
Abstract
Chemical investigations of higher plants, with particular attention paid to their steroidal glycosides, present a promising approach for generating anti-cancer agents from natural products. We conducted a systematic phytochemical investigation of nine higher plants—whole plants and rhizomes of Convallaria majalis, whole plants of Agave utahensis, roots of Adonis amurensis, seeds of Adonis aestivalis, bulbs of Bessera elegans, bulbs of Fritillaria meleagris, seeds of Digitalis purpurea, underground parts of Yucca glauca, and bulbs of Lilium pumilum—which led to the discovery of novel steroidal glycosides. The structures of these new constituents were determined based on spectroscopic data and chemical transformations. The identification of the monosaccharides including their absolute configurations was carried out by direct HPLC analysis of their hydrolysates using an optical rotation detector. Cytotoxicity of the isolated steroidal glycosides was evaluated against various tumor cells (A549, ACHN, HepG-2, HL-60, HSC-2, HSC-3, HSC-4, HSG, and SBC-3) and normal cells (Fa2 N-4, HK-2, and TIG-3 cells). Certain steroidal glycosides exhibit selective cytotoxicity and synergistic effects, making them potential lead compounds for use as anti-cancer agents. We document the isolation of 139 steroidal glycosides from higher plants and assessment their cytotoxic activities.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Steroidal glycosides constitute a group of natural products found in a limited range of plant families, including Agavaceae, Leguminosae, Liliaceae, Plantaginaceae, Ranunculaceae, and Solanaceae. Predominantly present as glycosides in nature, steroidal glycosides display a diverse array of chemical structures. These compounds are categorized as spirostanol, furostanol, pregnane, cardenolide, and cardiac glycosides, based on their aglycone structures. Furthermore, steroidal glycosides from higher plants have demonstrated various biological activities, including antitumor, antidiabetic, antitussive, and inhibition of platelet aggregation [1, 2]. In vitro studies have identified cytotoxic activities against tumor cell lines including A549, Caco-2, Hela, HepG-2, HL-60, HT-29, MCF-7, PC-3, SGC-7901, and U251, showing mechanisms, such as apoptosis, necrosis, autophagy, ferroptosis, and cell cycle arrest [3,4,5,6]. Terrestrosin D, a spirostanol glycoside isolated from Tribulus terrestris, has shown to induce cell cycle arrest in the G1 phase and stimulate the caspase-independent apoptosis of PC-3 cells [5]. A novel spirostanol glycoside T-17, isolated from Tupistra chinensis, induces apoptosis and triggers cytoprotective autophagy in human gastric cancer cell lines by activating the JNK pathway [7]. Thus, a large amount of cytotoxic data against a variety of tumor cells were performed and the molecular mechanism of steroidal glycosides was elucidated. Although in vivo experiments on their anti-cancer activities are limited, certain steroidal glycosides have been shown to exhibit anti-cancer effects. For instance, oleandrin (administered at 0.3 or 0.6 mg/kg, i.p., 7 days) suppressed tumor growth in mice bearing EMT6 murine mammary carcinoma [8]. In another study, cotreatment of glioma bearing mice with temozolomide (TMZ) and oleandrin (administered at 0.3 mg/kg, i.p., 100 days) strongly prolongs mice survival compared with TMZ treatment alone [9]. Garofalo et al. demonstrated the potential of oleandrin as a co-adjuvant drug in standard chemotherapeutics. Additionally, OSW-1, a steroidal cholestane diglycoside isolated from Ornithogalum saundersiae, significantly boosted the anti-metastatic ability of doxorubicin in 4T1 mice and enhanced CD8+ T cell infiltration into the immune microenvironment of the lungs [10].
Immune checkpoint inhibitors (ICIs) and molecular targeted therapeutic agents have emerged as promising tools for cancer therapy. However, resistance to ICIs can develop in some patients, leading to a lack of an objective response [11]. Between 1981 and 2019, 25% of all the anti-cancer drugs approved by the Food and Drug Administration were derived from natural products or natural product derivatives [12], highlighting the potential of novel anti-cancer agents in chemotherapy. Steroidal glycosides might be considered as lead compounds for preclinical studies in cancer chemotherapy.
This review presents our previous phytochemical studies on steroidal glycosides from nine plant sources: the whole plants and rhizomes of Convallaria majalis (Liliaceae) [13,14,15], whole plants of Agave utahensis (Agavaceae) [16,17,18], roots of Adonis amurensis (Ranunculaceae) [19, 20], seeds of Adonis aestivalis (Ranunculaceae) [21, 22], bulbs of Bessera elegans (Liliaceae) [23], bulbs of Fritillaria meleagris (Liliaceae) [24], seeds of Digitalis purpurea (Scrophulariaceae) [25,26,27,28], underground parts of Yucca glauca (Agavaceae)[29], and bulbs of Lilium pumilum (Liliaceae) [30]. These studies also investigated the cytotoxic activities of the isolated compounds against tumor cells and normal cells. Plants belonging to the families Liliaceae and Ranunculaceae are particularly rich sources of bioactive steroidal glycosides, such as OSW-1 or galtonioside A, which exhibit cytotoxic activity against tumor cells [31,32,33]. Furthermore, potent cytotoxic compounds have been discovered in popular ornamental garden plants, such as Bessera elegans and Fritillaria meleagris [23, 24]. These results indicate that garden plants with no proven medicinal backgrounds may also be potential sources of novel drugs.
Pregnane glycosides
Pregnane glycosides, initially isolated from Digitalis purpurea and termed “digitanol glycosides”, are now found in various Apocynaceae, Asclepiadaceae, Malpighiaceae, Ranunculaceae, and Zygophyllaceae species [34]. Structurally, pregnane glycosides consist of six-membered A–C rings and a five-membered D ring in the C21 steroid skeleton, commonly featuring deoxy sugars linked to C-3 of the aglycone. The C/D cis structure of pregnane glycoside was synthesized via the cardenolide route. Based on the substituents and degree of oxidative cracking of the aglycone ring, the skeleton of the aglycone structure is divided into diverse structures. Various types of pregnane skeletons show various biological activities, including antibacterial, antidiabetic, anti-inflammatory, anti-obesity, and antitumor activities [34].
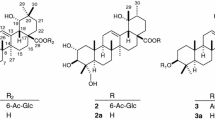
From the seeds of D. purpurea, three novel rearranged 11,12-secopregnane glycosides, digipregnosides A (1), B (2), and C (3), and two novel 12,20-epoxypregnane glycosides, digipregnosides D (4) and E (5), were isolated (Fig. 1) [28]. The chemical structures of 1–5 were determined based on extensive spectroscopic analyses, including long-range heteronuclear single quantum multiple bond correlation (HSQMBC) spectral data and hydrolytic cleavage results. The HSQMBC spectrum provides more than 3JC,H coupling correlations and suggested a novel skeleton of 1. A plausible biogenetic pathway of the novel rearranged 11,12-secopregnane skeleton is proposed (Fig. 2), wherein the aglycone moiety of 1 is presumed to be biosynthesized from that of 4 (4a). After the C-11/C-12 bond of ring C was oxidatively cleaved, the hydroxy group of the C-11 carboxy functionality attacks the C-15 carbonyl group, resulting in the formation of γ- and δ-lactone rings. Evaluation of 1 and 3–5 for their cytotoxic activities against SBC-3 small cell lung carcinoma cells and TIG-3 normal human fibroblast cells was performed using a modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. Compounds 1, 4, and 5 exhibited cytotoxic activity against SBC-3 cells, with IC50 values of 1.3, 0.17, and 2.1 μM, respectively. Notably, 1 and 4 showed tumor-selective cytotoxicity against SBC-3 cells at 0.001 μM, inducing apoptotic cell death through caspase-3/7 activation [28].

