
Abstract
Purpose
This investigation aimed to estimate and characterize the microbial diversity in soils with cadmium (Cd) at different concentrations and to evaluate whether Brassica napus can restore the soil microbial diversity.
Materials and methods
We conducted the pot experiment to analyze the composition of microbial communities in the soil contaminated with 0, 1, and 2 mg/kg Cd, as well as planted with oilseed rape. The bacterial and fungal communities were characterized via next-generation sequencing based on 16S and 18S rRNA gene fragments pyrosequencing, respectively.
Results and discussion
The results show that cadmium contamination decreased both the microbial richness and diversity in the soil, while the cultivation of oilseed rape increased the richness and diversity. In bacteria, Proteobacteria was the most abundant phylum in all the samples accounting for 39.62 to 46.14%, followed by Bacteroidetes, Actinobacteria, and Chloroflexi. These phyla collectively comprised more than 70% of all phyla. Ascomycota was the most abundant phylum in all samples in fungi (89.65 to 96.00%), and it was the only phylum whose abundance was increased with the rise of Cd concentration.
Conclusions
Microbial richness and diversity were affected by the combined action of Cd and B. napus. Cd contamination decreased the microbial richness and diversity, while cropping with oilseed rape increased the microbial richness and diversity, which alleviated the deleterious effect of the Cd pollution in soils. These reflected that oilseed rape played a positive role in maintaining species diversity of microorganism from the side.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Soil is both a crucial natural environment for human survival and a vital resource for agricultural production. It is widely known that the main functions of soil in the ecological system are to maintain biological productivity, sustain environmental quality, and improve animal and plant health (Doran and Parkin 1994). With the development of world industrialization, urbanization, and agricultural modernization, cadmium gradually entered into the agricultural soil. In China, heavy metal pollution in urban soils, urban road dusts, and agricultural soils also become increasingly serious related with the rapidly industrialization and urbanization during the last 2 decades. It has been reported that the distribution of Cd concentration throughout China is 0.082 to 1.31 mg/kg in agricultural soil (Niu et al. 2013). The maximum is higher than the grade II reference value of 1.0 mg/kg (pH > 7.5) (China soil environment quality standard, GB15618-2008). Cadmium accumulation within the soil can perform harmful impacts on soil biodiversity, soil quality, and crop growth, and even threatens human health via the food chain (Fu et al. 2011; Maron et al. 2011).
Soil function depends on microbial communities which mainly consist of fungi and bacteria. Crop growth is closely related to the soil microorganisms. Interactions between the microbial community and plant roots contribute to crop growth and production, nutrient circulation and energy flow, as well as phytoremediation (Lundberg et al. 2012; Croes et al. 2013). More and more evidences have suggested that metal contamination has been extensively shown to decrease species diversity and alter microbial community structure (Dahlin et al. 1997; Abaye et al. 2005). Hong et al. (2015) found that the variable degree of heavy metals in the soil in iron mining areas increased bacterial alpha diversity and changed the dominant genera. Cadmium pollution of soil reportedly can change the taxonomic diversity and functionality of the microbial communities, and further affect soil quality and plant growth (de Campos et al. 2013; Hill et al. 2000; Li et al. 2014; Tipayno et al. 2011).
Classical cultivation techniques and some recent techniques for analyzing microbial diversity (i.e. terminal restriction fragment length polymorphism, real-time PCR quantification) usually show many limitations such that they only reflect information on a few strains with high abundance so as to loss some ecologically significant species (Bruce et al. 2000). Therefore, in our work, the next-generation sequencing (NGS) technology of Illumina selected its availability to detect rare and even unknown species so as to obtain the total composition and relative richness of microbial communities in any given sample (Gilbert et al. 2013; Oberauner et al. 2013).
Brassica napus L. is one of the main oil-producing crops and is very important for biofuel production. Traditionally, it yields high biomass. A developed root system makes it to be easily cultivated and has the high capacity to assimilate heavy metals, which makes it an ideal crop for Cd phytoremediation (Marchiol et al. 2004; Vangronsveld et al. 2009). Many studies have verified that some certain oilseed rape varieties had tolerance of Cd and possessed the ability to accumulate it (De-chun and Wong 2002; Ru et al. 2004; Su et al. 2009). However, studies regarding interactions between microorganisms and B. napus in soil polluted with heavy mental are still at a preliminary stage (Germida et al. 1998; Dunfield and Germida 2003; Farina et al. 2012; Croes et al. 2013; de Campos et al. 2013). So far, no study has revealed the effects of varying Cd levels on both bacterial and fungal diversity in the soil cropped with oilseed rape via NGS technology.
The present study was aimed to estimate and characterize the microbial diversity under two different Cd concentration treatments and to evaluate whether B. napus can restore the soil microbial diversity. By means of high-throughput sequencing technology, we were able to show the composition of microbial community nearby the root in the Cd-contaminated soil. In the end, we hope to provide some ecological information for the use of B. napus to restore the microbial community diversity in order to alleviate the negative effect in agricultural production with Cd-polluted soils.
