
Abstract
Aging plays a pivotal role in the pathogenesis of cerebral small vessel disease (CSVD), contributing to the onset and progression of vascular cognitive impairment and dementia (VCID). In older adults, CSVD often leads to significant pathological outcomes, including blood–brain barrier (BBB) disruption, which in turn triggers neuroinflammation and white matter damage. This damage is frequently observed as white matter hyperintensities (WMHs) in neuroimaging studies. There is mounting evidence that older adults with atherosclerotic vascular diseases, such as peripheral artery disease, ischemic heart disease, and carotid artery stenosis, face a heightened risk of developing CSVD and VCID. This review explores the complex relationship between peripheral atherosclerosis, the pathogenesis of CSVD, and BBB disruption. It explores the continuum of vascular aging, emphasizing the shared pathomechanisms that underlie atherosclerosis in large arteries and BBB disruption in the cerebral microcirculation, exacerbating both CSVD and VCID. By reviewing current evidence, this paper discusses the impact of endothelial dysfunction, cellular senescence, inflammation, and oxidative stress on vascular and neurovascular health. This review aims to enhance understanding of these complex interactions and advocate for integrated approaches to manage vascular health, thereby mitigating the risk and progression of CSVD and VCID.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cerebral small vessel disease (CSVD) emerges as a critical yet frequently underappreciated component within the complex landscape of age-related neurovascular disorders [1,2,3,4,5]. This multifaceted spectrum involves a range of pathologies that affect the cerebral microcirculation, including small arteries, arterioles, capillaries, and postcapillary venules, and plays a substantial role in stroke and cognitive impairment and dementia associated with aging [2, 5,6,7,8,9]. CSVD stands as a key factor in the emergence and progression of vascular cognitive impairment and dementia (VCID) and contributes notably to the pathogenesis of dementias within the Alzheimer’s disease (AD) spectrum [2, 5, 7,8,9,10,11,12]. Neuropathologically, CSVD includes a spectrum of pathologies impacting perforating arteries, arterioles, capillaries, and veins in the brain, as well as the leptomeningeal vessels [6]. Histologically, it is characterized by arteriolosclerosis, lipohyalinosis, fibrinoid necrosis, and cerebral amyloid angiopathy (CAA), among other histopathological categorizations [6].
Functionally, CSVD leads to several critical consequences, such as endothelial dysfunction and cerebral blood flow dysregulation, leading to brain ischemia [8, 13,14,15,16,17]; increased microvascular fragility resulting in cerebral microhemorrhages (CMHs) [18]; and blood–brain barrier (BBB) disruption [19,20,21,22,23], which triggers neuroinflammation [24, 25]. Among these, the disruption of the BBB is particularly pivotal [24,25,26,27], serving as the focal point of this review. The BBB is a critical regulator of the cerebral microenvironment, that ensures the protection of neural tissue from systemic influences. Disruption of the BBB is a key feature of CSVD and contributes significantly to increased neuroinflammation, demyelination, impaired synaptic communication, neuronal damage, and cognitive decline [24, 25] (Fig. 1).

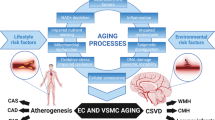
Bridging atherosclerosis and CSVD: unraveling the continuum of accelerated vascular aging. This figure presents a conceptual model illustrating how fundamental cellular and molecular aging mechanisms synergistically drive the progression of both macrovascular and microvascular aging. The upper section of the figure delineates the interconnected aging hallmarks, such as oxidative stress, mitochondrial dysfunction, cellular senescence, and increased inflammation. These aging processes synergistically induce functional and phenotypic alterations in endothelial cells (ECs) and vascular smooth muscle cells (VSMCs), laying the groundwork for various aging-associated vascular diseases. Lifestyle risk factors, notably unhealthy diets, exacerbate these vascular aging pathways, leading to the development of atherosclerosis in large arteries, manifesting as carotid artery stenosis (CAS), coronary artery disease (CAD), and peripheral artery disease (PAD), and CSVD within the cerebral microcirculation. The diagram proposes that atherosclerotic vascular diseases and CSVD share common aging origins, explaining their frequent co-occurrence in the elderly, including manifestations such as cerebral microhemorrhages (CMHs), lacunar infarcts, and white matter hyperintensities (WMHs). Aging-induced dysfunction of cerebromicrovascular endothelial cells culminates in BBB disruption, fostering neuroinflammation. This, alongside regional ischemia from reduced cerebral blood flow (CBF) due to capillary rarefaction and endothelial dysfunction, contributes to the development of WMHs
The aforementioned pathophysiological alterations of the cerebral microcirculation underpin the imaging signs of CSVD [1, 3, 28,29,30], which are crucial for its clinical diagnosis. These include white matter hyperintensities (WMHs), CMHs, enlarged perivascular spaces, and lacunar infarcts [1, 3, 28,29,30,31,32], with WMHs playing a particularly significant role as their pathogenesis involves BBB disruption [26, 33,34,35,36,37,38,39,40] (Fig. 1). Understanding CSVD, an age-related disease, necessitates a deep dive into the mechanisms driving and accelerating cerebromicrovascular aging, especially how this aging process promotes BBB disruption [24, 25, 41, 42].
Vascular aging represents a comprehensive, multifaceted process that affects the vascular system as a whole, ranging from the large arteries to the microvasculature [41, 42]. It is understood not as a series of isolated occurrences within various vessel sizes but as a continuum of interrelated changes across the vascular network [41,42,43]. In this context, the intricate link between systemic cardiovascular health and accelerated, premature development of age-related microvascular pathologies (“accelerated cerebromicrovascular aging”) becomes evident [14, 36, 44,45,46,47,48,49]. The age-associated pathological alterations in larger vessels, such as those seen in atherosclerosis, are fundamentally connected to changes in the microvasculature, contributing to CSVD development [44,45,46,47,48,49]. The risk factors known to accelerate cellular aging processes and thereby atherogenesis in larger arteries also play a role in accelerating microvascular aging, thus promoting CSVD [41, 42]. Risk factors accelerating microvascular aging are also known to promote BBB disruption [24, 50,51,52,53,54,55,56]. Here, the concept of the continuum of accelerated vascular aging, linking atherosclerosis—an age-related disease of larger vessels—to CSVD, is crucial [41, 44,45,46,47,48,49].
This review aims to synthesize the current knowledge and recent advancements in understanding the connection between peripheral atherosclerosis and BBB disruption and how this interaction contributes to CSVD and VCID. We will explore the mechanisms by which atherosclerosis may affect BBB integrity and examine the clinical implications of this relationship. Furthermore, this review will highlight emerging research, potential therapeutic targets, and future research directions. By providing a comprehensive overview of these interconnected pathways, we aim to deepen the understanding of the systemic nature of neurovascular disorders and contribute to the development of more effective prevention and treatment strategies.
CSVD, white mater damage, and VCID
Neuroimaging techniques and their role in diagnosis
Neuroimaging plays a crucial role in the diagnosis and management of CSVD and VCID [30, 57,58,59]. Computed tomography (CT) can provide a basic impression but magnetic resonance imaging (MRI) is the method of choice in identifying hallmark features of CSVD, including WMHs, lacunar infarcts, CMHs, lobar hemorrhages, superficial siderosis, perivascular space (PVS) enlargement, and cerebral atrophy [30, 57,58,59].
WMHs are commonly detected through advanced neuroimaging techniques. MRI has emerged as the gold standard for identifying WMHs. T2-weighted sequences are highly sensitive to changes in water content while fluid-attenuated inversion recovery (FLAIR) imaging can effectively differentiate WMHs with gliosis from the surrounding normal brain tissue and from PVSs by suppressing the signal from cerebrospinal fluid, enhancing the contrast and visibility of these lesions [30, 57,58,59]. CMHs and their specific locations play a crucial role in the differential diagnosis of CSVD, particularly in distinguishing between the most common sporadic types, such as arteriolosclerosis induced by hypertension, and cerebral amyloid angiopathy. While CT scans can readily identify larger lobar hemorrhages or those occurring in the basal ganglia and thalami, CMHs and superficial siderosis remain undetectable on both CT and standard MRI sequences. Their detection is reliant on hemosiderin-sensitive MRI sequences, such as gradient echo (GRE) T2* or, more effectively, susceptibility weighted imaging (SWI), which are specialized for identifying the hemosiderin deposits indicative of past bleeding events. Diffusion weighted imaging (DWI) is the primary tool to detect cytotoxic oedema resulting from recent ischemic events. Additionally, diffusion tensor imaging (DTI) provides insights into the microstructural integrity of white matter, offering complementary information about the extent and nature of WMHs. Advanced MRI techniques, such as functional MRI (fMRI), shed light on the functional connectivity of brain networks, offering further understanding of white matter integrity and the potential impact of WMHs on brain networks.
The severity of CSVD can be quantitatively assessed using MRI [30, 58,59,60,61,62,63,64,65]. The Fazekas grading system classifies CSVD into mild (grade 1), moderate (grade 2), and severe (grade 3) categories, based on the quantity, appearance, and distribution of WMHs. Additional diagnostic considerations include evaluating the extent of hyperintensities and lacunar infarcts that affect critical regions such as the basal ganglia, thalami, pons, and cerebellum. Furthermore, the number and spatial distribution of CMHs play a crucial role in determining the overall severity of CSVD, offering a comprehensive view of its impact.
Another significant capability of MRI is the detection of cerebral atrophy, allowing for the assessment of brain volume reduction. Visual grading scales serve as valuable tools in basic diagnostic procedures, while more detailed scientific evaluations rely on volumetric analyses or voxel-based morphometry, utilizing three-dimensional (3D) GRE T1 sequences. These advanced techniques offer precise measurements of the volumes of grey matter, white matter, and cerebrospinal fluid (CSF) compartments across various brain regions. By comparing data from follow-up MRIs, the rate of neurodegenerative processes can be accurately determined. Additionally, employing data from 3D FLAIR sequences, these methods can also quantify the extent of WMHs and track their progression over time, providing comprehensive insights into cerebral changes.
These imaging modalities not only facilitate the diagnosis of CSVD but also aid in understanding its progression and impact on cognitive function, thus serving as critical tools in the clinical assessment of VCID and in guiding therapeutic interventions directed at modifying underlying vascular risk factors.
Epidemiology
The prevalence of CSVD varies widely depending on the studied population, the diagnostic criteria used, and the sensitivity of the imaging techniques [66]. In general, CSVD is more common in older adults, with studies suggesting that signs of CSVD can be found in more than 90% of older adults when using sensitive MRI criteria [66]. Recent studies utilizing advanced imaging techniques suggest that around 50% of the older general population may exhibit CMHs [2, 67,68,69,70]. The occurrence of WMHs, both subcortical and periventricular, is more commonly observed in individuals over the age of 60, with their prevalence notably increasing with advancing age [66]. Data from the Rotterdam Scan Study highlighted that the prevalence of subcortical and periventricular WMHs rose by 0.2% and 0.4%, respectively, for each additional year of age [66]. Specifically, among those aged 60 to 70 years, 87% presented with subcortical WMHs and 68% with periventricular WMHs [66]. The figures escalated to 100% for subcortical and 95% for periventricular WMHs among individuals aged 80 to 90 years [66]. The prevalence also increases with risk factors such as hypertension, diabetes mellitus, smoking, and hyperlipidemia [66, 71, 72]. CSVD is increasingly recognized as a major contributor to age-related cognitive decline and VCID [71]. VCID, which ranges from mild cognitive impairment to fully developed vascular dementia, is believed to affect a considerable percentage of the elderly, though precise figures vary due to differences in diagnostic criteria [71]. These conditions not only impose a substantial burden on individuals and healthcare systems but also highlight the importance of early detection and intervention.
Pathogenesis of CSVD: role of white matter injury
The pathogenesis of CSVD and VCID is intricately linked to white matter injury, which plays a pivotal role in the disease’s progression and its clinical manifestations [13, 17, 26, 35, 71, 73,74,75,76,77,78,79] (Fig. 1). White matter injury in CSVD is primarily characterized by demyelination, axonal loss, and disruptions in white matter tract integrity, consequences of chronic hypoperfusion and BBB breakdown [26, 34, 35, 37, 38, 40]. These pathological changes result in impaired white matter connectivity, affecting the brain's ability to communicate effectively across different regions. The disruption of these neural pathways is a key contributor to the clinical symptoms of CSVD, including cognitive decline, gait disturbances, and mood changes [26, 35, 71, 74, 79,80,81,82,83,84,85,86,87,88,89,90,91]. Understanding the mechanisms underlying white matter injury in CSVD is crucial for developing targeted therapeutic strategies aimed at preserving white matter integrity and preventing disease progression. The pathogenesis of CSVD is complex and multifactorial, involving accelerated cellular and molecular mechanisms of aging, modulated by genetic, lifestyle, and environmental factors [8, 9, 33, 92].
BBB disruption as a manifestation of CSVD
Role in the pathogenesis of VCID
BBB disruption is a prominent manifestation of CSVD [24, 25] and plays a significant role in the pathogenesis of VCID [21, 93]. The BBB, a selective barrier crucial for maintaining cerebral homeostasis, when compromised, leads to an influx of neurotoxic substances, proteins, cytokines, metabolites, and bacterial breakdown products into the brain parenchyma [24]. BBB disruption is thought to contribute to microglia activation, neuroinflammation, neuronal, and synaptic dysfunction and white matter injury, all characteristic features of VCID [5, 24, 25, 32, 36, 43, 57, 94,95,96]. The resulting damage is closely linked to the cognitive decline seen in VCID, illustrating the critical role of BBB integrity in preserving cognitive function [5, 24, 25, 32, 36, 43, 57, 94,95,96]. Additionally, the interplay among CSVD, BBB disruption, and neurodegenerative processes, particularly in the context of Alzheimer’s disease, adds another layer of complexity to the pathogenesis of VCID [24, 25].