Structures of pregnane glycosides 1–5 isolated from the seeds of Digitalis purpurea and 6 and 7 from the whole plants of Convallaria majalis

A possible biosynthetic pathway of the aglycone moiety of 1 from 4a
Chemical transformations were conducted to support the chemical structure determination of steroidal glycosides, including spirostanol, furostanol, pseudo-furostanol, and pregnane (Fig. 3). The new pregnane glycoside (6), isolated from the whole plants of Convallaria majalis, was formed from a pseudo-furostanol glycoside through the oxidative cleavage of the C-20(22) double bond. This was confirmed by the fact that the peracetate (6b) of 6 was identical to the product obtained by treating furostanol glycoside (124) with acetic anhydride (Ac2O) in pyridine at 110 ℃ for 3 h followed by treatment with CrO3 in acetic acid (AcOH). Compound 6 was subjected to alkaline methanolysis with 7% sodium methoxide to obtain pregnane glycoside (7) and 5-[(β-D-glucopyranosyl)oxy]-4-methyl pentanoic acid (6a). Conversely, acid hydrolysis of 124 with 1 M HCl yielded a spirostanol steroid (107a) as the aglycone and D-galactose, D-glucose, and D-xylose as carbohydrate moieties [14]. Identification of the monosaccharides was conducted by direct HPLC analysis of the hydrolysate using a combination of refractive index and optical rotation detector. Compounds 6 and 7, isolated from the whole plants of Convallaria majalis, did not show cytotoxic activity against HL-60 human promyelocytic leukemia cells, A549 human lung adenocarcinoma cells, HSC-4, and HSC-2 human oral squamous cell carcinoma cells (IC50 > 12 μM).

Chemical transformations of 6
Amurensiosides A–K (8–18), isolated from the roots of Adonis amurensis, are newly discovered tetra-, hexa-, and heptaglycosides that contain deoxysugars (D-cymarose and D-diginose), characteristic pregnane and cardiotonic glycosides in plants (Fig. 4) [19]. The configuration of C-17 was confirmed via NOESY correlation between H-17 and Me-18. Aglycones of 13, 17, and 18 represent novel polyoxygenated pregnane derivatives. The aglycone of 15, adonilide, is a hexacyclic pregnane featuring a five-membered lactone group and an ether linkage between C-14 and C-20. While the aglycones of 15 (adonilide) and 16 (fukujusonorone) were previously reported in the 1960s [35, 36], their glycosides remain unreported. Among 8–18, compounds 8, 9, 12, and 13 exhibited cytotoxicity solely toward HSC-2 cells (IC50 66, 26, 47, and 58 μg/mL, respectively). This result indicates that replacing the benzoyl group with the nicotinoyl group at C-12 reduced the cytotoxic activity.

Structures of pregnane glycosides 8–18 isolated from the roots of Adonis amurensis
The aglycones of Aestivalosides A-L (19–30) were isolated from the seeds of Adonis aestivalis and previously undescribed adonilide derivatives (Fig. 5) [22]. Oligoglycosides attached to C-3 of the aglycone of Aestivalosides B-H (20–26) are novel di-, tri-, tetra-, and pentaglycosides containing the following deoxysugars: D- and L-cymarose, D-diginose, and D-oleandrose. Adonilide derivatives have been previously identified in a limited number of Adonis species, including A. amurensis and A. multiflora. This is the first report of pregnane glycosides from A. aestivalis [37,38,39]. Compounds 19–30 were not cytotoxic to HSC-2, HSC-3, HSC-4, and HL-60 cells, even at sample concentration of 90 μM.