2 Materials and methods
2.1 Experimental design
We selected the nutritious soil (bought from Sansheng Xiang Agricultural Co., Ltd. Chengdu) for the experiment. At the beginning of the experiment, we air-dried the soil and passed it through a 2-mm sieve as our initial soil. The physicochemical characteristics of the tested soil are shown in Table 1, and the background concentration of Cd is 0.17 ± 0.03 mg/kg, which is within the primary standard set by China soil environment quality standard, GB15618-1995. According to the investigation of Yuan et al. (2014), we subsequently added Cd applied as CdCl2 to the tested soils and mixed homogeneously to set three concentrations 0, 1, and 2 mg/kg, respectively, which exceeded the grade II reference value of 0.45 mg/kg (pH 6.5–7.5, China soil environment quality standard, GB15618-2008), except for the 0 Cd applied treatments. We kept the soil at room temperature for a month to ensure the complete cadmium assimilation. We divided our experiment into two groups: one group (C) planted with oilseed rape and the other unplanted group as check (CK). The plant line 84100-18 oilseed rape B. napus L. we used was bred by our laboratory. The experimental group was separately marked as C0, C1, and C2. The check group was labeled as CK0, CK1, and CK2, respectively. Each treatment was set in triplicates.
We placed the experimental pots outdoor to ensure that the treatment environment was as close to natural conditions as possible. We watered the plants daily with deionized water to keep a balanced water holding capacity until the maturity of oilseed rape. The experimental period ranged from October 29, 2014, to April 25, 2015, when the oilseed rape was matured. According to our weather record, the rain water was little during the period.
2.2 Sampling collection
We sampled in early May 2015 when oilseed rape matured. We gathered about 500 g rhizosphere soil adjacent with the root of oilseed rape with the topsoil removed (at a depth of 5–10 cm) and immediately transferred the samples into sterile plastic bags kept on ice. Half of the samples was immediately frozen in liquid nitrogen and then stored at − 80 °C until DNA extraction for further molecular analysis. The remaining half was air-dried for a week at room temperature and subsequently sieved through a 100-mesh sifter to remove stones and visible plant fragments. Then, the samples were stored at 4 °C for further physicochemical analysis.
2.3 Soil physicochemical analysis
In order to evaluate soil quality and microbial community, we closely followed the methods reported by Bloem et al. (2006) that suggested indicators for soil quality assessment and microbial analysis.
We determined the physicochemical properties of soil at the beginning of our experiment in order to have a baseline of soil characterization. We evaluated the following items: soil organic matter (SOM), pH, total phosphorous (TP), total nitrogen (TN), total potassium (TK), and Cd. The method for determining the contents of SOM and TN was reported by Sun et al. (2006). The soil pH was measured via an INESA pH meter (Shanghai REX Instrument Factory, Shanghai, China) in a 1:5 suspension of ultrapure water. The TP content was measured using the ammonium molybdate spectrometry method (Curtius et al. 1987). TK content via the method was introduced in the book Soil agricultural chemistry analysis (Bao 2000).
All determinations of Cd concentrations were via inductively coupled plasma spectrometry (ICP-MS).The soil samples were digested sequentially with 5 mL HCL at 150 °C for 60 min, 5 mL HNO3 at 150 °C for 60 min, 3 mL HF at 150 °C for 10 min, and 3 mL HClO4 at 190 °C for 100 min in a graphite digestion apparatus. All samples were run together with certified reference samples (GBW07603 GSV-2) for assurance control. All soil samples were tested three times and calculated the mean value.
2.4 DNA extraction and PCR analysis
We extracted microbial DNA from 0.5 g soil using the Power Soil DNA kit (MoBiol Laboratories, Solana Beach, CA), according to the manufacturer’s protocols. The extracted DNA was evaluated on a 2% agarose gel, and the concentration and quality (A260/A280) of the extracts were determined using a NanoDrop ND-2000 spectrophotometer (NanoDrop, Wilmington, DE, USA). To minimize the DNA extraction bias, three successive DNA extracts of each sample were pooled. Subsequent to the pre-experiment, we selected the V3-V4 regions of the bacterial 16S rRNA gene and amplified these via PCR (95 °C for 3 min, followed by 27 cycles at 95 °C for 30 s, 55 °C for 30 s, 72 °C for 45 s, and a final extension at 72 °C for 10 min) using the following primers 338-F (5′-barcode-ACTCCTACGGGAGGCAGGA-3′) and 806R (5′-barcode-GGACTA-CHVGGGTWTCTAAT-3′) according to Dennis et al. (2013). PCR reactions were conducted in a 20 μL mixture containing 2 μL of 10× buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.2 μL TaKaRa rTaq, 0.2 μL BSA, 10 ng of template DNA, and supplemented ddH2O to a total of 20 μL finally. The barcode for each sample is an eight-base sequence unique.
We selected the 0817F/1196R primer set to amplify the 18S ribosomal RNA gene region of the fungal DNA. This primer set was selected because it has been shown to be fungal-specific and target a region of the 18S rRNA gene, which is variable between major taxa and can be aligned (permitting phylogenetic analysis such as UniFrac). This specific region has been subjected to pyrosequencing for environmental samples, especially for fungi (Bates et al. 2013; Meiser et al. 2014). However, it should be noted that this region is not sufficiently variable to permit detailed taxonomic identification for fungal communities; thus, community analyses are restricted to the family level or above. We used an aliquot of the extracted DNA from each sample as an amplification template. We amplified the 18S rRNA gene region with the primer set 0817F/1196R that contained the A and B sequencing adaptors. We conducted the PCR reactions (95 °C for 3 min, followed by 27 cycles at 95 °C for 30 s, 55 °C for 30 s, 72 °C for 45 s, and a final extension at 72 °C for 10 min) in a 20 μL mixture containing 2 μL of 10× buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.2 μL TaKaRa rTaq, 0.2 μL BSA, 10 ng template DNA, and supplemented ddH2O to 20 μL. All PCR reactions were performed in an ABI Gene Amp 9700 (Applied Bio systems). All of the real-time PCR reactions were run in triplicate for each DNA extracted of soil sample.