In CSVD, BBB impairment is often attributed to endothelial cell dysfunction and age-related pathological changes in the cerebral microcirculation, often exacerbated by chronic hypertension [5, 8, 33, 36, 38, 69, 97,98,99], obesity, and/or metabolic diseases [51, 100,101,102,103,104]. Preclinical research further illuminates this understanding, showing that hallmarks of aging, such as endothelial senescence, increased presence of pro-inflammatory and pro-geronic circulating factors, and a decline in anti-geronic humoral factors contribute significantly to BBB disruption [43, 105,106,107,108,109,110,111]. These insights have been particularly highlighted by heterochronic parabiosis experiments and studies on transgenic animals, which demonstrate how aging-related systemic factors can influence the integrity of the BBB, thereby exacerbating the pathophysiological processes of CSVD and VCID [112,113,114].
Biomarkers for BBB dysfunction in CSVD
Detecting BBB dysfunction in CSVD is challenging, but given the importance of this multi-cellular phenotype in understanding connections between CSVD and brain degeneration, efforts have led to the development of several novel imaging approaches [32, 115, 116] and quantitative biofluid measures, such as CSF/plasma, CSF/serum albumin quotient, and CSF fibrinogen [26, 117,118,119,120,121]. Dynamic contrast-enhanced MRI (DCE-MRI) and dynamic susceptibility contrast MRI (DSC-MRI) capture leakage of BBB based on modeling of gadolinium entry into the brain parenchyma. Additional MRI methods, such as arterial spin labeling (ASL MRI), do not require contrast agents and measure cerebral blood flow [122]. Together, DCE-MRI and ASL MRI provide complementary perspectives on CSVD. This combination of techniques enhances the accuracy and depth of understanding of CSVD, perfusion and BBB integrity, crucial for diagnosing and monitoring neurological diseases that compromise BBB function. Beyond imaging, biomarkers in cerebrospinal fluid [23], and circulating markers of endothelial dysfunction and BBB permeability are under development [123,124,125,126,127,128]. Examples include CSF/plasma albumin ratio [117, 118], CSF fibrinogen levels [129], levels of matrix metallopeptidases 2 and 9 (MMP2 and MMP9) in serum and CSF [130,131,132], and soluble platelet-derived growth factor receptor beta (sPDGFRβ) content in CSF [133,134,135]. The combination of biomarkers of CSVD and BBB dysfunction and plasma and CSF proteomics will shed light on drivers of barrier dysfunction and mediators of CSVD-related brain degeneration in individuals harboring CSVD.
Continuum of vascular aging: peripheral atherosclerosis and its impact on the brain
The continuum of vascular aging presents a comprehensive framework that connects aging-associated pathophysiological alterations in large peripheral arteries, like those seen in atherosclerosis, with changes in the microvasculature that culminate in CSVD and BBB disruption [8, 41,42,43, 94, 109, 136, 137] (Fig. 1). This holistic perspective underscores the necessity of considering vascular health as an integrated system, where macrovascular and microvascular pathologies interact synergistically. A wealth of translational, experimental, and clinical evidence supports the linkage between peripheral atherosclerosis and CSVD, highlighting their collective impact on VCID as well as Alzheimer’s disease [138, 139]. This interconnectedness emphasizes the importance of a unified approach to understanding and addressing vascular aging and its implications for brain health.
Clinical evidence linking atherosclerosis to CSVD
Numerous studies have demonstrated a clear association between atherosclerosis and CSVD [44,45,46,47,48,49, 140]. Atherosclerotic vascular disease exhibits a diverse range of manifestations across various vascular territories, resulting in several clinical conditions such as carotid artery stenosis, acute myocardial infarction within coronary arteries, ischemic strokes from occlusion of intracerebral arteries, and peripheral arterial disease affecting the limbs. Clinical evidence suggests that atherosclerotic vascular disease, characterized by the build-up of plaques in large arteries in the aforementioned vascular beds, not only compromises systemic circulation but also has downstream effects on the cerebral microvasculature. Importantly, patients with atherosclerotic changes in carotid or coronary arteries are observed to have a higher prevalence of CSVD markers such as WMHs (Figs. 2, 3, and 4), CMHs, and lacunar infarcts, as revealed through neuroimaging studies [45, 46, 49, 140,141,142,143,144,145,146,147,148,149]. Estimates indicate that atherosclerosis in the peripheral circulation elevates the risk of CSVD by a factor of two to six [46, 49].

A case of converging pathways: atherosclerosis and CSVD in a senior patient. This figure presents MRI findings from a 77-year-old female with a history of hypertension, insulin-dependent diabetes mellitus, and Crohn’s disease, who exhibited symptoms of weakness and disorientation. CT angiography revealed severe stenosis of the proximal right middle cerebral artery (MCA) and its M2 branches (panel A, arrow), indicative of intracranial atherosclerosis. Concurrently, MRI FLAIR images showcased white matter hyperintensities (panel B, arrows), aligning with a diagnosis of moderate severity chronic CSVD, alongside an acute infarct in the right MCA watershed territory (not shown). This case vividly illustrates the interconnected nature of vascular pathologies, encapsulating the essence of accelerated vascular aging's impact on both large and small cerebral vessels

Vascular aging in full spectrum: a complex case of multisystem involvement. This figure features the case of an 87-year-old female with a comprehensive medical history, including asthma, insulin-dependent diabetes mellitus, hypertension, hyperlipidemia, atherosclerotic coronary artery disease, and stage 3 chronic kidney disease, who presented with abdominal pain and subsequently experienced transient slurred speech and right facial numbness. CT angiography highlighted calcified atherosclerotic plaques in the bulbs of the bilateral internal carotid arteries (A, arrows), illustrating the widespread impact of systemic atherosclerosis. MRI FLAIR images revealed white matter hyperintensities (B, arrows), suggestive of chronic CSVD, without evidence of acute ischemia, leading to a diagnosis of a transient ischemic attack. This case underscores the multifaceted nature of vascular aging, showcasing how systemic atherosclerotic changes and small vessel disease converge, affecting both cerebral and peripheral vascular health

The intersection of age-related peripheral and cerebral vascular disease: a case study. This figure displays the medical journey of a 67-year-old female with a history of hypertension, non-insulin-dependent diabetes mellitus, and significant infrarenal aortic atherosclerosis (A, arrow) leading to severe stenosis of the left femoral artery, who experienced dizziness and nausea. Imaging revealed an acute right cerebellar infarct and right vertebral artery occlusion, suggestive of acute atherosclerosis or dissection. CT angiography further identified bilateral intracranial carotid artery stenosis due to atherosclerosis (B), while MRI GRE sequences showed microhemorrhages (C, arrowheads), and FLAIR sequences highlighted white matter hyperintensities (D), indicative of chronic small vessel disease. This case encapsulates the intricate connection between systemic atherosclerotic disease and its cerebral manifestations, demonstrating how atherosclerosis can precipitate both acute cerebrovascular events and chronic small vessel disease
Evidence linking peripheral atherosclerosis to BBB disruption
Recent research highlights a significant connection between peripheral atherosclerosis, BBB disruption, and the subsequent pathological outcomes resulting from compromised BBB integrity [146, 149,150,151]. Risk factors of atherosclerosis, including hypertension, hypercholesterolemia, hyperlipidemia, and smoking, were also reported to associate with increased BBB permeability [16, 23]. Moreover, preclinical studies indicate that atherosclerosis in large arteries [152] and carotid artery stenosis are causally associated with BBB disruption [153]. These findings, both clinical and preclinical, underscore the crucial role of vascular health in maintaining neurovascular integrity and averting a series of complications associated with BBB damage. The mechanisms underlying atherosclerosis in larger vessels—characterized by pro-inflammatory and pro-oxidative changes—mirror those affecting the cerebral circulation, highlighting the interconnected nature of systemic and cerebral vascular health.
Shared pathomechanisms between atherosclerosis and BBB disruption
The intricate relationship between atherosclerosis and BBB disruption is underscored by shared pathomechanisms that highlight the interconnectedness of systemic vascular conditions and neurovascular health. This section explores the multifaceted roles of atherogenic diets, circulating factors—including hypercholesterolemia, inflammatory mediators, and endocrine influences—endothelial dysfunction, oxidative stress, inflammation, and endothelial senescence in contributing to both atherosclerosis and BBB integrity.
The role of atherogenic diets in BBB disruption: preclinical evidence
Atherogenic diets, particularly those high in fats and sugars, have been implicated in the disruption of the BBB [52, 102, 154, 155], presenting a significant link between dietary habits and neurovascular integrity. Preclinical studies utilizing animal models have provided compelling evidence on how these diets contribute to both atherosclerosis and BBB disruption, highlighting a shared pathomechanism in the progression of vascular and neurovascular diseases [51, 52, 100,101,102, 156,157,158,159,160,161,162,163].
High-fat diets (HFD) have been shown to induce systemic inflammation and oxidative stress, factors known to exacerbate atherosclerotic plaque formation [164, 165]. These systemic changes also affect cerebral vasculature, leading to increased BBB permeability [51, 52, 100, 101, 156,157,158,159,160,161,162,163]. Specifically, animal studies have demonstrated that prolonged exposure to HFD results in the upregulation of pro-inflammatory cytokines and MMPs and dysregulation of tight junction constituents in the brain, compromising BBB integrity [51, 102]. Importantly, the adverse effects of consumption of atherogenic diets both on BBB integrity [52] and atherogenesis [164] are exacerbated in aging, likely due to an age-related impairment of cellular oxidative stress resilience mechanisms [103, 164, 166,167,168].
Similarly, diets high in sugars contribute to metabolic dysregulation, including insulin resistance and hyperlipidemia, which are known risk factors for atherosclerosis. These metabolic alterations have been associated with increased BBB permeability in preclinical models [102, 163]. Moreover, the combined effects of high-fat and high-sugar diets not only amplify the risk factors for atherosclerosis but also pose a significant threat to BBB integrity [102], potentially accelerating the onset and progression of CSVD and related neurovascular complications. These preclinical findings underscore the importance of dietary habits in maintaining vascular and neurovascular health and highlight the need for further research to explore potential therapeutic interventions targeting diet-induced BBB disruption and white matter damage.
Circulating factors: from hypercholesterolemia and inflammatory mediators to endocrine influences
Circulating factors such as lipids, hormones, inflammatory cytokines, microRNAs, and activators of innate immunity are implicated in the pathology of both atherosclerosis and BBB disruption [45, 55, 107, 169,170,171,172,173,174,175,176, 114]. These factors can mediate vascular inflammation and endothelial damage, linking systemic vascular changes to microvascular alterations in the brain.
Preclinical studies have provided evidence that hypertriglyceridemia and hypercholesterolemia can lead to BBB disruption [55, 56, 140, 169]. This disruption is thought to stem from the direct impact of elevated triglyceride and/or cholesterol levels on the endothelial cells lining the cerebral vasculature, mirroring their detrimental effects on endothelial cells in large vessels, which are key contributors to the development of atherosclerosis [177,178,179].
The potential role of increased systemic levels of inflammatory cytokines during the progression of atherosclerosis is a critical area of study [180,181,182,183], given their known impact on the BBB. Inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1β), and interleukin 6 (IL-6), are pivotal in the inflammatory response that characterizes atherosclerosis. These cytokines contribute to endothelial dysfunction, a hallmark of atherosclerosis, by promoting the expression of adhesion molecules, attracting monocytes to the endothelium, and facilitating their transformation into foam cells within the arterial wall [180,181,182]. Moreover, the systemic elevation of these inflammatory mediators can also affect the cerebral vasculature, compromising the integrity of the BBB [184]. Preclinical studies confirm that systemic atherosclerosis is associated with significant cerebrovascular inflammation in mice, which is characterized by increased IL-1β [185]. Aging itself is characterized by increased levels of pro-inflammatory cytokines [186, 187]. Mounting preclinical and clinical data indicate the detrimental effect of these pro-inflammatory cytokines on BBB integrity [186,187,188,189,190,191,192]. Elevated levels of soluble P-selectin (sP-selectin), a biomarker for platelet/endothelial activation and a known risk factor for vascular disease, have also been shown to contribute directly to increased BBB permeability and heightened susceptibility to atherosclerosis in transgenic mouse models [193].
Apolipoprotein E (APOE), a lipid-transport protein, plays a significant role in the interplay between atherosclerosis and neurodegenerative disorders, notably dementia [194,195,196]. The protein's three isoforms—APOE2, APOE3, and APOE4—exhibit varied effects on lipid metabolism and neuroinflammation. APOE4, in particular, is distinguished by its association with adverse lipid profiles, increased neuroinflammation, and susceptibility to early cognitive decline and white matter damage [197,198,199]. This isoform significantly elevates the risk of Alzheimer's disease (AD), with heterozygotes experiencing more than twice the risk and homozygotes facing a risk increase of over ninefold [139, 197]. The risk extends to VCID, where APOE4 carriers are at a heightened risk [139]. APOE4’s influence on the BBB is profound, promoting the breakdown of essential tight junction proteins like occludin, claudin-5, and zonula occludens-1 via the low density lipoprotein receptor-related protein 1 (LRP1) signaling pathway, thus compromising BBB integrity [200]. This mechanism has been clinically correlated with BBB disruption in the limbic region among AD patients, as evidenced by MRI studies [201]. The role of APOE, especially the APOE4 allele, underscores the complex genetic factors contributing to both vascular and neurovascular pathologies.