Structures of pregnane glycosides 19–30 from the seeds of Adonis aestivalis
Cardenolide glycosides
Cardenolide glycosides are a group of naturally occurring compounds found in several plant families including Apocynaceae, Liliaceae, Ranunculaceae, and Scrophulariaceae. Cardenolide has α,β-unsaturated butyrolactone ring at C-17 of the aglycone, a 14β-hydroxy group at C-14, and a C/D cis ring connection in the C23 steroid skeleton. The oligoglycosides attached to the C-3 of the aglycones comprise deoxysugars, which are typical of plant pregnane and cardiotonic glycosides. Cardenolide glycosides selectively inhibit Na+/K+-ATPase pumps. Recent studies demonstrated the specific cytotoxicity of cardenolide glycosides against renal adenocarcinoma and hepatocellular carcinoma cells, indicating their potential as candidates for anti-cancer drug development. Strophanthidin, a cardiac glycoside isolated from Strophanthus kombe, induces apoptosis by attenuating multiple biochemical signaling pathways and arresting the cell cycle at the G2/M phase through p53-dependent and -independent mechanism in A549, HepG-2, and MCF-7 cells [40]. Furthermore, strophanthidin inhibited autophagy by inhibiting the expression of the LC3 and p62 complexes in A549 cells [3]. Oleandrin, a cardiac glycoside isolated from Nerium oleander, induces apoptosis in SW480 human colorectal cancer cells via the mitochondrial pathway, without significantly reducing the viability of NCM460 normal human colonic epithelial cells [4].
The structures of cardenolide glycosides (31–45), isolated from the seeds of Digitalis purpurea and including newly identified compounds (31, 38, and 41), contain digitoxigenin or gitoxigenin as the aglycone moiety, with D-glucose, D-digitoxose, and D-digitalose as sugar moieties (Fig. 6) [27]. Amurensiosides L-O (46–49), also newly discovered from the roots of Adonis amurensis, feature digitoxigenin as the aglycone, along with D-glucose, D-digitoxose, D-diginose, and D-cymarose as sugar moieties (Fig. 7) [20]. Among 31–49, compounds 32, 39, 41, 42, and 46–49 exhibit potent cytotoxic activity against HL-60 cells (IC50 0.034–0.069 μM). While the introduction of a hydroxy group at the C-16 position of the digitoxigenin aglycone reduced its cytotoxic activity (gitoxigenin > digitoxigenin), hydroxylation at the C-11α position shows no influence on the activity. 3-O-Methylation of the (inner) C-3’ hydroxy group at the fucopyranosyl moiety and 3-O-acetylation of the (inner) C-3’ hydroxy group at the digitoxopyranosyl moiety reduced cytotoxic activity. Conversely, glycosylation of the terminal glucosyl moiety of 35 and 39 had little effect on activity. As expected from a previous study [41], a steroidal aglycone (digitoxigenin) displayed weaker cytotoxic activity (IC50 0.22 μM) than corresponding glycoside 46 (IC50 0.057 μM) against HL-60 cells. Additionally, 32, 39, 41, and 42 exhibit greater cytotoxicity against ACHN human renal adenocarcinoma cells (IC50 0.02, 0.18, 0.08, and 0.47 μM, respectively) compared to HK-2 human embryonic kidney cells (IC50 0.16, 0.37, 0.24, and 0.76 μM, respectively) [25]. In addition, glucodigifucoside (32) demonstrated greater specificity against HepG-2 human liver cancer cells (IC50 0.21 μM) compared to that of Fa2 N-4 (IC50 1.25 μM). Notably, 32 did not induce typical apoptotic morphological changes or alter Bax or Bcl-2 expression levels in ACHN cells. Compound 32 exerted carcinoma-specific cytotoxicity via p53-induced p21/Cip1 expression, independent of the p53-evoked pro-apoptotic pathway. Compounds 46–49 (IC50 0.21–1.5μM) and digitoxigenin (IC50 0.35 μM) also showed potent cytotoxic activity against HSC-2 cells.

Structures of cardenolide glycosides 31–45 isolated from the seeds of Digitalis purpurea