2.5 Illumina MiSeq sequencing
We extracted amplicons from 2% agarose gels and purified them using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to the manufacturer’s instructions. For quantification, we used the QuantiFluor-ST (Promega, USA) blue fluorescence quantitative system. Purified amplicons were pooled in equimolar and paired-end sequenced (2 × 300) on an Illumina MiSeq platform (Major bio, Shanghai) according to standard protocols.
2.6 Processing and analysis of sequencing data
Raw FASTQ files were de-multiplexed and quality-filtered using Trimmomatic (Bolger et al. 2014) (version 0.63), FLASH, with the following steps: (1) We cut off the reads at any site to obtain an average quality score < 20 in a 50-bp sliding window and discarded the reads shorter than 50 bp, (2) exactly matched via the barcode and primers and then removed mismatch and reads that containing ambiguous characters, and (3) assembled the reads only when the overlapping sequences longer than 10 bp. Reads that could not be assembled were discarded. Operational taxonomic units (OTUs) with 97% similarity cutoff were clustered via Usearch (version 7.1), and chimeric sequences were identified and discarded via UCHIME.
We used the Qiime (version 1.17) platform and RDP Classifier (Wang et al. 2007) (version 2.2) to conduct the taxonomy analysis based on Silva (Quast et al. 2013) (http://www.arb-silva.de) and Unite (Kõljalg et al. 2013) (http://unite.ut.ee/index.php) with a 70% confidence threshold. We obtained the rarefaction analysis to assess the coverage of the microbial community by the datasets via Mothur (Schloss et al. 2009) (version 1.30.1) based on the OTU clustering results. Then, we conducted the beta diversity analysis via Mothur (version 1.30.1) based on the OTU clustering results, which revealed the diversity indices, including the Chao, ACE, and Shannon diversity indices. Venn diagrams were formed by Venn Diagram, while Heatmap figures were performed in Vegan packages in R-language.
3 Results
3.1 Sequencing results and diversity indices
For the bacterial community, we obtained 240,368 reads and 12,319 operational taxonomic units (OTUs) from six samples via PE300 Miseq sequencing analysis. Each sample contained 34,645 to 45,002 reads (mean = 40,061), with different phylogenetic OTUs ranging from 1767 to 2254 (mean = 2053). We used rarefaction curves to reflect the species richness of different samples and rationality of sequencing data. All rarefaction curves tended to the saturation plateau (Fig. 1a), demonstrating adequate volume of our sequencing data. Apparently, a large number of reads might account for a small contribution to the total number of OTUs. Moreover, we detected a large variation in the total number of OTUs at the 97% similarity level of different samples. The shapes of the rarefaction curves indicated more species richness in cropped soils (C) than in idle soils (CK). Different Cd concentrations in idle soil showed diverse OTU density (CK0 > CK2 > CK1). The OTUs of CK2 and CK1 indicated that Cd at the concentration of 2 mg/kg could concentrate some bacteria and increase their richness in soil compared to 1 mg/kg. Bacterial richness in cropped soils is consistent with the increasing of Cd concentrations (C2 & C1 > C0), and all higher than the idle soil. Our results suggested that cultivation of oilseed rape has the potential to increase the bacterial richness of Cd-contaminated soils.

Rarefaction curves of the OTU number at 97% similarity level of all samples. a Bacteria community. b Fungi community CK0, CK1, and CK2 non-plant control treatments with 0, 1, and 2 mg/kg Cd, respectively. C0, C1, and C2 experimental treatments cropped with oilseed rape in 0, 1, and 2 mg/kg Cd, respectively
Accurate taxonomic allocation and alpha-diversity estimations based on OTU cleared data were made of the species present in the soil at 97% similarity level (Table 2). Among samples of the cropped soil, the maximum predicted OTU at 97% similarity was C1 (2254). For the idle soils, the maximum predicted OTU was CK0 (1985). Non-parametric analysis of diversity indexes (ACE, Chao, Shannon) revealed similar trends than the 97% similarity level: Both the observed and estimated total richness were higher in cropped soil communities than in idle soil ones. For the data in crop groups, e.g., the ACE estimators of richness which were 2409 for C1 and 1921 for CK1, the Chao indices which were 2419 for C1 and 1954 for CK1, all the species richness of cropped soil increased by 12 to 24%. The bacterial diversity in cropped soil was also higher compared to that of idle soil, although this was only a little increase (1.82 to 4.57%). We found that cultivation of oilseed rape has the potential to increase the richness and diversity of bacterial in Cd-contaminated soil.
For the fungal community, we obtained 236,160 reads and 444 OTUs from six samples via PE300 Miseq sequencing analysis. Each sample contained 36,049 to 43,115 reads (mean = 39,360), with different phylogenetic OTUs ranging from 62 to 81 (mean = 74). The rarefaction curves indicated the species richness of different samples and the rationality of our sequencing data (Fig. 1b). All rarefaction curves tended to the saturation plateau, demonstrating that the volume of our sequencing data was reasonable. Similarly, a large number of reads might account for a small contribution to the total number of OTUs. The rarefaction curves revealed that the species richness of the cropped soils (C) was slightly elevated compared to the idle soils (CK). Fungi richness throughout all samples did not appear obvious changes with regard to the Cd concentration variations. CK0 displayed the largest richness of the idle soils (CK0 > CK1 > CK2). The results indicate that Cd has the potential to decrease the fungal species richness, while cultivation of oilseed rape has the potential to remediate this loss caused by Cd contamination.
