2 receptor coupling to GIRKs. EC50 values could not be determined for 7 or 8 at either receptor, and 9 and 10 showed only micromolar potency for CB1 (EC50 = 4080 and 1930 nM, respectively) and CB2 (EC50 = 4120 and 2980 nM, respectively).
Of the 2-alkyl-2H-indazoles, 7, 9, and 10, displayed a maximum effect at CB1 receptors comparable to 1 µM CP 55,940 at the highest concentration tested, 30 µM. At 30 µM, 8 achieved only 61 % of the CP 55,940 hyperpolarization. At CB2 receptors, 30 µM of 9 elicited a response equivalent to 1 µM CP 55,940, while 7, 8, and 10 at 30 µM achieved only 25–47 % of the maximum.
The dramatic difference in potency and efficacy of the 1-alkyl-1H-indazoles 3–6 and 2-alkyl-2H-indazoles 7–10 at CB1 and CB2 receptors is shown in Fig. 6a, b respectively. The non-linear regression fit of CP 55,940 is included as a dashed line for comparison. The 1-alkyl compounds 3–6 (white data points) were all more potent and efficacious than CP 55,940 at CB1 receptors, while 2-alkyl regioisomers 7–10 (black data points) were substantially less potent, with three reaching efficacy comparable to maximum CP 55,940 only at the highest concentration tested (Fig. 6a). At CB2 receptors, 3–6 demonstrate potency and efficacy comparable to CP 55,940, but 7–10, with the exception of 9, show efficacy approximately 20–40 % that of a maximally efficacious concentration of CP 55,940 (Fig. 6b).

Hyperpolarization of a CB1 and b CB2 receptors induced by 3–10 as a proportion of that produced by 1 µM CP 55,940. Membrane potential was measured using a fluorescent dye, as outlined in the text. Each point represents the mean ± standard error of at least five independent determinations, each performed in duplicate. Data was fitted with a four parameter logistic equation in GraphPad Prism
Conclusions
This study represents the first pharmacological characterization of the 2-alkyl-2H-indazole regioisomers of SC designer drugs AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA. A general synthetic route to both 1-alkyl-1H- and 2-alkyl-2H-indazole-3-carboxamides was demonstrated, and may be of use to cannabinoid researchers for the preparation of related analogues. Additionally, characteristic differences in the fragmentation patterns of these regioisomers were described, enabling their differentiation by GC–MS.
AB-CHMINACA, AB-FUBINACA, AB-PINACA, 5F-AB-PINACA, and the corresponding 2-alkyl-2H-indazole regioisomers 7–10 were evaluated for their activity at human CB1 and CB2 receptors in vitro using FLIPR membrane potential assays. AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA are potent, efficacious agonists of CB1 and CB2 receptors, while the corresponding 2-alkyl-2H-indazole regioisomers possessed only low potency as CB1/CB2 agonists. Having demonstrated the weak cannabimimetic properties of the 2-alky-2H-indazole regioisomers of AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA, and the synthesis of both regioisomers from a common precursor, it is possible that 2-alky-2H-indazole-3-carboxamide SCs occur in recreational products as synthesis byproducts of 1-alky-1H-indazole-3-carboxamides rather than analogues produced deliberately as SCs in their own right.