Endocrine influences, notably the hormonal shifts accompanying aging, significantly impact vascular health [107, 112, 113, 202, 203]. A critical hormonal change is the decline in circulating levels of insulin-like growth factor 1 (IGF-1), which is essential for vascular homeostasis [202]. This decline affects both the genesis of atherosclerosis [173, 204,205,206,207,208,209,210,211] and the development of microvascular pathologies [107, 202, 212,213,214,215,216,217,218,219,220,221,222,223]. IGF-1 serves to protect the endothelium, with its age-related reduction contributing to endothelial dysfunction, a key factor in atherogenesis [221]. The decrease in IGF-1 signaling not only impairs nitric oxide production [221] and vascular oxidative stress resilience [224] but also heightens pro-oxidative [208, 225] and pro-inflammatory [205, 208, 210, 226] states within the vascular system, thereby fostering atherosclerotic plaque progression [205, 208, 210, 226]. IGF-1 confers pro-angiogenic, anti-apoptotic, and anti-senescence effects on cerebromicrovascular endothelial cells, contributing to the maintenance of the functional and structural integrity of the cerebral microcirculation [107, 202, 212,213,214, 217,218,219, 221,222,223, 227,228,229,230]. IGF-1 deficiency is linked to detrimental changes in cerebromicrovascular health, such as microvascular rarefaction and reduced CBF, which reflect vascular aging [218, 223]. In older adults, lower IGF-1 levels are associated with impaired neurovascular coupling (NVC) responses [212], a connection further supported by experimental models showing significant NVC impairments with induced IGF-1 deficiency [221]. Genetic disruptions in IGF-1 signaling, including IGF1R knockdown, impair endothelial and astrocytic components of NVC [214, 215] and promote BBB disruption [107], emphasizing the crucial role of IGF-1 in maintenance of neurovascular health. IGF-1 deficient mouse models also exhibit increased microvascular fragility and a higher propensity for CMHs [213, 218], with vascular wall remodeling indicating compromised structural integrity.
Endothelial dysfunction
Both atherosclerosis and BBB disruption share endothelial dysfunction as a fundamental pathophysiological mechanism [98, 177, 231,232,233,234,235,236]. In the context of atherosclerosis, endothelial dysfunction sets off a cascade of events leading to the formation of atherosclerotic plaques [177, 232, 234, 235]. It promotes platelet aggregation and increases vasoconstriction, further exacerbating the condition by enhancing inflammation and pathological remodeling, contributing to the narrowing and stiffening of arteries. Similarly, within the cerebral vasculature, endothelial dysfunction significantly impacts BBB integrity [43].
Oxidative stress and inflammation
Oxidative stress and inflammation are critical factors in the progression of both atherosclerosis and CSVD, acting as intertwined pathological processes [33, 41,42,43]. The oxidative modification of lipoproteins and endothelial activation in atherosclerosis and the oxidative damage to cerebral endothelial cells in CSVD are examples of how oxidative stress serves as a common pathogenic pathway. Similar to the alterations seen in larger vessels during aging, the microcirculation experiences an increased cellular production of reactive oxygen species (ROS) [237,238,239,240,241,242,243,244]. This escalation in ROS production is primarily driven by age-related factors, such as a decline in cellular Nicotinamide adenine dinucleotide (NAD+) levels, dysregulation of sirtuin 1 (SIRT1), and a consequent increase in mitochondrial ROS generation [237, 240]. This heightened state of microvascular oxidative stress is a crucial contributor to endothelial dysfunction [237, 240], a central aspect of CSVD. It impairs the bioavailability of nitric oxide and fosters inflammatory responses, which are critical in the pathogenesis of CSVD [34, 231, 245, 246]. The activation of endothelial cells, marked by the expression of adhesion molecules and recruitment of inflammatory cells, also increases the propensity for thrombosis. These changes within the cerebral microcirculation have direct implications for several key pathological manifestations of CSVD. Oxidative stress and endothelial dysfunction are intimately associated with the disruption of the BBB [52, 247], the impairment of neurovascular coupling responses [237, 240, 241], formation of lacunar infarcts, and the pathogenesis of microvascular fragility and CMHs [244].
In the realm of inflammation, robust preclinical and clinical evidence indicates that systemic inflammation is a critical trigger for BBB disruption. The underlying mechanisms are complex and involve various circulating inflammatory mediators, including cytokines. Cellular components of the BBB, such as endothelial cells, pericytes, and astrocytes, are key players in this process. They express pattern recognition receptors and activate inflammasomes in response to blood-borne signals, including Pathogen-Associated Molecular Patterns (PAMPs) and Damage-Associated Molecular Patterns (DAMPs) [190, 191]. This activation leads to increased BBB permeability and the secretion of proinflammatory cytokines and chemokines like IL-1β, IL-1α, IL-6, monocyte chemoattractant protein-1 (MCP-1), and C-C motif chemokine ligand 5 (CCL5, also known as RANTES), which further exacerbate BBB disruption and promote leukocyte attraction, creating a self-perpetuating cycle of inflammation [189, 192].
There is strong preclinical and clinical evidence that systemic inflammation triggers BBB disruption [248, 249]. Chronic low-grade systemic inflammation associated with systemic atherosclerosis can also trigger these BBB-disrupting mechanisms. There is evidence that an elevated inflammatory status within atherosclerotic plaques is associated with increased BBB disruption and neuroinflammation [250, 251]. Therefore, chronic inflammatory states, as seen in peripheral atherosclerosis, might play a crucial role in BBB dysfunction. This relationship highlights the systemic nature of these pathologies, underscoring the need for a comprehensive approach to understanding and managing the interaction between peripheral atherosclerosis and cerebral microvascular health.
Endothelial senescence
As individuals age, there is a marked increase in the number of cells entering a state of senescence primarily due to the accumulation of oxidative stress-induced DNA damage [172, 252,253,254,255,256,257,258,259]. The accumulation of senescent cells is particularly prominent within atherosclerotic plaques, a phenomenon that is further accelerated by common cardiovascular risk factors such as hypertension and diabetes mellitus [172, 252, 254, 255, 260,261,262,263,264,265,266,267]. Within the vascular system, these senescent cells are known to secrete a range of pro-inflammatory and matrix-degrading molecules, collectively referred to as the senescence-associated secretory phenotype (SASP) [41, 42]. SASP factors contribute to chronic inflammation and can lead to increased plaque instability in atherosclerotic vascular diseases. Additionally, the shortening of telomeres in vascular cells is another factor that drives cellular senescence, further influencing the formation and progression of atherosclerotic plaques.
Accumulation of senescent cells is also crucial aspect of microvascular aging and pathology, especially in the context of CSVD [50, 107, 268, 269]. Oxidative DNA damage-mediated senescence is particularly pronounced in cerebromicrovascular endothelial cells [108, 269, 270]. As microvascular endothelial cells become senescent, they exhibit a SASP, characterized by the secretion of various pro-inflammatory cytokines and matrix-degrading enzymes [271]. This molecular cascade significantly exacerbates microvascular damage and plays a key role in the development of CSVD [106, 108, 268, 270, 272]. These findings underscore the intricate link between cellular aging processes and the development of both macrovascular and microvascular pathologies, highlighting the importance of understanding cellular senescence in the context of the continuum of accelerated vascular aging.
In recent years, research has shed light on the potential for rejuvenating the cerebral microcirculation by specifically targeting senescent cells [106, 108, 268, 270]. Studies employing pharmacological or genetic interventions to eliminate these cells in aging mouse models have yielded encouraging results. These outcomes include the restoration of endothelial function, improvements in neurovascular coupling responses, and bolstered integrity of the BBB [106, 108, 268, 270]. A notable example is the use of the BCL-2 inhibitor senolytic drug Navitoclax, which has been shown to reverse both BBB disruption [106] and mitigate the development of CMHs induced by hypertension in aged mice [268]. This discovery is particularly significant as it points towards a promising therapeutic approach to prevent microvascular changes that are instrumental in the pathogenesis of CSVD associated with systemic atherosclerosis. By targeting the senescent cells that contribute to accelerated microvascular aging and dysfunction, there is potential to address a fundamental aspect of CSVD and perhaps ameliorate its impact on brain health.
In summary, understanding the continuum of vascular aging, which links systemic and cerebral vascular health, is crucial. Recognizing the shared pathomechanisms between peripheral atherosclerosis and BBB disruption is essential for developing comprehensive strategies to mitigate CSVD progression, particularly in older adults with increased cardiovascular risk.
Clinical implications of BBB disruption in atherosclerosis and CSVD
Progressive BBB disruption facilitates the entry of neurotoxic substances, inflammatory cytokines, and cells into the brain parenchyma, contributing to neuroinflammation, neuronal damage, synaptic dysfunction, and, ultimately, cognitive decline [24, 273, 274]. It is likely that compromised BBB integrity and its sequalae are closely linked to the development of a spectrum of cognitive impairments ranging from mild cognitive deficits to severe dementia [24, 25]. The progression from age-related BBB disruption in the context of CSVD and atherosclerosis to cognitive impairment underscores the critical role of vascular health in maintaining cognitive function [24, 37, 132, 273,274,275,276].
Early identification of individuals at risk of BBB disruption and subsequent cognitive decline is essential for preventing or mitigating the impact of VCID [116, 274]. Risk assessment strategies may include the evaluation of vascular risk factors such as hypertension, diabetes mellitus, and hyperlipidemia, alongside the use of advanced neuroimaging techniques to detect early signs of BBB permeability in patients with peripheral atherosclerotic diseases. Biomarkers in blood and cerebrospinal fluid, reflecting endothelial dysfunction and BBB integrity may also hold promise for early detection and monitoring of disease progression.
Protecting or restoring BBB integrity offers a promising avenue for future therapeutic intervention. Targeting the underlying mechanisms of BBB disruption, such as inflammation, oxidative stress, and endothelial cell dysfunction, could mitigate the progression of VCID in high risk patients. Additionally, emerging research on senolytics and drugs that target specific pathways implicated in BBB disruption (e.g., the signaling pathways involving APOE4) highlights the potential for novel interventions [273]. Modulating lifestyle factors, such as consumption of a healthy diet and regular exercise, which have been shown to influence vascular health and BBB integrity, also presents a viable strategy for protecting cognitive function [277, 278].
Conclusion
In conclusion, the evidence compiled from both clinical and preclinical studies unequivocally underscores the intricate connection between atherosclerotic vascular diseases in the peripheral circulation and BBB disruption, shedding light on the significant impact of vascular health on neurovascular integrity. The findings reveal that risk factors commonly associated with atherosclerosis, such as hypertension, hypercholesterolemia, hyperlipidemia, and smoking, are also implicated in the pathogenesis of CSVD and increased BBB permeability, which, in turn, can lead to a cascade of neurocognitive consequences. This research highlights the critical role that systemic vascular health plays in preserving the BBB and, by extension, in preventing the myriad complications that stem from its breakdown. The shared mechanisms of pro-geronic, pro-inflammatory, and pro-oxidative changes across the vascular system emphasize the need for holistic approaches in managing vascular health to safeguard against CSVD and VCID. Moving forward, it is imperative that future research continues to explore the relationship between vascular pathology and BBB integrity to develop targeted interventions that can mitigate the risk of CSVD and maintain cognitive function in aging populations.
References
Cannistraro RJ, Badi M, Eidelman BH, Dickson DW, Middlebrooks EH, Meschia JF. CNS small vessel disease: a clinical review. Neurology. 2019;92:1146–56. https://doi.org/10.1212/WNL.0000000000007654.
Hainsworth AH, Markus HS, Schneider JA. Cerebral small vessel disease, hypertension, and vascular contributions to cognitive impairment and dementia. Hypertension. 2024;81:75–86. https://doi.org/10.1161/HYPERTENSIONAHA.123.19943.
Rosenberg GA, Wallin A, Wardlaw JM, Markus HS, Montaner J, Wolfson L, Iadecola C, Zlokovic BV, Joutel A, Dichgans M, et al. Consensus statement for diagnosis of subcortical small vessel disease. J Cereb Blood Flow Metab. 2016;36:6–25. https://doi.org/10.1038/jcbfm.2015.172.
Markus HS, de Leeuw FE. Cerebral small vessel disease: recent advances and future directions. Int J Stroke. 2023;18:4–14. https://doi.org/10.1177/17474930221144911.
Elahi FM, Wang MM, Meschia JF. Cerebral small vessel disease-related dementia: more questions than answers. Stroke. 2023;54:648–60. https://doi.org/10.1161/STROKEAHA.122.038265.
Craggs LJ, Yamamoto Y, Deramecourt V, Kalaria RN. Microvascular pathology and morphometrics of sporadic and hereditary small vessel diseases of the brain. Brain Pathol. 2014;24:495–509. https://doi.org/10.1111/bpa.12177.
Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701. https://doi.org/10.1016/S1474-4422(10)70104-6.