Accurate taxonomic allocation and alpha-diversity estimations based on OTU picker data were made of the species present in the soil at 97% similarity level (Table 3). Among the samples of cropped soils, the predicted OTUs at 97% similarity were virtually invariable. For the idle soils, the maximum predicted OTUs were gradually decreased with the increased Cd concentration. The non-parametric analysis of diversity and richness indexes (ACE, Chao, and Shannon) revealed similar trends at the 97% similarity level. Both the observed and estimated total richness in cropped soil communities were higher compared to that of idle soils. The fungal diversity of cropped soil was also higher compared to that of idle soil (except for CK0), and the size of this increase ranged from 22.75 to 33.04%. The fungal diversity in both CK and C distinctly declined with the rise of Cd concentration. The results revealed that cultivation of oilseed rape had a positive influence on fungal species richness and diversity in Cd-contaminated soil.
3.2 Bacterial community structure
After classification comparisons, we assigned the classification confidence threshold in a lineage without classified information in the database as unclassified. We assigned no rank to all sequences that could not be exactly classified into any known groups in a taxonomic level. All reads were assigned to bacteria with 38 different phyla, 325 families, or 563 genera. More than 95% of the total reads in every sample collectively comprised of 10 different phyla.
The classification of the sequences at the phylum and class levels is shown in Fig. 2. At the phylum level (Fig. 2a), Proteobacteria was the most abundant bacterial phylum in all of the six soil samples, and the percentage varied from 39.62% (CK2) to 46.14% (CK1). Bacteroidetes, Actinobacteria, and Chloroflexi followed in abundance with percentages varying from 13.45 to 16.63%, from 7.23 to 15.48%, and from 5.43 to 12.31%, respectively. Other major phyla were Acidobacteria, Gemmatimonadetes, and Candidate_division_TM7, which collectively comprised 17.63% of all reads. Firmicutes, Verrucomicrobia, and Cyanobacteria were much minor phyla, with relative abundance conjointly comprising 3.50% throughout all samples. At a moderate Cd contamination level (1 mg/kg), Proteobacteria and Bacteroidetes had a little more relative abundance than the other two levels (0 and 2 mg/kg), while Chloroflexi displayed a reverse result.

Bacteria community structures (a, b) and OTUs Venn diagrams (c, d) of all samples. a Phylum level, b class level of Proteobacteria phylum, c OTUs detected in CK, and d OTUs detected in C Venn diagrams are at 97% similarity level CK0, CK1, and CK2 non-plant control treatments with 0, 1, and 2 mg/kg Cd, respectively. C0, C1, and C2 experimental treatments cropped with oilseed rape in 0, 1, and 2 mg/kg Cd, respectively
Alphaproteobacteria, Gammaproteobacteria, Betaproteobacteria, and Deltaproteobacteria were the four most abundant classes in all samples (Fig. 2b), and belonging to Proteobacteria phylum. The idle soils contained a higher proportion of Alphaproteobacteria than the cropped soils and the relative abundance in unplanted groups (CK) decreased with increasing level of Cd (CK0 > CK1 > CK2). At a moderate Cd contamination level (1 mg/kg), the relative abundance of Gammaproteobacteria was higher than the other two groups (0 and 2 mg/kg). Identified classes belong to Betaproteobacteia and Deltaproteobacteria that showed an identical result. A 0.41% of the Proteobacteria phylum comprised of the subdivisions Proteobacteria_unclassified and Epsilonproteobacteria. MACA-EFT26, SB-1, and TA18 were the least abundant species in all the samples and even were completely missing in some of the samples (e.g. MACA-EFT26).
A Venn diagram can be used to calculate the common, unique number of OTUs in multiple samples. This results in a more intuitive performance number for the composition of OTUs for the similarity and community overlaps of environmental samples. In our experiment, the analysis was at 97% similarity level. The C groups (Fig. 2d) shared a higher number of OTUs than the CK groups (Fig. 2c). The total observed OTUs in the CK groups were 2212, and approximately 70% of the OTUs (1537 of the total OTUs) were shared among them. This indicates that they shared the majority of the OTUs and we only detected a small unique microbiome. In the CK group, any two of the three samples shared about 77% of the total OTUs, indicating that there was no significant difference between them. CK1 and CK2 displayed the maximum overlap of all (79% of the total). Similar to the CK group, the total observed OTUs in the C group were 2508 and about 74% of the OTUs (1867 of the total OTUs) were shared among them. This result indicates that the C group had a larger microbial species abundance compared to the CK. We found a similar result in the C group, where any two of the three samples shared about 80% of the total OTUs. C1 and C2 had the most OTUs in common (84% of the total), indicating that they had the least difference. Compared with the two groups, we found that the cropped soils had more bacterial species, showing that cultivating the B. napus on Cd-contaminated soil helps to restore the biodiversity of soil microorganisms.
3.3 Fungal community structure
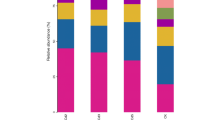
We assigned all the reads to fungal taxa. In the idle soils, Ascomycota were the most abundant phylum throughout all six samples (with 89.65 to 96.00%) (Fig. 3a). Chytridiomycota and Fungi_unclassified followed their abundance in idle soils, whereas Chytridiomycota and Fungi_norank were followed by Ascomycota in cropped soils except C1. Another major phylum was Basidiomycota, and the percentage varied from 0.47 to 3.14%. Therefore, we concluded that the four most abundant fungal phyla were members of Fungi_unclassified, Fungi_norank, Chytridiomycota, and Ascomycota. The abundance of Ascomycota increased with the rising Cd concentration throughout all samples, while phyla belonging to Basidiomycota and Fungi_unclassified were more abundant in the CK0 sample. The proportion of genera belonging Ascomycota in CK2 was obviously more than C2 while no distinct difference in other samples. The abundance of Basidiomycota in the C group was well above the CK especially in C1, while the abundance of Fungi_unclassified in the CK group was higher than the maximum of C group. If we ignore the treatment conditions, our results show that Ascomycota is undoubtedly the most widely distributed fungal phylum in the soil.