Structures of cardenolide glycosides 46–49 and 55 from the roots of Adonis amurensis, 50–54 from the seeds of Adonis aestivalis, and 56 and 57 from the rhizomes of Convallaria majalis
5α-Hydroxy cardenolides (50–52) and 5β-hydroxy cardenolides (53 and 54) were isolated from the seeds of Adonis aestivalis (Fig. 7) [21]. These cardenolides consist of cardenolide or strophanthidin as the aglycone and D-oleandrose, D-diginose, D-digitoxose, and D-glucose as the carbohydrate components. In the 50–52, the 17β-hydroxy configuration was confirmed by the following NOE correlations: from Me-18 to the two hydroxy groups at C-14 and C-17/H-21β, respectively. Cardenolides 50–54 were assessed for their cytotoxic activity against HSC-2, HSC-3, and HSC-4 human oral squamous cell carcinoma and HL-60 cells, as well as against human non-malignant HGF gingival fibroblasts, HPLF periodontal ligament fibroblasts, and HPC pulp cells. Compounds 51, 53, and 54 exhibited potent cytotoxic activity against HSC-2, HSC-3, HSC-4, and HL-60 cells (IC50 0.013–2.8) and demonstrated selective cytotoxicity toward malignant cells. Particularly, among the 5α-hydroxy cardenolides (50–52), only the monoglucoside (51) was cytotoxic to tumor cell lines. Cell death of HSC-2 and HL-60 caused by 51, 53, and 54 was partially mediated through the induction of apoptosis, along with observed nuclear chromatin condensation and cell shrinkage. However, the typical laddering of DNA fragmentation and activation of caspase-3 was not detected. Therefore, 51, 53, and 54 may trigger caspase-3 independent apoptotic cell death in HSC-2 and HL-60 cells.
Amurensioside P (55), which was isolated from the roots of Adonis amurensis, exhibited potent cytotoxic activity against HL-60 and HSC-2 cells (IC50 0.048 and 0.22 μM), respectively (Fig. 7) [20]. Consistent with previous findings, its steroidal aglycone, strophanthidin, displayed weaker cytotoxic activities against HL-60 and HSC-2 cells (IC50 0.19 and 0.32 μM).
Convallatoxin (56) and convallatoxol (57), which were isolated from the rhizomes of Convallaria majalis, displayed potent cytotoxic activity against HSG human submandibular gland carcinoma cells with IC50 values of 0.012 and 0.028 μg/mL, respectively (Fig. 7). However, at a sample concentration of 0.10μg/mL, they exhibited no cytotoxicity against normal HPLF human periodontal ligament fibroblasts cells [13].
Spirostanol glycosides
The aglycones of spirostanol glycosides are divided into the 5α(A/B-trans), 5β(A/B-cis), and 5(6)-ene types with A and B rings. 5β-steroidal glycosides, a minority among naturally occurring steroidal glycosides compared to 5α- and 5(6)-ene steroidal glycosides, are exclusively found in a limited number of higher plants belonging to the Agavaceae and Liliaceae families [42,43,44,45].
5α-spirostanol glycosides
Among 5α-spirostanol glycosides 58–63, which were isolated from the seeds of Digitalis purpurea, both tetra- and pentaglycosides (58–62) were cytotoxic to SBC-3 cells comparable to the positive control, etoposide (IC50 58–62: 1.0–1.7 μM; etoposide: 1.0 μM) (Fig. 8) [26]. Glucosylation at C-3 in the terminal galactopyranosyl moiety of 62 significantly diminished its cytotoxicity (IC50 63: > 10 μM). Compounds 60 and 62 showed high selectivity index (SI) values (> 5.9 and 10) between the SBC-3 tumor cells and TIG-3 normal cells. Compound 60 induced apoptotic cell death along with caspase-3 activation in SBC-3 cells, whereas 62 did not induce any apoptotic features in SBC-3 cells. Combining each of 58 and 60–62 (0.1 and 1.0 μM) with etoposide (0.01 and/or 0.1 μM) showed a synergistic effect (combination index (CI) values 0.17–0.50) against SBC-3 cells. Notably, the combination of gitonin (58) and etoposide achieved a strong synergistic effect (CI 0.27–0.43), although this synergistic effect was reduced by the introduction of a hydroxy group at the C-15 position (59). Among 58–63, gitonin (58) notably released high mobility group box (HMGB)-1 in the preliminary screening assay. HMGB-1 is a DNA-binding protein in the nucleus; when released by dying cells, it acts as a damage-associated molecular pattern (DAMP) to trigger a strong inflammatory response [46]. Furthermore, the combination of 58 and etoposide resulted in an increase in the extracellular release of DAMPs, including the release of HMGB-1, secretion of ATP, and exposure of calreticulin (CALR) in SBC-3 cells. DAMPs can induce immunogenic cell death (ICD), a type of tumor cell death, and play a major role in stimulating the immune system in cancer therapy. These data indicate that 58, either alone or in combination with etoposide, could induce immunogenic cell death (Fig. 9). Recently, Li et al. achieved the total synthesis of gitonin (58) for the first time from readily commercially available tigogenin and evaluated the cytotoxicity of gitonin and its structural analogs against A549, HepG-2, and MCF-7 cells [47]. The trisaccharide analog of gitonin exhibited cytotoxicity comparable to that of gitonin, indicating that the molecular structure of the branched tetrasaccharide of gitonin may not be essential for its cytotoxic activity.

Structures of 5α-spirostanol glycosides 58–63 from the seeds of Digitalis purpurea, 64–70 isolated from the bulbs of Bessera elegans, and 71 from the bulbs of Fritillaria meleagris

DAMPs-releasing activities of 58
Eight steroidal glycosides (64–71) isolated from the bulbs of B. elegans and F. meleagris, including five new compounds (64–68), were evaluated for their cytotoxic activities against HL-60 and A549 tumor cells, as well as TIG-3 normal cells [23, 24] (Fig. 8). Compounds 64, 68, and 69–70 showed cytotoxic activity against both HL-60 and A549 cells, demonstrating selective cytotoxicity toward tumor cells. Consistent with previous findings [48, 49], aglycones 64a, 65a, 67a, 68a, and 69a lacked cytotoxic activity against all tested cell lines. Comparisons of their cytotoxic activities indicated that introducing a hydroxy group to the C-14α position reduced cytotoxicity (64 vs. 65 and 66 vs. 70), whereas the presence of a C-2α hydroxy group or C-12 ketone group did not affect cytotoxic activity (61 vs. 62, 68 vs. 69). Preliminary analysis suggests that the presence of a C-6 hydroxy group or C-6 O-glucosyl group significantly reduces the cytotoxic activity of spirostanol glycosides [50, 51]. This study further supports the notion that the introduction of a hydroxy group to steroidal aglycones reduces their cytotoxic activity (Fig. 10).

Structure–activity relationships of 5α-spirostanol glycosides. ➡: Reduces the cytotoxicity
5β and 5(6)-ene-spirostanol glycosides
Agave utahensis Engelm. and Yucca glauca Nutt. ex J. Fraser are members of the Agavaceae family. Twenty 5β-spirostanol glycosides (72–91), comprising thirteen undescribed compounds (72–84) were isolated from A. utahensis and Y. glauca (Fig. 11) [17, 29]. Compounds 72–79 were evaluated for their cytotoxic activity against HL-60 cells, whereas 80–91 were evaluated against A549 and HL-60 cells. Compounds 72–74, 80, 84, 85, and 88–91 exhibited cytotoxicity against HL-60 cells, whereas 80, 85, 90, and 91 exhibited cytotoxicity against A549 cells. These findings suggest that the presence of C-2β hydroxy group, C-12 carbonyl group, and C-12 hydroxy group significantly diminishes the cytotoxic potential of these 5β-spirostanol glycosides. Compounds 72 and 80 induced apoptosis in HL-60 cells and conspicuously activated caspase-3.