De Silva TM, Faraci FM. Contributions of aging to cerebral small vessel disease. Annu Rev Physiol. 2020;82:275–95. https://doi.org/10.1146/annurev-physiol-021119-034338.
Chojdak-Lukasiewicz J, Dziadkowiak E, Zimny A, Paradowski B. Cerebral small vessel disease: a review. Adv Clin Exp Med. 2021;30:349–56. https://doi.org/10.17219/acem/131216.
Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–713. https://doi.org/10.1161/STR.0b013e3182299496.
Makedonov I, Black SE, MacIntosh BJ. Cerebral small vessel disease in aging and Alzheimer’s disease: a comparative study using MRI and SPECT. Eur J Neurol. 2013;20:243–50. https://doi.org/10.1111/j.1468-1331.2012.03785.x.
Schreiber S, Drukarch B, Garz C, Niklass S, Stanaszek L, Kropf S, Bueche C, Held F, Vielhaber S, Attems J, et al. Interplay between age, cerebral small vessel disease, parenchymal amyloid-beta, and tau pathology: longitudinal studies in hypertensive stroke-prone rats. J Alzheimers Dis. 2014;42(Suppl 3):S205-215. https://doi.org/10.3233/JAD-132618CH2G7H1517785JK5[pii].
Wang X, Wang Y, Gao D, Zhao Z, Wang H, Wang S, Liu S. Characterizing the penumbras of white matter hyperintensities in patients with cerebral small vessel disease. Jpn J Radiol. 2023;41:928–37. https://doi.org/10.1007/s11604-023-01419-w.
van Dinther M, Schram MT, Jansen JFA, Backes WH, Houben A, Berendschot T, Schalkwijk CG, Stehouwer CDA, van Oostenbrugge RJ, Staals J. Extracerebral microvascular dysfunction is related to brain MRI markers of cerebral small vessel disease: The Maastricht Study. Geroscience. 2022;44:147–57. https://doi.org/10.1007/s11357-021-00493-0.
Lu W, Yu C, Wang L, Wang F, Qiu J. Perfusion heterogeneity of cerebral small vessel disease revealed via arterial spin labeling MRI and machine learning. Neuroimage Clin. 2022;36: 103165. https://doi.org/10.1016/j.nicl.2022.103165.
Gyanwali B, Tan CS, Petr J, Escobosa LLT, Vrooman H, Chen C, Mutsaerts HJ, Hilal S. Arterial spin-labeling parameters and their associations with risk factors, cerebral small-vessel disease, and etiologic subtypes of cognitive impairment and dementia. AJNR Am J Neuroradiol. 2022;43:1418–23. https://doi.org/10.3174/ajnr.A7630.
Stewart CR, Stringer MS, Shi Y, Thrippleton MJ, Wardlaw JM. Associations between white matter hyperintensity burden, cerebral blood flow and transit time in small vessel disease: an updated meta-analysis. Front Neurol. 2021;12: 647848. https://doi.org/10.3389/fneur.2021.647848.
Ungvari Z, Tarantini S, Kirkpatrick AC, Csiszar A, Prodan CI. Cerebral microhemorrhages: mechanisms, consequences, and prevention. Am J Physiol Heart Circ Physiol. 2017;312:H1128–43. https://doi.org/10.1152/ajpheart.00780.2016ajpheart.00780.2016[pii].
Wallin A, Blennow K, Fredman P, Gottfries CG, Karlsson I, Svennerholm L. Blood brain barrier function in vascular dementia. Acta Neurol Scand. 1990;81:318–22. https://doi.org/10.1111/j.1600-0404.1990.tb01562.x.
Tayler H, Miners JS, Guzel O, MacLachlan R, Love S. Mediators of cerebral hypoperfusion and blood-brain barrier leakiness in Alzheimer’s disease, vascular dementia and mixed dementia. Brain Pathol. 2021;31: e12935. https://doi.org/10.1111/bpa.12935.
Taheri S, Gasparovic C, Huisa BN, Adair JC, Edmonds E, Prestopnik J, Grossetete M, Shah NJ, Wills J, Qualls C, Rosenberg GA. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke. 2011;42:2158–63. https://doi.org/10.1161/STROKEAHA.110.611731.
Li M, Li Y, Zuo L, Hu W, Jiang T. Increase of blood-brain barrier leakage is related to cognitive decline in vascular mild cognitive impairment. BMC Neurol. 2021;21:159. https://doi.org/10.1186/s12883-021-02189-6.
Lin Z, Sur S, Liu P, Li Y, Jiang D, Hou X, Darrow J, Pillai JJ, Yasar S, Rosenberg P, et al. Blood-brain barrier breakdown in relationship to alzheimer and vascular disease. Ann Neurol. 2021;90:227–38. https://doi.org/10.1002/ana.26134.
Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–50. https://doi.org/10.1038/nrneurol.2017.188.
Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci. 2018;21:1318–31. https://doi.org/10.1038/s41593-018-0234-x.
Freeze WM, Jacobs HIL, de Jong JJ, Verheggen ICM, Gronenschild E, Palm WM, Hoff EI, Wardlaw JM, Jansen JFA, Verhey FR, Backes WH. White matter hyperintensities mediate the association between blood-brain barrier leakage and information processing speed. Neurobiol Aging. 2020;85:113–22. https://doi.org/10.1016/j.neurobiolaging.2019.09.017.
Elahi FM, Casaletto KB, Altendahl M, Staffaroni AM, Fletcher E, Filshtein TJ, Glymour MM, Miller BL, Hinman JD, DeCarli C, et al. “Liquid biopsy” of white matter hyperintensity in functionally normal elders. Front Aging Neurosci. 2018;10:343. https://doi.org/10.3389/fnagi.2018.00343.
Zwanenburg JJM, van Osch MJP. Targeting cerebral small vessel disease with MRI. Stroke. 2017;48:3175–82. https://doi.org/10.1161/STROKEAHA.117.016996.
Blair GW, Hernandez MV, Thrippleton MJ, Doubal FN, Wardlaw JM. Advanced neuroimaging of cerebral small vessel disease. Curr Treat Options Cardiovasc Med. 2017;19:56. https://doi.org/10.1007/s11936-017-0555-1.
Duering M, Biessels GJ, Brodtmann A, Chen C, Cordonnier C, de Leeuw FE, Debette S, Frayne R, Jouvent E, Rost NS, et al. Neuroimaging standards for research into small vessel disease-advances since 2013. Lancet Neurol. 2023;22:602–18. https://doi.org/10.1016/S1474-4422(23)00131-X.
Brown R, Benveniste H, Black SE, Charpak S, Dichgans M, Joutel A, Nedergaard M, Smith KJ, Zlokovic BV, Wardlaw JM. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc Res. 2018;114:1462–73. https://doi.org/10.1093/cvr/cvy113.
Voorter PHM, van Dinther M, Jansen WJ, Postma AA, Staals J, Jansen JFA, van Oostenbrugge RJ, van der Thiel MM, Backes WH. Blood-brain barrier disruption and perivascular spaces in small vessel disease and neurodegenerative diseases: a review on MRI methods and insights. J Magn Reson Imaging. 2024;59:397–411. https://doi.org/10.1002/jmri.28989.
Low A, Mak E, Rowe JB, Markus HS, O’Brien JT. Inflammation and cerebral small vessel disease: a systematic review. Ageing Res Rev. 2019;53: 100916. https://doi.org/10.1016/j.arr.2019.100916.
Rudilosso S, Stringer MS, Thrippleton M, Chappell F, Blair GW, Jaime Garcia D, Doubal F, Hamilton I, Janssen E, Kopczak A, et al. Blood-brain barrier leakage hotspots collocating with brain lesions due to sporadic and monogenic small vessel disease. J Cereb Blood Flow Metab. 2023;43:1490–502. https://doi.org/10.1177/0271678X231173444.
Karvelas N, Elahi FM. White matter hyperintensities: complex predictor of complex outcomes. J Am Heart Assoc. 2023;12: e030351. https://doi.org/10.1161/JAHA.123.030351.
Ungvari Z, Toth P, Tarantini S, Prodan CI, Sorond F, Merkely B, Csiszar A. Hypertension-induced cognitive impairment: from pathophysiology to public health. Nat Rev Nephrol. 2021;17:639–54. https://doi.org/10.1038/s41581-021-00430-6.
Kerkhofs D, Wong SM, Zhang E, Uiterwijk R, Hoff EI, Jansen JFA, Staals J, Backes WH, van Oostenbrugge RJ. Blood-brain barrier leakage at baseline and cognitive decline in cerebral small vessel disease: a 2-year follow-up study. Geroscience. 2021;43:1643–52. https://doi.org/10.1007/s11357-021-00399-x.
Kerkhofs D, Wong SM, Zhang E, Staals J, Jansen JFA, van Oostenbrugge RJ, Backes WH. Baseline blood-brain barrier leakage and longitudinal microstructural tissue damage in the periphery of white matter hyperintensities. Neurology. 2021;96:e2192–200. https://doi.org/10.1212/WNL.0000000000011783.
Li Y, Li M, Zhang X, Shi Q, Yang S, Fan H, Qin W, Yang L, Yuan J, Jiang T, Hu W. Higher blood-brain barrier permeability is associated with higher white matter hyperintensities burden. J Neurol. 2017;264:1474–81. https://doi.org/10.1007/s00415-017-8550-8.
Huisa BN, Caprihan A, Thompson J, Prestopnik J, Qualls CR, Rosenberg GA. Long-term blood-brain barrier permeability changes in binswanger disease. Stroke. 2015;46:2413–8. https://doi.org/10.1161/STROKEAHA.115.009589.
Ungvari Z, Tarantini S, Sorond F, Merkely B, Csiszar A. Mechanisms of vascular aging, a geroscience perspective: JACC focus seminar. J Am Coll Cardiol. 2020;75:931–41. https://doi.org/10.1016/j.jacc.2019.11.061.
Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of vascular aging. Circ Res. 2018;123:849–67. https://doi.org/10.1161/CIRCRESAHA.118.311378.
Toth P, Tarantini S, Csiszar A, Ungvari Z. Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol. 2017;312:H1–20. https://doi.org/10.1152/ajpheart.00581.2016ajpheart.00581.2016[pii].
Dearborn JL, Schneider AL, Sharrett AR, Mosley TH, Bezerra DC, Knopman DS, Selvin E, Jack CR, Coker LH, Alonso A, et al. Obesity, insulin resistance, and incident small vessel disease on magnetic resonance imaging: atherosclerosis risk in communities study. Stroke. 2015;46:3131–6. https://doi.org/10.1161/STROKEAHA.115.010060.
Gustavsson AM, van Westen D, Stomrud E, Engstrom G, Nagga K, Hansson O. Midlife atherosclerosis and development of Alzheimer or vascular dementia. Ann Neurol. 2020;87:52–62. https://doi.org/10.1002/ana.25645.
Kim BJ, Lee SH, Kim CK, Ryu WS, Kwon HM, Choi SY, Yoon BW. Advanced coronary artery calcification and cerebral small vessel diseases in the healthy elderly. Circ J. 2011;75:451–6. https://doi.org/10.1253/circj.CJ-10-0762.
Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Niessen WJ, Hofman A, Krestin GP, Breteler MM. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology. 2008;70:1208–14. https://doi.org/10.1212/01.wnl.0000307750.41970.d970/14/1208[pii].
Conijn MM, Hoogduin JM, van der Graaf Y, Hendrikse J, Luijten PR, Geerlings MI. Microbleeds, lacunar infarcts, white matter lesions and cerebrovascular reactivity – a 7 T study. Neuroimage. 2012;59:950–6. https://doi.org/10.1016/j.neuroimage.2011.08.059.
Ding L, Hong Y, Peng B. Association between large artery atherosclerosis and cerebral microbleeds: a systematic review and meta-analysis. Stroke Vasc Neurol. 2017;2:7–14. https://doi.org/10.1136/svn-2016-000049.
Balasubramanian P, Kiss T, Tarantini S, Nyul-Toth A, Ahire C, Yabluchanskiy A, Csipo T, Lipecz A, Tabak A, Institoris A, et al. Obesity-induced cognitive impairment in older adults: a microvascular perspective. Am J Physiol Heart Circ Physiol. 2021;320:H740–61. https://doi.org/10.1152/ajpheart.00736.2020.
Ouyang S, Hsuchou H, Kastin AJ, Wang Y, Yu C, Pan W. Diet-induced obesity suppresses expression of many proteins at the blood-brain barrier. J Cereb Blood Flow Metab. 2014;34:43–51. https://doi.org/10.1038/jcbfm.2013.166.
Tucsek Z, Toth P, Sosnowska D, Gautam T, Mitschelen M, Koller A, Szalai G, Sonntag WE, Ungvari Z, Csiszar A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. 2014;69:1212–26. https://doi.org/10.1093/gerona/glt177glt177[pii].
Toth P, Tucsek Z, Sosnowska D, Gautam T, Mitschelen M, Tarantini S, Deak F, Koller A, Sonntag WE, Csiszar A, Ungvari Z. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab. 2013;33:1732–42. https://doi.org/10.1038/jcbfm.2013.143jcbfm2013143[pii].
Mazzone P, Tierney W, Hossain M, Puvenna V, Janigro D, Cucullo L. Pathophysiological impact of cigarette smoke exposure on the cerebrovascular system with a focus on the blood-brain barrier: expanding the awareness of smoking toxicity in an underappreciated area. Int J Environ Res Public Health. 2010;7:4111–26. https://doi.org/10.3390/ijerph7124111.