Fungal community structures (a) and OTUs Venn diagrams (b, c) of all samples. a Phylum level, b OTUs detected in CK, and c OTUs detected in C Venn diagrams are at 97% similarity level. CK0, CK1, and CK2 non-plant control treatments with 0, 1, and 2 mg/kg Cd, respectively. C0, C1, and C2 experimental treatments cropped with oilseed rape in 0, 1, and 2 mg/kg Cd, respectively
We depicted the community overlaps as Venn diagrams. The C group (Fig. 3c) shared more OTUs compared to the CK group (Fig. 3b). Surprisingly, when we did not take the physicochemical features and the plant vegetation between the samples into account, our analysis revealed that some of the OTUs were shared by all the six samples at 97% similarity level (52 OTUs). The percentage of sequences that belong to these OTUs reached 44 at 97% similarity level. We observed 85 OTUs in the CK groups; approximately 66% of the OTUs (56 of the total OTUs) were shared among the three groups (Fig. 3b), indicating that they shared the majority of the OTUs. CK1 and CK2 shared the most common OTUs (80% of the total), which indicates that they shared the most common fungi. Similar to the CK group, the total observed OTUs in the C group were 101 and about 65% of the OTUs (66 of the total OTUs) were shared between them (Fig. 3c), indicating that the C group had more fungal species compared with the CK group. C0 and C1 had the most common OTUs (73% of the total). In a comparison of the two groups, we found that the cropped soil had more fungal species, showing that the cultivation of oilseed rape on the contaminated soil is a valid technique to restore biodiversity.
We found 11 and 10 dominant genera in the CK (Fig. 4a) and C groups (Fig. 4b), respectively. The relative abundance of these genera was larger than 0.1% in at least one of the six (CK0, CK1, CK2, C0, C1, and C2). Combining the two figures, we discovered that Peziales_unclassified, Sordariales_unclassified, Ascomycota_unclassified, Mortierellaceae_uncultured, Chytridiomycetes_unclassified, and Fungi_unclassified were the most abundant of the dominant genera and distributed throughout all of the groups. On the whole, Cd contamination decreased the relative abundances of most dominant genera. But, a small number of individual genera were not the case. For example, the relative abundance of Peziales_unclassified increased with the rising of Cd levels in the two groups, showing that Peziales_unclassified may have a potential resistance to Cd. When cropped with oilseed rape (C group), the relative abundance of Sordariales_unclassified was significantly boosted than CK group. The result indicated that cultivation of oilseed rape on contaminated soil adds to the restoration of some microbial taxonomic richness and diversity.

Dominant fungi of the CK group (a) and C group (b) at the genus level. CK0, CK1, and CK2 non-plant control treatments with 0, 1, and 2 mg/kg Cd, respectively. C0, C1, and C2 experimental treatments cropped with oilseed rape in 0, 1, and 2 mg/kg Cd, respectively
In order to show the overlap between the communities throughout our samples, we used an incidence-based one (Bray-Curtis (B-C)) to calculate distances. We deployed the R Programming Language to generate community similarity heatmaps (Fig. 5). At a clustering level of 97% similarity, the abundant OTUs were similar at genus level in all samples, as well as the rare OTUs. Regardless of the treatments and environment, C0 and C1 appeared to be the most similar samples. CK1 and CK2 also were relatively similar, while CK0 showed little similarity to others, which was supported by the results in the Venn diagrams (Fig. 3).

Heatmaps of genus (a) and OTUs (b) at 97% similarity level (fungi community). CK0, CK1, and CK2 non-plant control treatments with 0, 1, and 2 mg/kg Cd, respectively. C0, C1, and C2 experimental treatments cropped with oilseed rape in 0, 1, and 2 mg/kg Cd, respectively
According to the results of our heatmaps, the largest shared OTUs predominantly belong to Pezizales_unclassifid (OTU107), Sordariales_unclassifid (OTU32), and Ascomycota_unclassifid (OTU27). In CK2, the percentage of members belonging to Pezizales_unclassifid was higher than CK1 (roughly 15%) which was nearly twice than CK0. We delineated the core microbiome of our samples and defined the set of OTUs distributed among all the samples and present at a percentage greater than 0.1%. We found 32 core OTUs, and all belonged to 51 genera and 6 phyla (Ascomycota, Basidiomycota, Chytridiomycota, Fungi_norank, Fungi_unclassified, and Zoopagales).