Structures of 5β-spirostanol glycosides 72–79 isolated from the whole plants of Agave utahensis and 80–91 isolated from the underground parts of Yucca glauca
5β and 5(6)-ene-spirostanol glycosides 92–99 were isolated from the bulbs of F. meleagris. The isolated compounds (92–99), including three novel compounds (92, 95, and 96), were evaluated for their cytotoxic activities against HL-60 and A549 cells (Fig. 12) [24, 32]. Among the steroidal alkaloids (92–94), only (22S)-spirosol glycosides 92 and 94 exhibited cytotoxic activity against HL-60 cells (IC50 5.0 and 4.4 μM), whereas (22R)-spirosol glycoside (93) demonstrated selective cytotoxicity toward A549 cells (IC50 7.9 μM) and induced apoptotic cell death without altering caspase-3 activity levels. Notably, 93 appears to induce apoptotic cell death in cultured tumor cells through distinct mechanisms of action. The absolute configuration of C-22 significantly influences the selective cytotoxicity of spirosol glycosides. The 5β-spirostanol glycoside (95) induced apoptotic cell death instead of cell cycle arrest in HL-60 cells in a time-dependent manner. Among the 5(6)-ene-spirostanol glycosides (96–99), neither 96 nor its aglycones (92a, 95a, and 96a) showed cytotoxicity against HL-60 and A549 cells. These results indicate that replacing the β-D-glucopyranosyl unit at C-3 of the aglycone with the β-D-xylopyranosyl unit reduced the cytotoxic activity.

Structures of 5β and 5(6)ene-spirostanol glycosides 92–99 isolated from the bulbs of Fritillaria meleagris
Convallasaponin A (100) and six 5β-polyhydroxylated spirostanol saponins (101–106) were isolated as novel compounds from C. majalis (Fig. 13). Dąbrowska-Balcerzak et al. also identified twelve 5β-polyhydroxylated spirostanol sapogenins and saponins from C. majalis [52]. Molecular docking simulations were conducted with the structures of the HER2 receptor and tubulin using in silico methods. Among these compounds, diols (less polar) exhibited higher affinities for the analyzed targets compared to tetrols and pentols (more polar), suggesting that the introduction of polar substituents to the steroidal nuclei reduced the binding effect [53, 54]. The 5(6)-ene-spirostanol glycosides (107–112), including three novel steroidal glycosides containing an O-β-D-glucopyranosyl-(1 → 2)-O-[β-D-xylopyranosyl-(1 → 3)]-O-β-D-glucopyranosyl-(1 → 4)-β-D-galactopyranose (lycotetrose) unit (110–112), were evaluated for their cytotoxic activity against HL-60, A549, HSC-4, and HSC-2 cells (Fig. 13) [14]. Compound 107 showed cytotoxic activity against all four tumor cell lines (IC50 0.96–3.15 μM) and led to necrotic cell death in a dose-dependent manner. As we previously reported, the introduction of polar substituents to the steroidal nuclei resulted in reduced cytotoxicity (107 vs. 108, 110–112).

Structures of 5β and 5(6)ene-spirostanol glycosides 100–112 isolated from the rhizomes of Convallaria majalis
Furostanol glycosides and pseudo-furostanol glycosides
Furostanol glycosides have hemiacetal structure and pseudo-furostanol glycosides have Δ20(22)-unsaturation at C-22, respectively. Numerous phytochemical investigations and biological assays have been performed using spirostanol glycosides. However, the corresponding furostanol or Δ20(22)-pseudo-furostanol glycosides have received insufficient attention [53]. Here, we present our phytochemical study of furostanol and Δ20(22)-pseudo-furostanol glycosides to elucidate their cytotoxic constituents.
Compounds 113–118 isolated from the bulbs of F. meleagris were evaluated for cytotoxicity against HL-60 and A549 cells (Fig. 14) [24]. 5β-Furostanol glycoside 113 displayed cytotoxic activity against both tumor cell lines (IC50 3.8 and 6.8 μM), whereas 114 and 118 were cytotoxic to A549 cells (IC50 7.6 and 4.5μM). Notably, pseudo-furostanol glycoside 118 demonstrated cytotoxic activity. Our findings revealed that substituting the D-glucose unit at C-3 of the aglycone with a D-xylose unit attenuated the cytotoxicity of 114.

Structures of furostanol and pseudo-furostanol glycosides 113–118 isolated from Fritillaria meleagris
Compounds 119–122, which were isolated from the bulbs of Lilium pumilum, comprise rare types of steroidal glycosides featuring a 2,3,4-trisubstituted β-D-glucopyranosyl moiety at C-3 of the aglycone (Fig. 15) [30]. Typically, α-L-arabinopyranosyl groups adopt a stable 4C1 conformation in glycosides (119 and 121). However, when the C-2 position of the arabinopyranosyl group is substituted with a 4C1 glycosyl group, such as D-glucose, D-galactose, and D-xylose, it assumes a 1C4 conformation to mitigate the steric hindrance from these C-2 substituents (120 and 122). The small 3JH-1,H-2 coupling constant (3.2–3.4 Hz), large 1JH-1,C-1 value, and three-bond coupled strong HMBC correlations from the anomeric proton to the C-3 and C-5 carbons of the arabinopyranosyl moiety suggest the conformation of the L-arabinopyranosyl group as 1C4 with an α-orientation of the anomeric center (Fig. 16). Notably, the 1C4 α-L-arabinopyranosyl moiety in 122 converts to the 4C1 conformation on peracetylation (122a). This interesting steric behavior of arabinopyranose remains unexplained due to steric hindrance. Recent study reports indicate that the bulky disaccharide of the rhamnosyl-(1 → 2)-glucosyl group attached to C-3 of the inner xylosyl moiety forces the α-L-arabinosyl group linked to C-4 of the same xylosyl moiety to adopt a 1C4 conformation in triterpene glycosides [55]. Therefore, future studies should gather additional data. The isolated compounds 119–122 exhibited no cytotoxicity against HL-60 cells at sample concentrations up to 15 μM.