References
Brandt SD, King LA, Evans-Brown M (2014) The new drug phenomenon. Drug Test Anal 6:587–597
Auwärter V, Dresen S, Weinmann W, Muller M, Putz M, Ferreiros N (2009) ‘Spice’ and other herbal blends: harmless incense or cannabinoid designer drugs? J Mass Spectrom 44:832–837
Uchiyama N, Kikura-Hanajiri R, Kawahara N, Goda Y (2009) Identification of a cannabimimetic indole as a designer drug in a herbal product. Forensic Toxicol 27:61–66
European Monitoring Centre for Drugs and Drug Addiction (2015) EMCDDA–Europol 2014 annual report on the implementation of council decision 2005/387/JHA, implementation reports. Publications Office of the European Union, Luxembourg. doi:10.2810/112317. Accessed 1 Feb 1, 2016
Kikura-Hanajiri R, Uchiyama N, Kawamura M, Goda Y (2014) Changes in the prevalence of new psychoactive substances before and after the introduction of the generic scheduling of synthetic cannabinoids in Japan. Drug Test Anal 6:832–839
Zawilska JB, Andrzejczak D (2015) Next generation of novel psychoactive substances on the horizon—a complex problem to face. Drug Alcohol Depend 157:1–17
Blakey K, Boyd S, Atkinson S, Wolf J, Slottje PM, Goodchild K, McGowan J (2015) Identification of the novel synthetic cannabimimetic 8-quinolinyl 4-methyl-3-(1-piperidinylsulfonyl)benzoate (QMPSB) and other designer drugs in herbal incense. Forensic Sci Int 260:40–53
Kondrasenko AA, Goncharov EV, Dugaev KP, Rubaylo AI (2015) CBL-2201. Report on a new designer drug: naphth-1-yl 1-(5-fluoropentyl)-1H-indole-3-carboxylate. Forensic Sci Int 257:209–213
McLaughlin G, Morris N, Kavanagh PV, Power JD, Twamley B, O’Brien J, Talbot B, Dowling G, Brandt SD (2015) The synthesis and characterization of the ‘research chemical’ N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(cyclohexylmethyl)-3-(4-fluorophenyl)-1H-pyrazole-5-carboxamide (3,5-AB-CHMFUPPYCA) and differentiation from its 5,3-regioisomer. Drug Test Anal. doi:10.1002/dta.1864
Uchiyama N, Asakawa K, Kikura-Hanajiri R, Tsutsumi T, Hakamatsuka T (2015) A new pyrazole-carboxamide type synthetic cannabinoid AB-CHFUPYCA [N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(cyclohexylmethyl)-3-(4-fluorophenyl)-1H-pyrazole-5-carboxamide] identified in illegal products. Forensic Toxicol 33:367–373
Westphal F, Sönnichsen FD, Knecht S, Auwärter V, Huppertz L (2015) Two thiazolylindoles and a benzimidazole: novel compounds on the designer drug market with potential cannabinoid receptor activity. Forensic Sci Int 249:133–147
Qian Z, Hua Z, Liu C, Jia W (2016) Four types of cannabimimetic indazole and indole derivatives, ADB-BINACA, AB-FUBICA, ADB-FUBICA, and AB-BICA, identified as new psychoactive substances. Forensic Toxicol 34:133–143
Shevyrin V, Melkozerov V, Eltsov O, Shafran Y, Morzherin Y (2016) Synthetic cannabinoid 3-benzyl-5-[1-(2-pyrrolidin-1-ylethyl)-1H-indol-3-yl]-1,2,4-oxadiazole. The first detection in illicit market of new psychoactive substances. Forensic Sci Int 259:95–100
Lobo Vicente J, Chassaigne H, Holland MV, Reniero F, Kolar K, Tirendi S, Vandecasteele I, Vinckier I, Guillou C (2016) Systematic analytical characterization of new psychoactive substances: a case study. Forensic Sci Int 265:107–115
Buchler IP, Hayes MJ, Hedge SG, Hockerman SL, Jones DE, Kortum SW, Rico JG, Tenbrink RE, Wu KK (2009) Indazole derivatives as CB1 receptor modulators and their preparation and use in treatment of diseases. Patent WO 2009/106980
Buchler IP, Hayes MJ, Hedge SG, Hockerman SL, Jones DE, Kortum SW, Rico JG, Tenbrink RE, Wu KK (2009) Indazole derivatives as CB1 receptor modulators and their preparation and use in the treatment of CB1-mediated diseases. Patent WO 2009/106982