Kalayci R, Kaya M, Uzun H, Bilgic B, Ahishali B, Arican N, Elmas I, Kucuk M. Influence of hypercholesterolemia and hypertension on the integrity of the blood-brain barrier in rats. Int J Neurosci. 2009;119:1881–904. https://doi.org/10.1080/14647270802336650.
Acharya NK, Levin EC, Clifford PM, Han M, Tourtellotte R, Chamberlain D, Pollaro M, Coretti NJ, Kosciuk MC, Nagele EP, et al. Diabetes and hypercholesterolemia increase blood-brain barrier permeability and brain amyloid deposition: beneficial effects of the LpPLA2 inhibitor darapladib. J Alzheimers Dis. 2013;35:179–98. https://doi.org/10.3233/JAD-122254.
van den Brink H, Doubal FN, Duering M. Advanced MRI in cerebral small vessel disease. Int J Stroke. 2023;18:28–35. https://doi.org/10.1177/17474930221091879.
Ren B, Tan L, Song Y, Li D, Xue B, Lai X, Gao Y. Cerebral small vessel disease: neuroimaging features, biochemical markers, influencing factors, pathological mechanism and treatment. Front Neurol. 2022;13: 843953. https://doi.org/10.3389/fneur.2022.843953.
Chen X, Wang J, Shan Y, Cai W, Liu S, Hu M, Liao S, Huang X, Zhang B, Wang Y, Lu Z. Cerebral small vessel disease: neuroimaging markers and clinical implication. J Neurol. 2019;266:2347–62. https://doi.org/10.1007/s00415-018-9077-3.
Thrippleton MJ, Backes WH, Sourbron S, Ingrisch M, van Osch MJP, Dichgans M, Fazekas F, Ropele S, Frayne R, van Oostenbrugge RJ, et al. Quantifying blood-brain barrier leakage in small vessel disease: review and consensus recommendations. Alzheimers Dement. 2019;15:840–58. https://doi.org/10.1016/j.jalz.2019.01.013.
The LSG, Poggesi A, Pantoni L, Inzitari D, Fazekas F, Ferro J, O’Brien J, Hennerici M, Scheltens P, Erkinjuntti T, et al. 2001–2011: A decade of the LADIS (Leukoaraiosis And DISability) study: what have we learned about white matter changes and small-vessel disease? Cerebrovasc Dis. 2011;32:577–88. https://doi.org/10.1159/000334498.
Schmidt R, Scheltens P, Erkinjuntti T, Pantoni L, Markus HS, Wallin A, Barkhof F, Fazekas F. White matter lesion progression: a surrogate endpoint for trials in cerebral small-vessel disease. Neurology. 2004;63:139–44. https://doi.org/10.1212/01.wnl.0000132635.75819.e5.
Pinter D, Enzinger C, Fazekas F. Cerebral small vessel disease, cognitive reserve and cognitive dysfunction. J Neurol. 2015;262:2411–9. https://doi.org/10.1007/s00415-015-7776-6.
Fazekas F, Ropele S, Schmidt R. Can small-vessel disease-related cerebral abnormalities be used as a surrogate marker for vascular dementia trials? J Neural Transm Suppl. 2002:61–67 https://doi.org/10.1007/978-3-7091-6139-5_6
De Guio F, Jouvent E, Biessels GJ, Black SE, Brayne C, Chen C, Cordonnier C, De Leeuw FE, Dichgans M, Doubal F, et al. Reproducibility and variability of quantitative magnetic resonance imaging markers in cerebral small vessel disease. J Cereb Blood Flow Metab. 2016;36:1319–37. https://doi.org/10.1177/0271678X16647396.
de Leeuw FE, de Groot JC, Oudkerk M, Witteman JC, Hofman A, van Gijn J, Breteler MM. Hypertension and cerebral white matter lesions in a prospective cohort study. Brain. 2002;125:765–72. https://doi.org/10.1093/brain/awf077.
Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C, Medical Research Council Cognitive F, Ageing S. Age, neuropathology, and dementia. N Engl J Med. 2009;360:2302–9. https://doi.org/10.1056/NEJMoa0806142.
Neuropathology Group. Medical Research Council Cognitive F, Aging S. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet. 2001;357:169–75.
Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, Geurts JJ. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry. 2011;82:126–35. https://doi.org/10.1136/jnnp.2009.204685.
Oveisgharan S, Kim N, Agrawal S, Yu L, Leurgans S, Kapasi A, Arfanakis K, Bennett DA, Schneider JA, Buchman AS. Brain and spinal cord arteriolosclerosis and its associations with cerebrovascular disease risk factors in community-dwelling older adults. Acta Neuropathol. 2023;145:219–33. https://doi.org/10.1007/s00401-022-02527-z.
Alber J, Alladi S, Bae HJ, Barton DA, Beckett LA, Bell JM, Berman SE, Biessels GJ, Black SE, Bos I, et al. White matter hyperintensities in vascular contributions to cognitive impairment and dementia (VCID): Knowledge gaps and opportunities. Alzheimers Dement (N Y). 2019;5:107–17. https://doi.org/10.1016/j.trci.2019.02.001.
Gons RA, van Norden AG, de Laat KF, van Oudheusden LJ, van Uden IW, Zwiers MP, Norris DG, de Leeuw FE. Cigarette smoking is associated with reduced microstructural integrity of cerebral white matter. Brain. 2011;134:2116–24. https://doi.org/10.1093/brain/awr145.
Benedictus MR, Goos JD, Binnewijzend MA, Muller M, Barkhof F, Scheltens P, Prins ND, van der Flier WM. Specific risk factors for microbleeds and white matter hyperintensities in Alzheimer’s disease. Neurobiol Aging. 2013;34:2488–94. https://doi.org/10.1016/j.neurobiolaging.2013.04.023.
Benedictus MR, van Harten AC, Leeuwis AE, Koene T, Scheltens P, Barkhof F, Prins ND, van der Flier WM. White matter hyperintensities relate to clinical progression in subjective cognitive decline. Stroke. 2015;46:2661–4. https://doi.org/10.1161/STROKEAHA.115.009475.
Freeze WM, Jacobs HI, Gronenschild EH, Jansen JF, Burgmans S, Aalten P, Clerx L, Vos SJ, van Buchem MA, Barkhof F, et al. White matter hyperintensities potentiate hippocampal volume reduction in non-demented older individuals with abnormal amyloid-beta. J Alzheimers Dis. 2017;55:333–42. https://doi.org/10.3233/JAD-160474.
Gyanwali B, Lui B, Tan CS, Chong EJY, Vrooman H, Chen C, Hilal S. Cerebral microbleeds and white matter hyperintensities are associated with cognitive decline in an asian memory clinic study. Curr Alzheimer Res. 2021;18:399–413. https://doi.org/10.2174/1567205018666210820125543.
Hommet C, Mondon K, Constans T, Beaufils E, Desmidt T, Camus V, Cottier JP. Review of cerebral microangiopathy and Alzheimer’s disease: relation between white matter hyperintensities and microbleeds. Dement Geriatr Cogn Disord. 2011;32:367–78. https://doi.org/10.1159/000335568000335568[pii].
Joutel A, Monet-Lepretre M, Gosele C, Baron-Menguy C, Hammes A, Schmidt S, Lemaire-Carrette B, Domenga V, Schedl A, Lacombe P, Hubner N. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest. 2010;120:433–45. https://doi.org/10.1172/JCI39733.
Prins ND, Scheltens P. White matter hyperintensities, cognitive impairment and dementia: an update. Nat Rev Neurol. 2015;11:157–65. https://doi.org/10.1038/nrneurol.2015.10.
Baezner H, Blahak C, Poggesi A, Pantoni L, Inzitari D, Chabriat H, Erkinjuntti T, Fazekas F, Ferro JM, Langhorne P, et al. Association of gait and balance disorders with age-related white matter changes: the LADIS study. Neurology. 2008;70:935–42. https://doi.org/10.1212/01.wnl.0000305959.46197.e670/12/935[pii].
Callisaya ML, Beare R, Phan T, Blizzard L, Thrift AG, Chen J, Srikanth VK. Progression of white matter hyperintensities of presumed vascular origin increases the risk of falls in older people. J Gerontol A Biol Sci Med Sci. 2015;70:360–6. https://doi.org/10.1093/gerona/glu148glu148[pii].
Choi P, Ren M, Phan TG, Callisaya M, Ly JV, Beare R, Chong W, Srikanth V. Silent infarcts and cerebral microbleeds modify the associations of white matter lesions with gait and postural stability: population-based study. Stroke. 2012;43:1505–10. https://doi.org/10.1161/STROKEAHA.111.647271STROKEAHA.111.647271[pii].
Hajjar I, Quach L, Yang F, Chaves PH, Newman AB, Mukamal K, Longstreth W Jr, Inzitari M, Lipsitz LA. Hypertension, white matter hyperintensities, and concurrent impairments in mobility, cognition, and mood: the Cardiovascular Health Study. Circulation. 2011;123:858–65. https://doi.org/10.1161/CIRCULATIONAHA.110.978114.
Siejka TP, Srikanth VK, Hubbard RE, Moran C, Beare R, Wood A, Phan T, Balogun S, Callisaya ML. White matter hyperintensities and the progression of frailty-the tasmanian study of cognition and gait. J Gerontol A Biol Sci Med Sci. 2020;75:1545–50. https://doi.org/10.1093/gerona/glaa024.
Zheng JJ, Lord SR, Close JC, Sachdev PS, Wen W, Brodaty H, Delbaere K. Brain white matter hyperintensities, executive dysfunction, instability, and falls in older people: a prospective cohort study. J Gerontol A Biol Sci Med Sci. 2012. https://doi.org/10.1093/gerona/gls063.
Camerino I, Sierpowska J, Reid A, Meyer NH, Tuladhar AM, Kessels RPC, de Leeuw FE, Piai V. White matter hyperintensities at critical crossroads for executive function and verbal abilities in small vessel disease. Hum Brain Mapp. 2021;42:993–1002. https://doi.org/10.1002/hbm.25273.
de Groot JC, de Leeuw FE, Oudkerk M, van Gijn J, Hofman A, Jolles J, Breteler MM. Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann Neurol. 2000;47:145–51. https://doi.org/10.1002/1531-8249(200002)47:2%3c145::aid-ana3%3e3.3.co;2-g.
de Groot JC, de Leeuw FE, Oudkerk M, Hofman A, Jolles J, Breteler MM. Cerebral white matter lesions and depressive symptoms in elderly adults. Arch Gen Psychiatry. 2000;57:1071–6. https://doi.org/10.1001/archpsyc.57.11.1071.
de Laat KF, Tuladhar AM, van Norden AG, Norris DG, Zwiers MP, de Leeuw FE. Loss of white matter integrity is associated with gait disorders in cerebral small vessel disease. Brain. 2011;134:73–83. https://doi.org/10.1093/brain/awq343.
Leeuwis AE, Weaver NA, Biesbroek JM, Exalto LG, Kuijf HJ, Hooghiemstra AM, Prins ND, Scheltens P, Barkhof F, van der Flier WM, et al. Impact of white matter hyperintensity location on depressive symptoms in memory-clinic patients: a lesion-symptom mapping study. J Psychiatry Neurosci. 2019;44:E1–10. https://doi.org/10.1503/jpn.180136.
van der Holst HM, Tuladhar AM, Zerbi V, van Uden IWM, de Laat KF, van Leijsen EMC, Ghafoorian M, Platel B, Bergkamp MI, van Norden AGW, et al. White matter changes and gait decline in cerebral small vessel disease. Neuroimage Clin. 2018;17:731–8. https://doi.org/10.1016/j.nicl.2017.12.007.
Parodi L, Mayerhofer E, Narasimhalu K, Yechoor N, Comeau ME, Rosand J, Langefeld CD, Anderson CD. Social determinants of health and cerebral small vessel disease: is epigenetics a key mediator? J Am Heart Assoc. 2023;12: e029862. https://doi.org/10.1161/JAHA.123.029862.
Skoog I, Wallin A, Fredman P, Hesse C, Aevarsson O, Karlsson I, Gottfries CG, Blennow K. A population study on blood-brain barrier function in 85-year-olds: relation to Alzheimer’s disease and vascular dementia. Neurology. 1998;50:966–71. https://doi.org/10.1212/wnl.50.4.966.
Banks WA, Reed MJ, Logsdon AF, Rhea EM, Erickson MA. Healthy aging and the blood-brain barrier. Nat Aging. 2021;1:243–54. https://doi.org/10.1038/s43587-021-00043-5.
Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011. https://doi.org/10.1161/STROKEAHA.110.608257.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–38. https://doi.org/10.1038/nrn3114nrn3114[pii].
Liao FF, Lin G, Chen X, Chen L, Zheng W, Raghow R, Zhou FM, Shih AY, Tan XL. Endothelial nitric oxide synthase-deficient mice: a model of spontaneous cerebral small-vessel disease. Am J Pathol. 2021;191:1932–45. https://doi.org/10.1016/j.ajpath.2021.02.022.