Structures of furostanol and pseudo-furostanol glycosides 119–122 from the bulbs of Lilium pumilum and 123–129 from the bulbs of Convallaria majalis

α-L-Arabinopyranosyl moieties as 4C1 conformation in 119 and 121 and1C4 conformation in 120 and 122
Compounds 123–129 were isolated from the bulbs of C. majalis and their cytotoxicity was evaluated (Fig. 15). Compound 124, a furostanol lycotetroside of 107, exhibited cytotoxicity exclusively toward adherent cell lines of A549, HSC-4, and HSC-2 cells (IC50 2.97–11.04 μM), inducing apoptotic cell death in A549 cells via caspase-3/7 activity in a time-dependent manner [14]. The cytotoxicity mechanism predominantly involves 3-O-lycotetroside, with the introduction of polar substituents to the steroidal nuclei, diminishing the cytotoxic potential of furostanol and spirostanol glycosides [56].
Compounds 130–136 were isolated from the bulbs of B. elegans (Fig. 17). Compounds 130, 133, 134, and 136 exhibited selective cytotoxicity toward HL-60 and A549 cells (IC50 0.5–6.2 μM) while demonstrating no significant impact on cell growth in TIG-3 normal cells (IC50 > 10 μM) [23]. The findings suggest that the presence of a C-2α hydroxy group does not influence cytotoxic activity, whereas the introduction of a hydroxy group to the C-14α position decreases cytotoxicity. Notably, pseudo-furostanol glycoside 136 selectively inhibited tumor cell growth in a time-dependent manner and induced apoptosis in HL-60 and A549 cells. Furthermore, 136 induced cell cycle arrest at the G0/G1 phase in A549 cells.