Centers for Disease Control and Prevention (2013) Notes from the field: severe illness associated with synthetic cannabinoid use-Brunswick, Georgia, 2013. Morb Mortal Wkly Rep 62:939
Centers for Disease Control and Prevention (2013) Notes from the field: severe illness associated with reported use of synthetic marijuana-Colorado, August–September 2013. Morb Mortal Wkly Rep 62:1016–1017
Hasegawa K, Wurita A, Minakata K, Gonmori K, Nozawa H, Yamagishi I, Watanabe K, Suzuki O (2014) Postmortem distribution of AB-CHMINACA, 5-fluoro-AMB, and diphenidine in body fluids and solid tissues in a fatal poisoning case: usefulness of adipose tissue for detection of the drugs in unchanged forms. Forensic Toxicol 33:45–53
Monte AA, Bronstein AC, Cao DJ, Heard KJ, Hoppe JA, Hoyte CO, Iwanicki JL, Lavonas EJ (2014) An outbreak of exposure to a novel synthetic cannabinoid. N Engl J Med 370:389–390
Hess C, Stockhausen S, Kernbach-Wighton G, Madea B (2015) Death due to diabetic ketoacidosis: induction by the consumption of synthetic cannabinoids? Forensic Sci Int 257:e6–e11
Schwartz MD, Trecki J, Edison LA, Steck AR, Arnold JK, Gerona RR (2015) A common source outbreak of severe delirium associated with exposure to the novel synthetic cannabinoid ADB-PINACA. J Emerg Med 48:573–580
Shevyrin V, Melkozerov V, Nevero A, Eltsov O, Shafran Y, Morzherin Y, Lebedev AT (2015) Identification and analytical characteristics of synthetic cannabinoids with an indazole-3-carboxamide structure bearing a N-1-methoxycarbonylalkyl group. Anal Bioanal Chem 407:6301–6315
Labay LM, Caruso JL, Gilson TP, Phipps RJ, Knight LD, Lemos NP, McIntyre IM, Stoppacher R, Tormos LM, Wiens AL, Williams E, Logan BK (2016) Synthetic cannabinoid drug use as a cause or contributory cause of death. Forensic Sci Int 260:31–39
Shanks KG, Clark W, Behonick G (2016) Death associated with the use of the synthetic cannabinoid ADB-FUBINACA. J Anal Toxicol. doi:10.1093/jat/bkv142
Uchiyama N, Shimokawa Y, Kawamura M, Kikura-Hanajiri R, Hakamatsuka T (2014) Chemical analysis of a benzofuran derivative, 2-(2-ethylaminopropyl)benzofuran (2-EAPB), eight synthetic cannabinoids, five cathinone derivatives, and five other designer drugs newly detected in illegal products. Forensic Toxicol 32:266–281
Langer N, Lindigkeit R, Schiebel H-M, Papke U, Ernst L, Beuerle T (2015) Identification and quantification of synthetic cannabinoids in “spice-like” herbal mixtures: update of the German situation for the spring of 2015. Forensic Toxicol 34:94–107
Uchiyama N, Matsuda S, Wakana D, Kikura-Hanajiri R, Goda Y (2013) New cannabimimetic indazole derivatives, N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-pentyl-1H-indazole-3-carboxamide (AB-PINACA) and N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(4-fluorobenzyl)-1H-indazole-3-carboxamide (AB-FUBINACA) identified as designer drugs in illegal products. Forensic Toxicol 31:93–100
European Monitoring Centre for Drugs and Drug Addiction (2014) EMCDDA–Europol 2013 annual report on the implementation of council decision 2005/387/JHA, implementation reports. Publications Office of the European Union, Luxembourg. doi:10.2810/51986. Accessed 1 Feb 2016
Wilkinson SM, Banister SD, Kassiou M (2015) Bioisosteric fluorine in the clandestine design of synthetic cannabinoids. Aust J Chem 68:4–8
Banister SD, Moir M, Stuart J, Kevin RC, Wood KE, Longworth M, Wilkinson SM, Beinat C, Buchanan AS, Glass M, Connor M, McGregor IS, Kassiou M (2015) Pharmacology of indole and indazole synthetic cannabinoid designer drugs AB-FUBINACA, ADB-FUBINACA, AB-PINACA, ADB-PINACA, 5F-AB-PINACA, 5F-ADB-PINACA, ADBICA, and 5F-ADBICA. ACS Chem Neurosci 6:1546–1559