4 Discussion
In our work, we find that in high levels of Cd contamination soil, the species richness, diversity, and evenness of bacterial communities are lower compared to the unpolluted ones in the idle soil group, which matches those reported by Nacke et al. (2011) and Will et al. (2010). According to the rarefaction curves in Fig. 1, both bacterial and fungal species richness in all C samples is more abundant than CK samples. As for OTUs of bacterial communities, the OTUs in CK2 are more than CK1, which suggests that high level of Cd concentration (2 mg/kg) can increase some bacterial richness to some extent. Refer to the diversity indexes (ACE, Chao, Shannon) in Table 2, both the bacterial and fungal richness and diversity in C is elevated than CK except fungal diversity in CK0. That is to say, when soils cropped with oilseed rape, the species richness, diversity, and evenness of bacterial communities in Cd-contaminated soil are higher than the unpolluted and idle soil. This is mainly because substrates secreted by plant roots performed a direct effect on microbial richness (Inceoglu et al. 2010). In the Venn diagrams (Figs. 2 and 3), 77 and 66% common OTUs are shared in bacteria and fungi, respectively. CK1 and CK2, and C1 and C2 display the maximum overlap in bacteria, which demonstrates that the commonality of bacterial communities was improved with the rise of Cd concentration. The extra OTUs in C group than CK group suggest that oilseed rape can promote or control bacterial or fungal diversity, which have been also reported before (Lottmann et al. 1999; Heuer et al. 2002; Larkin 2003; Rasche et al. 2006). So, this reflects that B. napus is able to alleviate the Cd toxicity to microbes so that the cultivation of B. napus is a valid method to restoring the soil bacterial diversity and enrich bacterial population structure.
The bacterial community structure at the phylum level displays a similar trend throughout all samples (Fig. 2a), such as Acidobacteria, Actinobacteria, and Chloroflexi. Their abundance decreases at first and then rises with the increase of Cd concentrations, while Proteobacteria and Bacteroidetes displayed a contrary trend. This result was similar to that found in other soils analyzed via the 16S rDNA pyrosequencing method (Golebiewski et al. 2014), even though the sites and conditions were not the same. Obviously, Proteobacteria is the most prominent phylum in our study followed by Chloroflexi, Acidobacteria, Actinobacteria, Gemmatimonadetes, Candidate_division_TM7, Firmicutes, Verrucomicrobia, and Bacteroidetes. The first three phyla account for 60 to 70% throughout all our samples, and the first nine phyla collectively account for approximately 95% of all the bacteria. Actinobacteria in CK group is all higher than C group, which may be a relatively more sensitive phylum affected by oilseed rape. In order to assess differences in soil bacterial communities in detail, we chose the class level of Proteobacteria (Fig. 2b) to analyze the influence on bacteria community in the condition of Cd exposure and cultivating with oilseed rape. Alphaproteobacteria in CK is higher than C, while Betaproteobacteria in C was more abundant than CK. As for fungal community structure (Fig. 3a), Ascomycota in both CK and C groups is coincident with the increased Cd concentration, which can be a key phylum of fungi to cope with Cd pollution. Kickxellomycotina and Blastocaladiales are mainly found in C group, so they may be the significant fungal phylum responding to oilseed rape. As for dominant genus in fungi, Pezizales_unclassified and Sordariales_unclassified may be the key genus in fungi due to its distinct increase (Fig. 4) when cropped with oilseed rape. Overall, at the lower Cd levels (1 mg/kg), we find that the abundance of all the classes is affected by the double action of Cd and oilseed rape. However, the influence of B. napus plays a leading role on microbial structure, which has the ability to remedy the loss of richness and diversity caused by Cd pollution. We suspect that this is due to the so called host-genotype-dependent differences in patterns of microbial associations (Inceoglu et al. 2010; Inceoglu et al. 2011). The remedial effect of B. napus seems largely independent with the degree of Cd pollution. According to the Figs. 3a and 4, we can observe a similar result that the fungal community is significantly affected by the double action of Cd and B. napus. In general, cultivation of oilseed rape plays a leading role in fungal richness and diversity, while its impact on bacteria community structure and species diversity is not as distinct. This indicates that the cultivation of oilseed rape is an effective method to restore the fungi richness and diversity in Cd-contaminated soil.
5 Conclusions
In conclusion, the results of our pot-experiment indicated that the analysis at the 97% similarity level was adequate to assess the influence of both Cd and B. napus on microbial diversity and community structure. The results indicate that Cd contamination has the potential to decrease the taxonomic richness and diversity of microorganisms in soil, and further alters the microbial structures. Especially the classes belonging to Alphaproteobacteria in bacteria and the phylum belonging to fungi_unclassified in fungi are more susceptible. However, our results also reveal that the cultivation of oilseed rape has a positive impact on these parameters and possesses the potential to restore the loss of soil microbial community caused by Cd pollution. We find that the relative abundances of classes in bacteria including Betaproteobacteria, Gammaproteobacteria, MACA-EFT26, Proteobacteria_unclassified, TA18, and genuses including Pezizales_unclassified and Sordariales_unclassified all increase after cropping with oilseed rape. These findings illustrate that the cultivation of oilseed rape can be used to restore the soil microbial diversity and enrich the microbial population, and furthermore, enhance the stability of the soil micro-ecological community structure so as to mitigate the deleterious effect of Cd-polluted soil on ecological environment and agricultural production ultimately.