Structures of furostanol and pseudo-furostanol glycosides 130–136 from the bulbs of Bessera elegans and 137–139 from the seeds of Digitalis purpurea
Furostanol glycosides 137–139, which were isolated from the seeds of D. purpurea, did not show cytotoxic activity against SBC-3 cells (IC50 > 10 μM) and their combination with etoposide did not demonstrate synergistic effects (Fig. 17) [26].
Conclusions and discussions
We conducted chemical investigations of higher plants, focusing on steroidal glycosides. Our ongoing research has led to the discovery of several steroidal glycosides in ornamental garden plants and medicinal herbs that exhibit cytotoxic activity or possess novel skeletons. Screening and systematic chemical analysis of plant extracts are thus promising avenues for uncovering the structural diversity of steroidal glycoside, despite the time investment required. Structure–activity relationships (SAR) are frequently observed for spirostanol and furostanol glycosides. The introduction of polar substituents, such as hydroxy, carbonyl, and glucosyl groups, to the aglycone moiety has been observed to diminish cytotoxicity. Notably, the presence of a hydroxy group in the axial position of the aglycone moiety may influence the cytotoxicity. While replacement of the terminal sugar in the sugar sequence at the C-3β hydroxy group of the aglycone had minimal impact on activity, the inner sugar attached to C-3 of the aglycone played a significant role in cytotoxicity. In our study, steroidal glycosides demonstrated cytotoxicity against various tumor cells (A549, ACHN, HepG-2, HL-60, HSC-2, HSC-3, HSC-4, HSG, and SBC-3) through diverse mechanisms, including necrosis, caspase-dependent or -independent apoptosis, cell proliferation arrest, and the induction of DAMPs release. Notably, the spirostanol glycosides showed synergistic cytotoxicity when combined with etoposide, whereas this synergistic effect was not observed for the corresponding furostanol glycosides. The identification of potential novel anti-cancer agents requires precise SAR determination and in vivo evaluation of steroidal glycosides. In addition, cytotoxicity assays against normal cell lines have not been sufficiently reported, and the selectivity of steroidal glycosides toward cancer cells needs to be studied. Further detailed studies on the cytotoxicity mechanisms, such as autophagy and ferroptosis, molecular targets, and chemical structures, are warranted.
References
Elekofehinti OO, Iwaloye O, Olawale F, Ariyo EO (2021) Saponins in cancer treatment: current progress and future prospects. Pathophysiology 28:250–272
Munafo JP Jr, Gianfagna TJ (2015) Chemistry and biological activity of steroidal glycosides from the Lilium genus. Nat Prod Rep 32:454–477
Reddy D, Ghosh P, Kumavath R (2020) Strophanthidin attenuates MAPK, PI3K/AKT/mTOR, and Wnt/β-catenin signaling pathways in human cancers. Front Oncol 9:1469
Pan L, Zhang Y, Zhao W, Zhou X, Wang C, Deng F (2017) The cardiac glycoside oleandrin induces apoptosis in human colon cancer cells via the mitochondrial pathway. Cancer Chemother Pharmacology 80:91–100
Sobolewska D, Galanty A, Grabowska K, Makowska-WJ W-B, Podolak I (2020) Saponins as cytotoxic agents: an update (2010–2018). Part I—steroidal saponins. Phytochem Rev 19:139–189
Bouabdallah S, A-Maktoum A, Amin A, (2023) Steroidal saponins: naturally occurring compounds as inhibitors of the hallmarks of cancer. Cancers 15:3900
Xu J, Wang Z, Huang Y, Wang Y, Xiang L, He X (2020) A spirostanol saponin isolated from Tupistra chinensis Baker simultaneously induces apoptosis and autophagy by regulating the JNK pathway in human gastric cancer cells. Steroids 164:108737
Francischini CRD, Mendonça CR, Barcelos KA, Silva MAM, Botelho AFM (2022) Antitumor effects of oleandrin in different types of cancers: systematic review. Toxicon 216:15–27
Garofalo S, Grimaldi A, Chece G, Porzia A, Morrone S, Mainiero F, Limatola C (2017) The glycoside oleandrin reduces glioma growth with direct and indirect effects on tumor cells. J Neurosci 37:3926–3939
Wu M, Huang Q, Liao M, Wu X, Xi H, Ma H, Xia Y (2022) OSW-1 induces apoptosis and cyto-protective autophagy, and synergizes with chemotherapy on triple negative breast cancer metastasis. Cell Oncol 45:1255–1275
Vafaei S, Zekiy AO, Khanamir RA, Zaman BA, Ghayourvahdat A, Azimizonuzi H, Zamani M (2022) Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell Int 22:1–27
Stone S, Newman DJ, Colletti SL, Tan DS (2022) Cheminformatic analysis of natural product-based drugs and chemical probes. Nat Prod Rep 39:20–32
Higano T, Kuroda M, Sakagami H, Mimaki Y (2007) Convallasaponin A, a new 5β-spirostanol triglycoside from the rhizomes of Convallaria majalis. Chem Pharm Bull 55:337–339
Matsuo Y, Shinoda D, Nakamaru A, Kamohara K, Sakagami H, Mimaki Y (2017) Steroidal glycosides from Convallaria majalis whole plants and their cytotoxic activity. Int J Mol Sci 18:2358
Higano T, Kuroda M, Jitsuno M, Mimaki Y (2007) Polyhydroxylated steroidal saponins from the rhizomes of Convallaria majalis. Nat Prod Comm 2:1934578X0700200
Yokosuka A, Mimaki Y (2009) Steroidal saponins from the whole plants of Agave utahensis and their cytotoxic activity. Phytochemistry 70:807–815
Yokosuka A, Jitsuno M, Yui S, Yamazaki M, Mimaki Y (2009) Steroidal glycosides from Agave utahensis and their cytotoxic activity. J Nat Prod 72:1399–1404
Yokosuka A, Mimaki Y (2007) Steroidal glycosides from Agave utahensis. Chem Pharm Bull 55:145–149
Kuroda M, Kubo S, Uchida S, Sakagami H, Mimaki Y (2010) Amurensiosides A-K, 11 new pregnane glycosides from the roots of Adonis amurensis. Steroids 75:83–94
Kubo S, Kuroda M, Yokosuka A, Sakagami H, Mimaki Y (2015) Amurensiosides L-P, five new cardenolide glycosides from the roots of Adonis amurensis. Nat Prod Comm 10:1934578X1501000
Kubo S, Kuroda M, Matsuo Y, Masatani D, Sakagami H, Mimaki Y (2012) New cardenolides from the seeds of Adonis aestivalis. Chem Pharm Bull 60:1275–1282
Kuroda M, Kubo S, Masatani D, Matsuo Y, Sakagami H, Mimaki Y (2018) Aestivalosides A-L, twelve pregnane glycosides from the seeds of Adonis aestivalis. Phytochemistry 150:75–84
Matsuo Y, Akagi N, Hashimoto C, Tachikawa F, Mimaki Y (2013) Steroidal glycosides from the bulbs of Bessera elegans and their cytotoxic activities. Phytochemistry 96:244–256
Matsuo Y, Shinoda D, Nakamaru A, Mimaki Y (2013) Steroidal glycosides from the bulbs of Fritillaria meleagris and their cytotoxic activities. Steroids 78:670–682