Rajani RM, Quick S, Ruigrok SR, Graham D, Harris SE, Verhaaren BFJ, Fornage M, Seshadri S, Atanur SS, Dominiczak AF, et al. Reversal of endothelial dysfunction reduces white matter vulnerability in cerebral small vessel disease in rats. Sci Transl Med. 2018;10. https://doi.org/10.1126/scitranslmed.aam9507
Wallin A, Roman GC, Esiri M, Kettunen P, Svensson J, Paraskevas GP, Kapaki E. Update on vascular cognitive impairment associated with subcortical small-vessel disease. J Alzheimers Dis. 2018;62:1417–41. https://doi.org/10.3233/JAD-170803.
de Paula GC, Brunetta HS, Engel DF, Gaspar JM, Velloso LA, Engblom D, de Oliveira J, de Bem AF. Hippocampal function is impaired by a short-term high-fat diet in mice: increased blood-brain barrier permeability and neuroinflammation as triggering events. Front Neurosci. 2021;15: 734158. https://doi.org/10.3389/fnins.2021.734158.
Salameh TS, Mortell WG, Logsdon AF, Butterfield DA, Banks WA. Disruption of the hippocampal and hypothalamic blood-brain barrier in a diet-induced obese model of type II diabetes: prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate. Fluids Barriers CNS. 2019;16:1. https://doi.org/10.1186/s12987-018-0121-6.
Sheikh MH, Errede M, d’Amati A, Khan NQ, Fanti S, Loiola RA, McArthur S, Purvis GSD, O’Riordan CE, Ferorelli D, et al. Impact of metabolic disorders on the structural, functional, and immunological integrity of the blood-brain barrier: Therapeutic avenues. FASEB J. 2022;36: e22107. https://doi.org/10.1096/fj.202101297R.
Tarantini S, Valcarcel-Ares MN, Yabluchanskiy A, Tucsek Z, Hertelendy P, Kiss T, Gautam T, Zhang XA, Sonntag WE, de Cabo R, et al. Nrf2 deficiency exacerbates obesity-induced oxidative stress, neurovascular dysfunction, blood brain barrier disruption, neuroinflammation, amyloidogenic gene expression and cognitive decline in mice, mimicking the aging phenotype. J Gerontol A Biol Sci Med Sci. 2018:in press.
Tucsek Z, Toth P, Sosnowsk D, Gautam T, Mitschelen M, Koller A, Szalai G, Sonntag WE, Ungvari Z, Csiszar A. Obesity in aging exacerbates blood brain barrier disruption, neuroinflammation and oxidative stress in the mouse hippocampus: effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. 2014;69:1212–26.
de Montgolfier O, Pouliot P, Gillis MA, Ferland G, Lesage F, Thorin-Trescases N, Thorin E. Systolic hypertension-induced neurovascular unit disruption magnifies vascular cognitive impairment in middle-age atherosclerotic LDLr(-/-):hApoB(+/+) mice. Geroscience. 2019;41:511–32. https://doi.org/10.1007/s11357-019-00070-6.
Gulej R, Nyul-Toth A, Ahire C, DelFavero J, Balasubramanian P, Kiss T, Tarantini S, Benyo Z, Pacher P, Csik B, et al. Elimination of senescent cells by treatment with Navitoclax/ABT263 reverses whole brain irradiation-induced blood-brain barrier disruption in the mouse brain. Geroscience. 2023;45:2983–3002. https://doi.org/10.1007/s11357-023-00870-x.
Gulej R, Csik B, Faakye J, Tarantini S, Shanmugarama S, Chandragiri SS, Mukli P, Conley S, Csiszar A, Ungvari Z, et al. Endothelial deficiency of insulin-like growth factor-1 receptor leads to blood-brain barrier disruption and accelerated endothelial senescence in mice, mimicking aspects of the brain aging phenotype. Microcirculation. 2023:e12840. https://doi.org/10.1111/micc.12840
Ahire C, Nyul-Toth A, DelFavero J, Gulej R, Faakye JA, Tarantini S, Kiss T, Kuan-Celarier A, Balasubramanian P, Ungvari A, et al. Accelerated cerebromicrovascular senescence contributes to cognitive decline in a mouse model of paclitaxel (Taxol)-induced chemobrain. Aging Cell. 2023:e13832 https://doi.org/10.1111/acel.13832
Knopp RC, Erickson MA, Rhea EM, Reed MJ, Banks WA. Cellular senescence and the blood-brain barrier: implications for aging and age-related diseases. Exp Biol Med (Maywood). 2023;248:399–411. https://doi.org/10.1177/15353702231157917.
Stamatovic SM, Martinez-Revollar G, Hu A, Choi J, Keep RF, Andjelkovic AV. Decline in Sirtuin-1 expression and activity plays a critical role in blood-brain barrier permeability in aging. Neurobiol Dis. 2019;126:105–16. https://doi.org/10.1016/j.nbd.2018.09.006.
Yamazaki Y, Baker DJ, Tachibana M, Liu CC, van Deursen JM, Brott TG, Bu G, Kanekiyo T. Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke. 2016;47:1068–77. https://doi.org/10.1161/STROKEAHA.115.010835.
Kiss T, Tarantini S, Csipo T, Balasubramanian P, Nyul-Toth A, Yabluchanskiy A, Wren JD, Garman L, Huffman DM, Csiszar A, Ungvari Z. Circulating anti-geronic factors from heterochonic parabionts promote vascular rejuvenation in aged mice: transcriptional footprint of mitochondrial protection, attenuation of oxidative stress, and rescue of endothelial function by young blood. Geroscience. 2020;42:727–48. https://doi.org/10.1007/s11357-020-00180-6.
Gulej R, Nyul-Toth A, Csik B, Petersen B, Faakye J, Negri S, Chandragiri SS, Mukli P, Yabluchanskiy A, Conley S, et al. Rejuvenation of cerebromicrovascular function in aged mice through heterochronic parabiosis: insights into neurovascular coupling and the impact of young blood factors. Geroscience. 2023. https://doi.org/10.1007/s11357-023-01039-2.
Gulej R, Nyúl-Tóth Á, Csik B, Patai R, Petersen B, Negri S, Chandragiri SS, Shanmugarama S, Mukli P, Yabluchanskiy A, Conley S, Huffman D, Tarantini S, Csiszar A, Ungvari Z. Young blood-mediated cerebromicrovascular rejuvenation through heterochronic parabiosis: enhancing blood-brain barrier integrity and capillarization in the aged mouse brain. GeroScience. 2024. https://doi.org/10.1007/s11357-024-01154-8
van den Kerkhof M, Voorter PHM, Canjels LPW, de Jong JJA, van Oostenbrugge RJ, Kroon AA, Jansen JFA, Backes WH. Time-efficient measurement of subtle blood-brain barrier leakage using a T(1) mapping MRI protocol at 7 T. Magn Reson Med. 2021;85:2761–70. https://doi.org/10.1002/mrm.28629.
Montagne A, Barnes SR, Nation DA, Kisler K, Toga AW, Zlokovic BV. Imaging subtle leaks in the blood-brain barrier in the aging human brain: potential pitfalls, challenges, and possible solutions. Geroscience. 2022;44:1339–51. https://doi.org/10.1007/s11357-022-00571-x.
Musaeus CS, Gleerup HS, Hogh P, Waldemar G, Hasselbalch SG, Simonsen AH. Cerebrospinal fluid/plasma albumin ratio as a biomarker for blood-brain barrier impairment across neurodegenerative dementias. J Alzheimers Dis. 2020;75:429–36. https://doi.org/10.3233/JAD-200168.
Musaeus CS, Gleerup HS, Hasselbalch SG, Waldemar G, Simonsen AH. Progression of blood-brain barrier leakage in patients with Alzheimer’s disease as measured with the cerebrospinal fluid/plasma albumin ratio over time. J Alzheimers Dis Rep. 2023;7:535–41. https://doi.org/10.3233/ADR-230016.
Kay AD, May C, Papadopoulos NM, Costello R, Atack JR, Luxenberg JS, Cutler NR, Rapoport SI. CSF and serum concentrations of albumin and IgG in Alzheimer’s disease. Neurobiol Aging. 1987;8:21–5. https://doi.org/10.1016/0197-4580(87)90053-4.
Garton MJ, Keir G, Lakshmi MV, Thompson EJ. Age-related changes in cerebrospinal fluid protein concentrations. J Neurol Sci. 1991;104:74–80. https://doi.org/10.1016/0022-510x(91)90218-v.
Jesse S, Brettschneider J, Sussmuth SD, Landwehrmeyer BG, von Arnim CA, Ludolph AC, Tumani H, Otto M. Summary of cerebrospinal fluid routine parameters in neurodegenerative diseases. J Neurol. 2011;258:1034–41. https://doi.org/10.1007/s00415-010-5876-x.
Binnie LR, Pauls MMH, Benjamin P, Dhillon MK, Betteridge S, Clarke B, Ghatala R, Hainsworth FAH, Howe FA, Khan U, et al. Test-retest reliability of arterial spin labelling for cerebral blood flow in older adults with small vessel disease. Transl Stroke Res. 2022;13:583–94. https://doi.org/10.1007/s12975-021-00983-5.
Terlecki P, Przywara S, Terlecki K, Janczak D, Antkiewicz M, Zubilewicz T. Effect of Reconstructive procedures of the extracranial segment of the carotid arteries on damage to the blood-brain barrier. Int J Environ Res Public Health. 2022;19 https://doi.org/10.3390/ijerph19106210
Hosoki S, Hansra GK, Jayasena T, Poljak A, Mather KA, Catts VS, Rust R, Sagare A, Kovacic JC, Brodtmann A, et al. Molecular biomarkers for vascular cognitive impairment and dementia. Nat Rev Neurol. 2023;19:737–53. https://doi.org/10.1038/s41582-023-00884-1.
Janes F, Cifu A, Pessa ME, Domenis R, Gigli GL, Sanvilli N, Nilo A, Garbo R, Curcio F, Giacomello R, et al. ADMA as a possible marker of endothelial damage A study in young asymptomatic patients with cerebral small vessel disease. Sci Rep. 2019;9:14207. https://doi.org/10.1038/s41598-019-50778-w.
Ihara M, Washida K, Yoshimoto T, Saito S. Adrenomedullin: a vasoactive agent for sporadic and hereditary vascular cognitive impairment. Cereb Circ Cogn Behav. 2021;2: 100007. https://doi.org/10.1016/j.cccb.2021.100007.
Hinman JD, Elahi F, Chong D, Radabaugh H, Ferguson A, Maillard P, Thompson JF, Rosenberg GA, Sagare A, Moghekar A, et al. Placental growth factor as a sensitive biomarker for vascular cognitive impairment. Alzheimers Dement. 2023;19:3519–27. https://doi.org/10.1002/alz.12974.
Cordon J, Duggan MR, Gomez GT, Pucha KA, Peng Z, Dark HE, Davatzikos C, Erus G, Lewis A, Moghekar A, et al. Identification of clinically relevant brain endothelial cell biomarkers in plasma. Stroke. 2023;54:2853–63. https://doi.org/10.1161/STROKEAHA.123.043908.
McAleese KE, Graham S, Dey M, Walker L, Erskine D, Johnson M, Johnston E, Thomas AJ, McKeith IG, DeCarli C, Attems J. Extravascular fibrinogen in the white matter of Alzheimer’s disease and normal aged brains: implications for fibrinogen as a biomarker for Alzheimer’s disease. Brain Pathol. 2019;29:414–24. https://doi.org/10.1111/bpa.12685.
Wang J, Fan DY, Li HY, He CY, Shen YY, Zeng GH, Chen DW, Yi X, Ma YH, Yu JT, Wang YJ. Dynamic changes of CSF sPDGFRbeta during ageing and AD progression and associations with CSF ATN biomarkers. Mol Neurodegener. 2022;17:9. https://doi.org/10.1186/s13024-021-00512-w.
Sagare AP, Sweeney MD, Makshanoff J, Zlokovic BV. Shedding of soluble platelet-derived growth factor receptor-beta from human brain pericytes. Neurosci Lett. 2015;607:97–101. https://doi.org/10.1016/j.neulet.2015.09.025S0304-3940(15)30151-8[pii].
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. https://doi.org/10.1016/j.neuron.2014.12.032S0896-6273(14)01141-6[pii].
Bjerke M, Zetterberg H, Edman A, Blennow K, Wallin A, Andreasson U. Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in vascular dementia and Alzheimer’s disease. J Alzheimers Dis. 2011;27:665–76. https://doi.org/10.3233/JAD-2011-110566.
Candelario-Jalil E, Thompson J, Taheri S, Grossetete M, Adair JC, Edmonds E, Prestopnik J, Wills J, Rosenberg GA. Matrix metalloproteinases are associated with increased blood-brain barrier opening in vascular cognitive impairment. Stroke. 2011;42:1345–50. https://doi.org/10.1161/STROKEAHA.110.600825.
Adair JC, Charlie J, Dencoff JE, Kaye JA, Quinn JF, Camicioli RM, Stetler-Stevenson WG, Rosenberg GA. Measurement of gelatinase B (MMP-9) in the cerebrospinal fluid of patients with vascular dementia and Alzheimer disease. Stroke. 2004;35:e159-162. https://doi.org/10.1161/01.STR.0000127420.10990.76.
Erdo F, Denes L, de Lange E. Age-associated physiological and pathological changes at the blood-brain barrier: a review. J Cereb Blood Flow Metab. 2017;37:4–24. https://doi.org/10.1177/0271678X16679420.
Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease–systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–52. https://doi.org/10.1016/j.neurobiolaging.2007.07.015.
Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK, Buxton AE, et al. Heart Disease and Stroke Statistics-2023 Update: a report from the American Heart Association. Circulation. 2023;147:e93–621. https://doi.org/10.1161/CIR.0000000000001123.
Duong MT, Nasrallah IM, Wolk DA, Chang CCY, Chang TY. Cholesterol, atherosclerosis, and APOE in vascular contributions to cognitive impairment and dementia (VCID): Potential mechanisms and therapy. Front Aging Neurosci. 2021;13: 647990. https://doi.org/10.3389/fnagi.2021.647990.
Baradaran H, Mtui EE, Richardson JE, Delgado D, Dunning A, Marshall RS, Sanelli PC, Gupta A. White matter diffusion abnormalities in carotid artery disease: a systematic review and meta-analysis. J Neuroimaging. 2016;26:481–8. https://doi.org/10.1111/jon.12347.
Komura S, Nomura T, Imaizumi T, Inamura S, Kanno A, Honda O, Hashimoto Y, Mikami T, Nonaka T. Asymptomatic cerebral findings on 3-Tesla MRI in patients with severe carotid artery stenoses. J Clin Neurosci. 2022;101:106–11. https://doi.org/10.1016/j.jocn.2022.05.004.
Baradaran H, Culleton S, Stoddard G, Alexander MD, Romero JR, Hadley JR, Kim SE, Parker DL, McNally JS. Association between high-risk extracranial carotid plaque and covert brain infarctions and cerebral microbleeds. Neuroradiology. 2023;65:287–95. https://doi.org/10.1007/s00234-022-03062-0.
Bos D, Ikram MA, Elias-Smale SE, Krestin GP, Hofman A, Witteman JC, van der Lugt A, Vernooij MW. Calcification in major vessel beds relates to vascular brain disease. Arterioscler Thromb Vasc Biol. 2011;31:2331–7. https://doi.org/10.1161/ATVBAHA.111.232728.
Zhao FF, Gao HY, Gao Y, Zhao Z, Li J, Ning FB, Zhang XN, Wang ZG, Yu AL, Guo YY, Sun BL. A correlational study on cerebral microbleeds and carotid atherosclerosis in patients with ischemic stroke. J Stroke Cerebrovasc Dis. 2018;27:2228–34. https://doi.org/10.1016/j.jstrokecerebrovasdis.2018.04.009.
Vidal JS, Sigurdsson S, Jonsdottir MK, Eiriksdottir G, Thorgeirsson G, Kjartansson O, Garcia ME, van Buchem MA, Harris TB, Gudnason V, Launer LJ. Coronary artery calcium, brain function and structure: the AGES-Reykjavik Study. Stroke. 2010;41:891–7. https://doi.org/10.1161/STROKEAHA.110.579581.
Choi J, Kim JY, Kwon HJ, Choi HJ, Kim SH, Kim S, Lee J, Park JE. Association of cerebral white matter hyperintensities with coronary artery calcium in a healthy population: a cross-sectional study. Sci Rep. 2022;12:21562. https://doi.org/10.1038/s41598-022-25654-9.
Jin H, Qin X, Zhao F, Yan Y, Meng Y, Shu Z, Gong X. Is coronary artery calcium an independent risk factor for white matter hyperintensity? BMC Neurol. 2023;23:313. https://doi.org/10.1186/s12883-023-03364-7.
Ozeren A, Acarturk E, Koc F, Demir M, Sarica Y, Eroglu H. Silent cerebral lesions on magnetic resonance imaging in subjects with coronary artery disease. Jpn Heart J. 1998;39:611–8. https://doi.org/10.1536/ihj.39.611.
Johansen MC, Gottesman RF, Kral BG, Vaidya D, Yanek LR, Becker LC, Becker DM, Nyquist P. Association of coronary artery atherosclerosis with brain white matter hyperintensity. Stroke. 2021;52:2594–600. https://doi.org/10.1161/STROKEAHA.120.032674.
Szarmach A, Halena G, Kaszubowski M, Piskunowicz M, Studniarek M, Lass P, Szurowska E, Winklewski PJ. Carotid artery stenting and blood-brain barrier permeability in subjects with chronic carotid artery stenosis. Int J Mol Sci. 2017;18 https://doi.org/10.3390/ijms18051008
de Leeuw FE, De Groot JC, Oudkerk M, Witteman JC, Hofman A, van Gijn J, Breteler MM. Aortic atherosclerosis at middle age predicts cerebral white matter lesions in the elderly. Stroke. 2000;31:425–9. https://doi.org/10.1161/01.str.31.2.425.
Van Skike CE, Jahrling JB, Olson AB, Sayre NL, Hussong SA, Ungvari Z, Lechleiter JD, Galvan V. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer’s disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol. 2018;314:H693–703. https://doi.org/10.1152/ajpheart.00570.2017.
Roberts JM, Maniskas ME, Bix GJ. Bilateral carotid artery stenosis causes unexpected early changes in brain extracellular matrix and blood-brain barrier integrity in mice. PLoS ONE. 2018;13: e0195765. https://doi.org/10.1371/journal.pone.0195765.
Contu L, Nizari S, Heath CJ, Hawkes CA. Pre- and post-natal high fat feeding differentially affects the structure and integrity of the neurovascular unit of 16-month old male and female mice. Front Neurosci. 2019;13:1045. https://doi.org/10.3389/fnins.2019.01045.
Badaut J, Copin JC, Fukuda AM, Gasche Y, Schaller K, da Silva RF. Increase of arginase activity in old apolipoprotein-E deficient mice under Western diet associated with changes in neurovascular unit. J Neuroinflammation. 2012;9:132. https://doi.org/10.1186/1742-2094-9-1321742-2094-9-132[pii].
Davanzo GG, Castro G, Monteiro LB, Castelucci BG, Jaccomo VH, da Silva FC, Marques AM, Francelin C, de Campos BB, de Aguiar CF, et al. Obesity increases blood-brain barrier permeability and aggravates the mouse model of multiple sclerosis. Mult Scler Relat Disord. 2023;72: 104605. https://doi.org/10.1016/j.msard.2023.104605.
Elahy M, Lam V, Pallebage-Gamarallage MM, Giles C, Mamo JC, Takechi R. Nicotine attenuates disruption of blood-brain barrier induced by saturated-fat feeding in wild-type mice. Nicotine Tob Res. 2015;17:1436–41. https://doi.org/10.1093/ntr/ntv044.
Li C, Shi L, Wang Y, Peng C, Wu L, Zhang Y, Du Z. High-fat diet exacerbates lead-induced blood-brain barrier disruption by disrupting tight junction integrity. Environ Toxicol. 2021;36:1412–21. https://doi.org/10.1002/tox.23137.
Liu Z, Hua W, Jin S, Wang Y, Pang Y, Wang B, Zhao N, Qi J, Song Y. Canagliflozin protects against hyperglycemia-induced cerebrovascular injury by preventing blood-brain barrier (BBB) disruption via AMPK/Sp1/adenosine A2A receptor. Eur J Pharmacol. 2024:176381 https://doi.org/10.1016/j.ejphar.2024.176381
Mamo JC, Lam V, Brook E, Mooranian A, Al-Salami H, Fimognari N, Nesbit M, Takechi R. Probucol prevents blood-brain barrier dysfunction and cognitive decline in mice maintained on pro-diabetic diet. Diab Vasc Dis Res. 2019;16:87–97. https://doi.org/10.1177/1479164118795274.
Mamo JCL, Lam V, Giles C, Coulson SH, Fimognari N, Mooranian A, Al-Salami H, Takechi R. Antihypertensive agents do not prevent blood-brain barrier dysfunction and cognitive deficits in dietary-induced obese mice. Int J Obes (Lond). 2017;41:926–34. https://doi.org/10.1038/ijo.2017.57.
Ogata S, Ito S, Masuda T, Ohtsuki S. Changes of blood-brain barrier and brain parenchymal protein expression levels of mice under different insulin-resistance conditions induced by high-fat diet. Pharm Res. 2019;36:141. https://doi.org/10.1007/s11095-019-2674-8.
Ramirez-Cruz A, Gomez-Gonzalez B, Baiza-Gutman LA, Manuel-Apolinar L, Angeles-Mejia S, Lopez-Cervantes SP, Ortega-Camarillo C, Cruz-Lopez M, Gomez-Olivares JL, Diaz-Flores M. Nicotinamide, an acetylcholinesterase uncompetitive inhibitor, protects the blood-brain barrier and improves cognitive function in rats fed a hypercaloric diet. Eur J Pharmacol. 2023;959: 176068. https://doi.org/10.1016/j.ejphar.2023.176068.
Collins AR, Lyon CJ, Xia X, Liu JZ, Tangirala RK, Yin F, Boyadjian R, Bikineyeva A, Pratico D, Harrison DG, Hsueh WA. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ Res. 2009;104:e42-54. https://doi.org/10.1161/CIRCRESAHA.108.188771.
Schreyer SA, Wilson DL, LeBoeuf RC. C57BL/6 mice fed high fat diets as models for diabetes-accelerated atherosclerosis. Atherosclerosis. 1998;136:17–24. https://doi.org/10.1016/S0021-9150(97)00165-2.
Fulop GA, Kiss T, Tarantini S, Balasubramanian P, Yabluchanskiy A, Farkas E, Bari F, Ungvari Z, Csiszar A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience. 2018;40:513–21. https://doi.org/10.1007/s11357-018-0047-6.
Ungvari Z, Bailey-Downs L, Sosnowska D, Gautam T, Koncz P, Losonczy G, Ballabh P, de Cabo R, Sonntag WE, Csiszar A. Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of Nrf2-mediated antioxidant response. Am J Physiol Heart Circ Physiol. 2011;301:363–72.
Ungvari Z, Bailey-Downs L, Gautam T, Sosnowska D, Wang M, Monticone RE, Telljohann R, Pinto JT, de Cabo R, Sonntag WE, et al. Age-associated vascular oxidative stress, Nrf2 dysfunction and NF-kB activation in the non-human primate Macaca mulatta. J Gerontol A Biol Sci Med Sci. 2011;66:866–75.
Barabasi B, Barna L, Santa-Maria AR, Harazin A, Molnar R, Kincses A, Vigh JP, Dukay B, Santha M, Toth ME, et al. Role of interleukin-6 and interleukin-10 in morphological and functional changes of the blood-brain barrier in hypertriglyceridemia. Fluids Barriers CNS. 2023;20:15. https://doi.org/10.1186/s12987-023-00418-3.
Gottesman RF, Albert MS, Alonso A, Coker LH, Coresh J, Davis SM, Deal JA, McKhann GM, Mosley TH, Sharrett AR, et al. Associations between midlife vascular risk factors and 25-year incident dementia in the Atherosclerosis Risk in Communities (ARIC) Cohort. JAMA Neurol. 2017;74:1246–54. https://doi.org/10.1001/jamaneurol.2017.1658.
Feinberg MW, Moore KJ. MicroRNA regulation of atherosclerosis. Circ Res. 2016;118:703–20. https://doi.org/10.1161/CIRCRESAHA.115.306300.
Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111:245–59. https://doi.org/10.1161/CIRCRESAHA.111.261388.
Higashi Y, Sukhanov S, Anwar A, Shai SY, Delafontaine P. Aging, atherosclerosis, and IGF-1. J Gerontol A Biol Sci Med Sci. 2012;67:626–39. https://doi.org/10.1093/gerona/gls102gls102[pii].
Kiss MG, Binder CJ. The multifaceted impact of complement on atherosclerosis. Atherosclerosis. 2022;351:29–40. https://doi.org/10.1016/j.atherosclerosis.2022.03.014.
Propson NE, Roy ER, Litvinchuk A, Kohl J, Zheng H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J Clin Invest. 2021;131 https://doi.org/10.1172/JCI140966
Hartmann P, Zhou Z, Natarelli L, Wei Y, Nazari-Jahantigh M, Zhu M, Grommes J, Steffens S, Weber C, Schober A. Endothelial Dicer promotes atherosclerosis and vascular inflammation by miRNA-103-mediated suppression of KLF4. Nat Commun. 2016;7:10521. https://doi.org/10.1038/ncomms10521.
Cooper A, Heagerty AM. Endothelial dysfunction in human intramyocardial small arteries in atherosclerosis and hypercholesterolemia. Am J Physiol. 1998;275:H1482-1488. https://doi.org/10.1152/ajpheart.1998.275.4.H1482.
Matsumoto S, Gotoh N, Hishinuma S, Abe Y, Shimizu Y, Katano Y, Ishihata A. The role of hypertriglyceridemia in the development of atherosclerosis and endothelial dysfunction. Nutrients. 2014;6:1236–50. https://doi.org/10.3390/nu6031236.
Walker LN, Bowyer DE. Endothelial healing in the rabbit aorta and the effect of risk factors for atherosclerosis. Hypercholesterolemia Arteriosclerosis. 1984;4:479–88. https://doi.org/10.1161/01.atv.4.5.479.
Su C, Lu Y, Wang Z, Guo J, Hou Y, Wang X, Qin Z, Gao J, Sun Z, Dai Y, et al. Atherosclerosis: the involvement of immunity, cytokines and cells in pathogenesis, and potential novel therapeutics. Aging Dis. 2023;14:1214–42. https://doi.org/10.14336/AD.2022.1208.