References
Abaye DA, Lawlor K, Hirsch PR, Brookes PC (2005) Changes in the microbial community of an arable soil caused by long-term metal contamination. Eur J Soil Sci 56(1):93–102. https://doi.org/10.1111/j.1365-2389.2004.00648.x
Bao SD (2000) Soil and agricultural chemistry analysis. China Agriculture Press, Beijing
Bates ST, Ahrendt S, Bik HM, Bruns TD, Caporaso JG, Cole J, Dwan M, Fierer N, Gu D, Houston S, Knight R, Leff J, Lewis C, Maestre JP, McDonald D, Nilsson RH, Porras-Alfaro A, Robert V, Schoch C, Scott J, Taylor DL, Parfrey LW, Stajich JE (2013) Meeting report: fungal ITS workshop (October 2012). Stand Genomic Sci 8(1):118–123. https://doi.org/10.4056/sigs.3737409
Bloem J, Hopkins DW, Benedetti A (2006) Microbiological methods for assessing soil quality. Microbiological Methods for Assessing Soil Quality 36:249–259
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Bruce KD, Jones TH, Bezemer TM, Thompson LJ, Ritchie DA (2000) The effect of elevated atmospheric carbon dioxide levels on soil bacterial communities. Glob Chang Biol 6(4):427–434. https://doi.org/10.1046/j.1365-2486.2000.00320.x
Croes S, Weyens N, Janssen J, Vercampt H, Colpaert JV, Carleer R, Vangronsveld J (2013) Bacterial communities associated with Brassica napus L. grown on trace element-contaminated and non-contaminated fields: a genotypic and phenotypic comparison. Microb Biotechnol 6(4):371–384. https://doi.org/10.1111/1751-7915.12057
Curtius AJ, Schlemmer G, Welz B (1987) Determination of phosphorus by graphite furnace atomic absorption spectrometry. Part 2. Comparison of different modifiers. J Anal Atom Spectrom 2(2):115–124. https://doi.org/10.1039/ja9870200115
Dahlin S, Witter E, Mårtensson A, Turner A, Bååth E (1997) Where’s the limit? Changes in the microbiological properties of agricultural soils at low levels of metal contamination. Soil Biol Biochem 29(9-10):1405–1415. https://doi.org/10.1016/S0038-0717(97)00048-5
de Campos SB, Youn JW, Farina R, Jaenicke S, Jünemann S, Szczepanowski R, Beneduzi A, Vargas LK, Goesmann A, Wendisch VF, Passaglia LMP (2013) Changes in root bacterial communities associated to two different development stages of canola (Brassica napus L. var oleifera) evaluated through next-generation sequencing technology. Microb Ecol 65(3):593–601. https://doi.org/10.1007/s00248-012-0132-9
De-chun S, Wong JWC (2002) The phytoremediation potential of oilseed rape (B. juncea) as a hyperaccumulator for cadmium contaminated soil China. Environ Sci 22:48–51
Dennis KL, Wang Y, Blatner NR, Wang S, Saadalla A, Trudeau E, Roers A, Weaver CT, Lee JJ, Gilbert JA, Chang EB, Khazaie K (2013) Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res 73(19):5905–5913. https://doi.org/10.1158/0008-5472.CAN-13-1511
Doran JW, Parkin TB (1994) Defining and assessing soil quality defining soil quality for a sustainable environment. Soil Science Society of America, Madison, pp 1–21
Dunfield KE, Germida JJ (2003) Seasonal changes in the rhizosphere microbial communities associated with field-grown genetically modified canola (Brassica napus). Appl Environ Microbiol 69(12):7310–7318. https://doi.org/10.1128/AEM.69.12.7310-7318.2003
Farina R, Beneduzi A, Ambrosini A, de Campos SB, Lisboa BB, Wendisch V, Vargas LK, Passaglia LMP (2012) Diversity of plant growth-promoting rhizobacteria communities associated with the stages of canola growth. Appl Soil Ecol 55:44–52. https://doi.org/10.1016/j.apsoil.2011.12.011
Fu X, Dou C, Chen Y, Chen X, Shi J, Yu M, Xu J (2011) Subcellular distribution and chemical forms of cadmium in Phytolacca americana L. J Hazard Mater 186(1):103–107. https://doi.org/10.1016/j.jhazmat.2010.10.122
Germida JJ, Siciliano SD, de Freitas JR, Seib AM (1998) Diversity of root-associated bacteria associated with field-grown canola (Brassica napus L.) and wheat (Triticum aestivum L.) FEMS Microbiol Ecol 26(1):43–50. https://doi.org/10.1111/j.1574-6941.1998.tb01560.x
Gilbert JA et al (2013) Functional assays and metagenomic analyses reveals differences between the microbial communities inhabiting the soil horizons of a Norway spruce plantation. PLoS One 8:e55929
Golebiewski M, Deja-Sikora E, Cichosz M, Tretyn A, Wrobel B (2014) 16S rDNA pyrosequencing analysis of bacterial community in heavy metals polluted soils. Microb Ecol 67(3):635–647. https://doi.org/10.1007/s00248-013-0344-7
Heuer H, Kroppenstedt RM, Lottmann J, Berg G, Smalla K (2002) Effects of T4 lysozyme release from transgenic potato roots on bacterial rhizosphere communities are negligible relative to natural factors. Appl Environ Microbiol 68(3):1325–1335. https://doi.org/10.1128/AEM.68.3.1325-1335.2002
Hill G et al (2000) Methods for assessing the composition and diversity of soil microbial communities. Appl Soil Ecol 15(1):25–36. https://doi.org/10.1016/S0929-1393(00)00069-X
Hong C, Si Y, Xing Y, Li Y (2015) Illumina MiSeq sequencing investigation on the contrasting soil bacterial community structures in different iron mining areas. Environ Sci Pollut Res 22(14):10788–10799. https://doi.org/10.1007/s11356-015-4186-3
Inceoglu O, Salles JF, van Overbeek L, van Elsas JD (2010) Effects of plant genotype and growth stage on the betaproteobacterial communities associated with different potato cultivars in two fields. Appl Environ Microbiol 76(11):3675–3684. https://doi.org/10.1128/AEM.00040-10