Fujino T, Kuroda M, Matsuo Y, Kubo S, Tamura C, Sakamoto N, Hayakawa M (2015) Cardenolide glycosides from the seeds of Digitalis purpurea exhibit carcinoma-specific cytotoxicity toward renal adenocarcinoma and hepatocellular carcinoma cells. Biosci Biotechnol Biochem 79:177–184
Matsuo Y, Tsuchihashi H, Takatori K, Fukaya H, Kuroda M, Mimaki Y (2022) Cytotoxic triterpene and steroidal glycosides from the seeds of Digitalis purpurea and the synergistic cytotoxicity of steroidal glycosides and etoposide in SBC-3 cells. Bioorg Chem 122:105697
Kuroda M, Kubo S, Matsuo Y, Atou T, Satoh J, Fujino T, Mimaki Y (2013) New cardenolide glycosides from the seeds of Digitalis purpurea and their cytotoxic activity. Biosci Biotechnol Biochem 77:1186–1192
Takatori K, Kuroda M, Mishima M, Matsuo Y, Mimaki Y (2021) Digipregnosides A-C, three novel rearranged 11, 12-secopregnane glycosides, and digipregnosides D and E, 12, 20-epoxypregnane glycosides from the seeds of Digitalis purpurea. Tetrahedron Lett 70:153020
Yokosuka A, Suzuki T, Tatsuno S, Mimaki Y (2014) Steroidal glycosides from the underground parts of Yucca glauca and their cytotoxic activities. Phytochemistry 101:109–115
Matsuo Y, Takaku R, Mimaki Y (2015) Novel steroidal glycosides from the bulbs of Lilium pumilum. Molecules 20:16255–16265
Mimaki Y (2006) Structures and biological activities of plant glycosides: cholestane glycosides from Ornithogalum saundersiae, O. thyrsoides and Galtonia candicans, and their cytotoxic and antitumor activities. Nat Prod Comm 1:1934578X0600100312
Mimaki Y, Yokosuka A, Kuroda M, Sashida Y (2001) Cytotoxic activities and structure-cytotoxic relationships of steroidal saponins. Biol Pharm Bull 24:1286–1289
Kuroda M, Mimaki Y, Sashida Y, Yamori T, Tsuruo T (2000) Galtonioside A, a novel cytotoxic cholestane glycoside from Galtonia candicans. Tetrahedron Lett 41:251–255
Si Y, Sha XS, Shi LL, Wei HY, Jin YX, Ma GX, Zhang J (2022) Review on pregnane glycosides and their biological activities. Phytochem Lett 47:1–17
Shimizu Y, Sato Y, Mitsuhashi H (1967) Isolation and structure of adonilide. Chem Pharm Bull 15:2005–2006
Shimizu Y, Sato Y, Mitsuhashi H (1969) Isolation and characterization of fukujusonorone, a 18-norpregnane derivative from Adonis amurensis Regel et Radd. Experientia 25:1129–1130
Yan Y, Zhou D, Li X, Feng Y, Wen X, Wang Y, Li N (2022) Pregnane glycosides from Adonis amurensis and their bioactivity. Phytochemistry 194:113046
Baek YS, Jung JW, Lee SH, Baek NI, Park JH (2015) A new pregnane hexaglycoside from Adonis multiflora. J Korean Soc Appl Biol Chem 58:895–899
Shang X, Miao X, Yang F, Wang C, Li B, Wang W, Zhang J (2019) The genus Adonis as an important cardiac folk medicine: a review of the ethnobotany, phytochemistry and pharmacology. Front Pharmacol 10:25
Akinwumi IA, Ambali OA (2024) Cardiac glycosides from african medicinal plants as promising therapeutics. Trop J Phytochem Pharm Sci 3:158–167
Xue R, Han N, Sakurai H, Saiki I, Ye C, Yin J (2013) Cytotoxic cardiac glycosides from the roots of Streptocaulon juventas. Planta Med 29:157–162
Iguchi T, Kuroda M, Naito R, Watanabe T, Matsuo Y, Yokosuka A, Mimaki Y (2019) Cholestane glycosides from Ornithogalum saundersiae bulbs and the induction of apoptosis in HL-60 cells by OSW-1 through a mitochondrial-independent signaling pathway. J Nat Med 73:131–145
Simmons-Boyce JL, Tinto WF (2007) Steroidal saponins and sapogenins from the Agavaceae family. Nat Prod Comm 2:1934578X0700200120
Zhang Y, Zhang YJ, Jacob MR, Li XC, Yang CR (2008) Steroidal saponins from the stem of Yucca elephantipes. Phytochemistry 69:264–270
Sidana J, Singh B, Sharma OP (2016) Saponins of Agave: chemistry and bioactivity. Phytochemistry 130:22–46
Sansone C, Bruno A, Piscitelli C, Baci D, Fontana A, Brunet C, Albini A (2021) Natural compounds of marine origin as inducers of immunogenic cell death (ICD): potential role for cancer interception and therapy. Cells 10:231
Li Y, Lv X, Liu J, Du Y (2024) Total synthesis and cytotoxicity evaluation of spirostanol saponin gitonin. Org Biomol Chem 22:2081–2090
Harmatha J, Buděšínský M, Zídek Z, Kmoníčková E (2021) Spirostanol saponins from flowers of Allium Porrum and related compounds indicating cytotoxic Activity and affecting nitric oxide production inhibitory effect in peritoneal macrophages. Molecules 26:6533
Jing S, Wang Y, Li X, Man S, Gao W (2017) Chemical constituents and antitumor activity from Paris polyphylla Smith var. yunnanensis. Nat Prod Res 31:660–666
Yokosuka A, Sano T, Hashimoto K, Sakagami H, Mimaki Y (2009) Steroidal glycosides from Furcraea foetida and their cytotoxic activity. Chem Pharm Bull 57:1161–1166
Lu Y, Luo J, Kong L (2011) Steroidal alkaloid saponins and steroidal saponins from Solanum surattense. Phytochemistry 72:668–673
Dąbrowska-B K, Nartowska J, Wawer I, Siudem P, Paradowska K (2021) Spirostanol sapogenins and saponins from Convallaria majalis L. structural characterization by 2D NMR, theoretical GIAO DFT calculations and molecular modeling. Molecules 26:2999
Zhao Y, Kang LP, Liu YX, Liang YG, Tan DW, Yu ZY, Ma BP (2009) Steroidal saponins from the rhizome of Paris polyphylla and their cytotoxic activities. Planta Med 75:356–363
Qin XJ, Zhang LJ, Zhang Y, Ni W, Yang XZ, Yu Q, Liu HY (2020) Polyphyllosides A-F, six new spirostanol saponins from the stems and leaves of Paris polyphylla var. chinensis. Bioorg Chem 99:103788
Yokosuka A, Kawakami S, Haraguchi M, Mimaki Y (2008) Stryphnosides A-F, six new triterpene glycosides from the pericarps of Stryphnodendron fissuratum. Tetrahedron 64:1474–1481
Lee KR, Kozukue N, Han JS, Park JH, Chang EY, Baek EJ, Friedman M (2004) Glycoalkaloids and metabolites inhibit the growth of human colon (HT29) and liver (HepG2) cancer cells. J Agric Food Chem 52:2832–2839
Acknowledgements
We are grateful to all collaborators and coworkers involved in the research project. The authors thank their collaborators for their technical assistance with the experiments.
Funding
This work was supported by JSPS KAKENHI (Grant Number JP21590023, JP25460134, JP16K18901, JP17K08346, and JP20K16033).
Author information
Authors and Affiliations
Contributions
The manuscript was written based on the contributions of all authors. All the authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matsuo, Y., Mimaki, Y. Search for new steroidal glycosides with anti-cancer potential from natural resources. J Nat Med 78, 807–827 (2024). https://doi.org/10.1007/s11418-024-01830-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-024-01830-1