Wiley JL, Marusich JA, Lefever TW, Antonazzo KR, Wallgren MT, Cortes RA, Patel PR, Grabenauer M, Moore KN, Thomas BF (2015) AB-CHMINACA, AB-PINACA, and FUBIMINA: affinity and potency of novel synthetic cannabinoids in producing Δ9-tetrahydrocannabinol-like effects in mice. J Pharmacol Exp Ther 354:328–339
Gatch MB, Forster MJ (2015) Δ9-Tetrahydrocannabinol-like effects of novel synthetic cannabinoids found on the gray market. Behav Pharmacol 26:460–468
Thornton SL, Akpunonu P, Glauner K, Hoehn KS, Gerona R (2015) Unintentional pediatric exposure to a synthetic cannabinoid (AB-PINACA) resulting in coma and intubation. Ann Emerg Med 66:343–344
Trecki J, Gerona RR, Schwartz MD (2015) Synthetic cannabinoid-related illnesses and deaths. N Engl J Med 373:103–107
Tyndall JA, Gerona R, De Portu G, Trecki J, Elie MC, Lucas J, Slish J, Rand K, Bazydlo L, Holder M, Ryan MF, Myers P, Iovine N, Plourde M, Weeks E, Hanley JR, Endres G, St Germaine D, Dobrowolski PJ, Schwartz M (2015) An outbreak of acute delirium from exposure to the synthetic cannabinoid AB-CHMINACA. Clin Toxicol 53:950–956
Drug Enforcement Administration, Department of Justice (2014) Schedules of controlled substances: temporary placement of four synthetic cannabinoids into Schedule I. Final order. Fed Regist 79:7577–7582
Drug Enforcement Administration, Department of Justice (2015) Schedules of controlled substances: temporary placement of three synthetic cannabinoids into schedule I. Final order. Fed Regist 80:5042–5047
Erratico C, Negreira N, Norouzizadeh H, Covaci A, Neels H, Maudens K, van Nuijs AL (2015) In vitro and in vivo human metabolism of the synthetic cannabinoid AB-CHMINACA. Drug Test Anal 7:866–876
Wurita A, Hasegawa K, Minakata K, Gonmori K, Nozawa H, Yamagishi I, Suzuki O, Watanabe K (2016) Identification and quantification of metabolites of AB-CHMINACA in a urine specimen of an abuser. Leg Med 19:113–118
Takayama T, Suzuki M, Todoroki K, Inoue K, Min JZ, Kikura-Hanajiri R, Goda Y, Toyo’oka T (2014) UPLC/ESI-MS/MS-based determination of metabolism of several new illicit drugs, ADB-FUBINACA, AB-FUBINACA, AB-PINACA, QUPIC, 5F-QUPIC and α-PVT, by human liver microsome. Biomed Chromatogr 28:831–838
Castaneto MS, Wohlfarth A, Pang S, Zhu M, Scheidweiler KB, Kronstrand R, Huestis MA (2015) Identification of AB-FUBINACA metabolites in human hepatocytes and urine using high-resolution mass spectrometry. Forensic Toxicol 33:295–310
Vikingsson S, Green H, Brinkhagen L, Mukhtar S, Josefsson M (2015) Identification of AB-FUBINACA metabolites in authentic urine samples suitable as urinary markers of drug intake using liquid chromatography quadrupole tandem time of flight mass spectrometry. Drug Test Anal. doi:10.1002/dta.1896
Hsin-Hung Chen M, Dip A, Ahmed M, Tan ML, Walterscheid JP, Sun H, Teng BB, Mozayani A (2016) Detection and characterization of the effect of AB-FUBINACA and its metabolites in a rat model. J Cell Biochem 117:1033–1043
Jang M, Shin I, Kim J, Yang W (2015) Simultaneous quantification of 37 synthetic cannabinoid metabolites in human urine by liquid chromatography-tandem mass spectrometry. Forensic Toxicol 33:221–234
Wohlfarth A, Castaneto MS, Zhu M, Pang S, Scheidweiler KB, Kronstrand R, Huestis MA (2015) Pentylindole/pentylindazole synthetic cannabinoids and their 5-fluoro analogs produce different primary metabolites: metabolite profiling for AB-PINACA and 5F-AB-PINACA. AAPS J 17:660–677
Namera A, Kawamura M, Nakamoto A, Saito T, Nagao M (2015) Comprehensive review of the detection methods for synthetic cannabinoids and cathinones. Forensic Toxicol 33:175–194