Inceoglu O, Al-Soud WA, Salles JF, Semenov AV, van Elsas JD (2011) Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing. PLoS One 6(8):e23321. https://doi.org/10.1371/journal.pone.0023321
Kõljalg U et al (2013) Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22:5271–5277
Larkin RP (2003) Characterization of soil microbial communities under different potato cropping systems by microbial population dynamics, substrate utilization, and fatty acid profiles. Soil Biol Biochem 35(11):1451–1466. https://doi.org/10.1016/S0038-0717(03)00240-2
Li Y, Chen L, Wen H (2014) Changes in the composition and diversity of bacterial communities 13 years after soil reclamation of abandoned mine land in eastern China. Ecol Res 30:357–366
Lottmann J, Heuer H, Smalla K, Berg G (1999) Influence of transgenic T4-lysozyme-producing potato plants on potentially beneficial plant-associated bacteria. FEMS Microbiol Ecol 29(4):365–377. https://doi.org/10.1111/j.1574-6941.1999.tb00627.x
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, Rio TG, Edgar RC, Eickhorst T, Ley RE, Hugenholtz P, Tringe SG, Dangl JL (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488(7409):86–90. https://doi.org/10.1038/nature11237
Marchiol L, Sacco P, Assolari S, Zerbi G (2004) Reclamation of polluted soil: phytoremediation potential of crop-related BRASSICA species. Water Air Soil Pollut 158(1):345–356. https://doi.org/10.1023/B:WATE.0000044862.51031.fb
Maron PA, Mougel C, Ranjard L (2011) Soil microbial diversity: methodological strategy, spatial overview and functional interest. C R Biol 334(5-6):403–411. https://doi.org/10.1016/j.crvi.2010.12.003
Meiser A, Bálint M, Schmitt I (2014) Meta-analysis of deep-sequenced fungal communities indicates limited taxon sharing between studies and the presence of biogeographic patterns. New Phytol 201(2):623–635. https://doi.org/10.1111/nph.12532
Nacke H, Thürmer A, Wollherr A, Will C, Hodac L, Herold N, Schöning I, Schrumpf M, Daniel R (2011) Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS One 6(2):e17000. https://doi.org/10.1371/journal.pone.0017000
Niu L, Yang F, Xu C, Yang H, Liu W (2013) Status of metal accumulation in farmland soils across China: from distribution to risk assessment. Environ Pollut 176:55–62. https://doi.org/10.1016/j.envpol.2013.01.019
Oberauner L, Zachow C, Lackner S, Hogenauer C, Smolle KH, Berg G (2013) The ignored diversity: complex bacterial communities in intensive care units revealed by 16S pyrosequencing. Sci Rep 3(1):1413. https://doi.org/10.1038/srep01413
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acid Res 41(D1):D590–D596. https://doi.org/10.1093/nar/gks1219
Rasche F, Hodl V, Poll C, Kandeler E, Gerzabek MH, van Elsas JD, Sessitsch A (2006) Rhizosphere bacteria affected by transgenic potatoes with antibacterial activities compared with the effects of soil, wild-type potatoes, vegetation stage and pathogen exposure. FEMS Microbiol Ecol 56(2):219–235. https://doi.org/10.1111/j.1574-6941.2005.00027.x
Ru SH, Wang JQ, Su DC (2004) Characteristics of Cd uptake and accumulation in two Cd accumulator oilseed rape species. J Environ Sci 16:594–598
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537–7541. https://doi.org/10.1128/AEM.01541-09
Su DC, Xing JP, Jiao WP, Wong WC (2009) Cadmium uptake and speciation changes in the rhizosphere of cadmium accumulator and non-accumulator oilseed rape varieties. J Environ Sci 21(8):1125–1128. https://doi.org/10.1016/S1001-0742(08)62391-8
Sun J-Y, Li M-Z, Zheng L-H, Hu Y-G, Zhang X-J (2006) Real-time analysis of soil moisture, soil organic matter, and soil total nitrogen with NIR spectra. Guang Pu Xue Yu Guang Pu Fen Xi 26:426–429
Tipayno SC, Chauhan PS, Woo S-M, Hong B-H, Park K-W, Chung J-B, Sa T-M (2011) Effects of metal and metalloid contamination on microbial diversity and activity in agricultural soils. Korean J Soil Sci Fert 44(1):146–159. https://doi.org/10.7745/KJSSF.2011.44.1.146
Vangronsveld J, Herzig R, Weyens N, Boulet J, Adriaensen K, Ruttens A, Thewys T, Vassilev A, Meers E, Nehnevajova E, van der Lelie D, Mench M (2009) Phytoremediation of contaminated soils and groundwater: lessons from the field. Environ Sci Pollut Res 16(7):765–794. https://doi.org/10.1007/s11356-009-0213-6
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267. https://doi.org/10.1128/AEM.00062-07
Will C, Thurmer A, Wollherr A, Nacke H, Herold N, Schrumpf M, Gutknecht J, Wubet T, Buscot F, Daniel R (2010) Horizon-specific bacterial community composition of German grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microbiol 76(20):6751–6759. https://doi.org/10.1128/AEM.01063-10
Yuan M, He H, Xiao L, Zhong T, Liu H, Li S, Deng P, Ye Z, Jing Y (2014) Enhancement of cd phytoextraction by two Amaranthus species with endophytic Rahnella sp JN27. Chemosphere 103:99–104. https://doi.org/10.1016/j.chemosphere.2013.11.040
Acknowledgements
This work was supported by the Science and Technology Department of Sichuan Province, the People’s Republic of China (Grant No. 2016NYZ0031 and 2017SZ0181).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Huaiying Yao
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, X., Bai, J., Wei, T. et al. Oilseed rape cultivation increases the microbial richness and diversity in soils contaminated with cadmium. J Soils Sediments 18, 2451–2462 (2018). https://doi.org/10.1007/s11368-018-1938-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-018-1938-y