DeRuiter J, Smith FT, Abdel-Hay K, Clark CR (2014) Analytical differentiation of 1-alkyl-3-acylindoles and 1-acyl-3-alkylindoles: isomeric synthetic cannabinoids. Anal Chem 86:3801–3808
Shevyrin V, Melkozerov V, Nevero A, Eltsov O, Morzherin Y, Shafran Y (2014) 3-Naphthoylindazoles and 2-naphthoylbenzoimidazoles as novel chemical groups of synthetic cannabinoids: chemical structure elucidation, analytical characteristics and identification of the first representatives in smoke mixtures. Forensic Sci Int 242:72–80
Girreser U, Rosner P, Vasilev A (2015) Structure elucidation of the designer drug N-(1-amino-3,3-dimethyl-1-oxobutan-2-yl)-1-(5-fluoropentyl)-3-(4-fluorophenyl)-pyrazole-5-carboxamide and the relevance of predicted C NMR shifts—a case study. Drug Test Anal. doi:10.1002/dta.1820
Uchiyama N, Shimokawa Y, Kikura-Hanajiri R, Demizu Y, Goda Y, Hakamatsuka T (2015) A synthetic cannabinoid FDU-NNEI, two 2-indazole isomers of synthetic cannabinoids AB-CHMINACA and NNEI indazole analog (MN-18), a phenethylamine derivative N-OH-EDMA, and a cathinone derivative dimethoxy-α-PHP, newly identified in illegal products. Forensic Toxicol 33:244–259
Jia W, Meng X, Qian Z, Hua Z, Li T, Liu C (2016) Identification of three cannabimimetic indazole and pyrazole derivatives, APINACA 2H-indazole analogue, AMPPPCA, and 5F-AMPPPCA. Drug Test Anal. doi:10.1002/dta.1967
Choi H, Heo S, Choe S, Yang W, Park Y, Kim E, Chung H, Lee J (2013) Simultaneous analysis of synthetic cannabinoids in the materials seized during drug trafficking using GC-MS. Anal Bioanal Chem 405:3937–3944
Banister SD, Wilkinson SM, Longworth M, Stuart J, Apetz N, English K, Brooker L, Goebel C, Hibbs DE, Glass M, Connor M, McGregor IS, Kassiou M (2013) The synthesis and pharmacological evaluation of adamantane-derived indoles: cannabimimetic drugs of abuse. ACS Chem Neurosci 4:1081–1092
Knapman A, Santiago M, Du YP, Bennallack PR, Christie MJ, Connor M (2013) A continuous, fluorescence-based assay of µ-opioid receptor activation in AtT-20 cells. J Biomol Screen 18:269–276
Yet L (2009) Chapter 5.4: five-membered ring systems: with more than one N atom. In: Gordon WG, John AJ (eds) Progress in heterocyclic chemistry, vol 20. Elsevier, Amsterdam, pp 190–219
Hunt KW, Moreno DA, Suiter N, Clark CT, Kim G (2009) Selective synthesis of 1-functionalized-alkyl-1H-indazoles. Org Lett 11:5054–5057
Saenz J, Mitchell M, Bahmanyar S, Stankovic N, Perry M, Craig-Woods B, Kline B, Yu S, Albizati K (2007) Process development and scale-up of AG035029. Org Process Res Dev 11:30–38
Grimsey NL, Graham ES, Dragunow M, Glass M (2010) Cannabinoid receptor 1 trafficking and the role of the intracellular pool: implications for therapeutics. Biochem Pharmacol 80:1050–1062
Acknowledgments
Work was supported in part by the European Union’s Seventh Framework Programme [FP7/2007-2013] INMiND (Grant Agreement No. HEALTH-F2-2011-278850) and NHMRC Project Grant 1107088.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no financial or other relations that could lead to a conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
M. Longworth and S. D. Banister contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Longworth, M., Banister, S.D., Mack, J.B.C. et al. The 2-alkyl-2H-indazole regioisomers of synthetic cannabinoids AB-CHMINACA, AB-FUBINACA, AB-PINACA, and 5F-AB-PINACA are possible manufacturing impurities with cannabimimetic activities. Forensic Toxicol 34, 286–303 (2016). https://doi.org/10.1007/s11419-016-0316-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-016-0316-y