Bedekovic D, Bosnjak I, Saric S, Kirner D, Novak S. Role of inflammatory cytokines in rheumatoid arthritis and development of atherosclerosis: a review. Medicina (Kaunas). 2023;59 https://doi.org/10.3390/medicina59091550
Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:969–79. https://doi.org/10.1161/ATVBAHA.110.207415.
Young JL, Libby P, Schonbeck U. Cytokines in the pathogenesis of atherosclerosis. Thromb Haemost. 2002;88:554–67.
Hillmer L, Erhardt EB, Caprihan A, Adair JC, Knoefel JE, Prestopnik J, Thompson J, Hobson S, Rosenberg GA. Blood-brain barrier disruption measured by albumin index correlates with inflammatory fluid biomarkers. J Cereb Blood Flow Metab. 2023;43:712–21. https://doi.org/10.1177/0271678X221146127.
Denes A, Drake C, Stordy J, Chamberlain J, McColl BW, Gram H, Crossman D, Francis S, Allan SM, Rothwell NJ. Interleukin-1 mediates neuroinflammatory changes associated with diet-induced atherosclerosis. J Am Heart Assoc. 2012;1: e002006. https://doi.org/10.1161/JAHA.112.002006.
Wilhelm I, Nyul-Toth A, Kozma M, Farkas AE, Krizbai IA. Role of pattern recognition receptors of the neurovascular unit in inflamm-aging. Am J Physiol Heart Circ Physiol. 2017;313:H1000–12. https://doi.org/10.1152/ajpheart.00106.2017.
Meszaros A, Molnar K, Nogradi B, Hernadi Z, Nyul-Toth A, Wilhelm I, Krizbai IA. Neurovascular inflammaging in health and disease. Cells. 2020;9 https://doi.org/10.3390/cells9071614
Serna-Rodriguez MF, Bernal-Vega S, de la Barquera JAO, Camacho-Morales A, Perez-Maya AA. The role of damage associated molecular pattern molecules (DAMPs) and permeability of the blood-brain barrier in depression and neuroinflammation. J Neuroimmunol. 2022;371: 577951. https://doi.org/10.1016/j.jneuroim.2022.577951.
Kozma M, Meszaros A, Nyul-Toth A, Molnar K, Costea L, Hernadi Z, Fazakas C, Farkas AE, Wilhelm I, Krizbai IA. Cerebral pericytes and endothelial cells communicate through inflammasome-dependent signals. Int J Mol Sci. 2021;22 https://doi.org/10.3390/ijms22116122
Nagyoszi P, Wilhelm I, Farkas AE, Fazakas C, Dung NT, Hasko J, Krizbai IA. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int. 2010;57:556–64. https://doi.org/10.1016/j.neuint.2010.07.002.
Nagyoszi P, Nyul-Toth A, Fazakas C, Wilhelm I, Kozma M, Molnar J, Hasko J, Krizbai IA. Regulation of NOD-like receptors and inflammasome activation in cerebral endothelial cells. J Neurochem. 2015;135:551–64. https://doi.org/10.1111/jnc.13197.
Nyul-Toth A, Kozma M, Nagyoszi P, Nagy K, Fazakas C, Hasko J, Molnar K, Farkas AE, Vegh AG, Varo G, et al. Expression of pattern recognition receptors and activation of the non-canonical inflammasome pathway in brain pericytes. Brain Behav Immun. 2017;64:220–31. https://doi.org/10.1016/j.bbi.2017.04.010.
Kisucka J, Chauhan AK, Zhao BQ, Patten IS, Yesilaltay A, Krieger M, Wagner DD. Elevated levels of soluble P-selectin in mice alter blood-brain barrier function, exacerbate stroke, and promote atherosclerosis. Blood. 2009;113:6015–22. https://doi.org/10.1182/blood-2008-10-186650.
Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:9649–53. https://doi.org/10.1073/pnas.90.20.9649.
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–81. https://doi.org/10.1073/pnas.90.5.1977.
Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:8098–102. https://doi.org/10.1073/pnas.90.17.8098.
Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Absolute 10-year risk of dementia by age, sex and APOE genotype: a population-based cohort study. CMAJ. 2018;190:E1033–41. https://doi.org/10.1503/cmaj.180066.
Tzioras M, Davies C, Newman A, Jackson R, Spires-Jones T. Invited Review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2019;45:327–46. https://doi.org/10.1111/nan.12529.
Koizumi K, Hattori Y, Ahn SJ, Buendia I, Ciacciarelli A, Uekawa K, Wang G, Hiller A, Zhao L, Voss HU, et al. Apoepsilon4 disrupts neurovascular regulation and undermines white matter integrity and cognitive function. Nat Commun. 2018;9:3816. https://doi.org/10.1038/s41467-018-06301-2.
Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, Zlokovic BV. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab. 2016;36:216–27. https://doi.org/10.1038/jcbfm.2015.44.
Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, Pachicano M, Joe E, Nelson AR, D’Orazio LM, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581:71–6. https://doi.org/10.1038/s41586-020-2247-3.
Bickel MA, Csik B, Gulej R, Ungvari A, Nyul-Toth A, Conley SM. Cell non-autonomous regulation of cerebrovascular aging processes by the somatotropic axis. Front Endocrinol (Lausanne). 2023;14:1087053. https://doi.org/10.3389/fendo.2023.1087053.
Kiss T, Nyul-Toth A, Gulej R, Tarantini S, Csipo T, Mukli P, Ungvari A, Balasubramanian P, Yabluchanskiy A, Benyo Z, et al. Old blood from heterochronic parabionts accelerates vascular aging in young mice: transcriptomic signature of pathologic smooth muscle remodeling. Geroscience. 2022;44:953–81. https://doi.org/10.1007/s11357-022-00519-1.
von der Thusen JH, Borensztajn KS, Moimas S, van Heiningen S, Teeling P, van Berkel TJ, Biessen EA. IGF-1 has plaque-stabilizing effects in atherosclerosis by altering vascular smooth muscle cell phenotype. Am J Pathol. 2011;178:924–34. https://doi.org/10.1016/j.ajpath.2010.10.007S0002-9440(10)00099-4[pii].
Shai SY, Sukhanov S, Higashi Y, Vaughn C, Rosen CJ, Delafontaine P. Low circulating insulin-like growth factor i increases atherosclerosis in Apoe-deficient Mice. Am J Physiol Heart Circ Physiol. 2011. https://doi.org/10.1152/ajpheart.01081.2010.
Hirai H, Kanaya R, Maeda M, Ina K, Hayashi T. The role of insulin growth factor on atherosclerosis and endothelial function: the effect on hyperlipidemia and aging. Life Sci. 2011. https://doi.org/10.1016/j.lfs.2010.12.021.
Shai SY, Sukhanov S, Higashi Y, Vaughn C, Kelly J, Delafontaine P. Smooth muscle cell-specific insulin-like growth factor-1 overexpression in Apoe-/- mice does not alter atherosclerotic plaque burden but increases features of plaque stability. Arterioscler Thromb Vasc Biol. 2010;30:1916–24. https://doi.org/10.1161/ATVBAHA.110.210831.
Higashi Y, Sukhanov S, Anwar A, Shai SY, Delafontaine P. IGF-1, oxidative stress and atheroprotection. Trends Endocrinol Metab. 2010;21:245–54. https://doi.org/10.1016/j.tem.2009.12.005.
Abbas A, Grant PJ, Kearney MT. Role of IGF-1 in glucose regulation and cardiovascular disease. Expert Rev Cardiovasc Ther. 2008;6:1135–49. https://doi.org/10.1586/14779072.6.8.1135.
Sukhanov S, Higashi Y, Shai SY, Vaughn C, Mohler J, Li Y, Song YH, Titterington J, Delafontaine P. IGF-1 reduces inflammatory responses, suppresses oxidative stress, and decreases atherosclerosis progression in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:2684–90. https://doi.org/10.1161/ATVBAHA.107.156257.
Delafontaine P, Song YH, Li Y. Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels. Arterioscler Thromb Vasc Biol. 2004;24:435–44. https://doi.org/10.1161/01.ATV.0000105902.89459.09.
Toth L, Czigler A, Hegedus E, Komaromy H, Amrein K, Czeiter E, Yabluchanskiy A, Koller A, Orsi G, Perlaki G, et al. Age-related decline in circulating IGF-1 associates with impaired neurovascular coupling responses in older adults. Geroscience. 2022;44:2771–83. https://doi.org/10.1007/s11357-022-00623-2.
Miller LR, Tarantini S, Nyul-Toth A, Johnston MP, Martin T, Bullen EC, Bickel MA, Sonntag WE, Yabluchanskiy A, Csiszar A, et al. Increased susceptibility to cerebral microhemorrhages is associated with imaging signs of microvascular degeneration in the retina in an insulin-like growth factor 1 deficient mouse model of accelerated aging. Front Aging Neurosci. 2022;14: 788296. https://doi.org/10.3389/fnagi.2022.788296.
Tarantini S, Nyul-Toth A, Yabluchanskiy A, Csipo T, Mukli P, Balasubramanian P, Ungvari A, Toth P, Benyo Z, Sonntag WE, et al. Endothelial deficiency of insulin-like growth factor-1 receptor (IGF1R) impairs neurovascular coupling responses in mice, mimicking aspects of the brain aging phenotype. Geroscience. 2021;43:2387–94. https://doi.org/10.1007/s11357-021-00405-2.
Tarantini S, Balasubramanian P, Yabluchanskiy A, Ashpole NM, Logan S, Kiss T, Ungvari A, Nyul-Toth A, Schwartzman ML, Benyo Z, et al. IGF1R signaling regulates astrocyte-mediated neurovascular coupling in mice: implications for brain aging. Geroscience. 2021;43:901–11. https://doi.org/10.1007/s11357-021-00350-0.
Farias Quipildor GE, Mao K, Hu Z, Novaj A, Cui MH, Gulinello M, Branch CA, Gubbi S, Patel K, Moellering DR, et al. Central IGF-1 protects against features of cognitive and sensorimotor decline with aging in male mice. Geroscience. 2019;41:185–208. https://doi.org/10.1007/s11357-019-00065-3.
Fulop GA, Ramirez-Perez FI, Kiss T, Tarantini S, Valcarcel Ares MN, Toth P, Yabluchanskiy A, Conley SM, Ballabh P, Martinez-Lemus LA, et al. IGF-1 deficiency promotes pathological remodeling of cerebral arteries: a potential mechanism contributing to the pathogenesis of intracerebral hemorrhages in aging. J Gerontol A Biol Sci Med Sci. 2018. https://doi.org/10.1093/gerona/gly144.
Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, Springo Z, Fulop GA, Ashpole N, Gautam T, Giles CB, Wren JD, Sonntag WE, et al. Insulin-like growth factor 1 deficiency exacerbates hypertension-induced cerebral microhemorrhages in mice, mimicking the aging phenotype. Aging Cell. 2017;16:469–79. https://doi.org/10.1111/acel.12583.
Tarantini S, Tucsek Z, Valcarcel-Ares MN, Toth P, Gautam T, Giles CB, Ballabh P, Wei JY, Wren JD, Ashpole NM, et al. Circulating IGF-1 deficiency exacerbates hypertension-induced microvascular rarefaction in the mouse hippocampus and retrosplenial cortex: implications for cerebromicrovascular and brain aging. Age (Dordr). 2016;38:273–89. https://doi.org/10.1007/s11357-016-9931-0.
Tarantini S, Giles CB, Wren JD, Ashpole NM, Valcarcel-Ares MN, Wei JY, Sonntag WE, Ungvari Z, Csiszar A. IGF-1 deficiency in a critical period early in life influences the vascular aging phenotype in mice by altering miRNA-mediated post-transcriptional gene regulation: implications for the developmental origins of health and disease hypothesis. Age (Dordr). 2016;38:239–58. https://doi.org/10.1007/s11357-016-9943-9.
Toth P, Tarantini S, Ashpole NM, Tucsek Z, Milne GL, Valcarcel-Ares NM, Menyhart A, Farkas E, Sonntag WE, Csiszar A, Ungvari Z. IGF-1 deficiency impairs neurovascular coupling in mice: implications for cerebromicrovascular aging. Aging Cell. 2015;14:1034–44. https://doi.org/10.1111/acel.12372.
Toth P, Tucsek Z, Tarantini S, Sosnowska D, Gautam T, Mitschelen M, Koller A, Sonntag WE, Csiszar A, Ungvari Z. IGF-1 deficiency impairs cerebral myogenic autoregulation in hypertensive mice. J Cereb Blood Flow Metab. 2014;34:1887–97. https://doi.org/10.1038/jcbfm.2014.156.
Sonntag WE, Deak F, Ashpole N, Toth P, Csiszar A, Freeman W, Ungvari Z. Insulin-like growth factor-1 in CNS and cerebrovascular aging. Front Aging Neurosci. 2013;5:27. https://doi.org/10.3389/fnagi.2013.00027.
Bailey-Downs LC, Mitschelen M, Sosnowska D, Toth P, Pinto JT, Ballabh P, Valcarcel-Ares MN, Farley J, Koller A, Henthorn JC, et al. Liver-specific knockdown of IGF-1 decreases vascular oxidative stress resistance by impairing the Nrf2-dependent antioxidant response: a novel model of vascular aging. J Gerontol Biol Med Sci. 2012;67:313–29.
Csiszar A, Labinskyy N, Perez V, Recchia FA, Podlutsky A, Mukhopadhyay P, Losonczy G, Pacher P, Austad SN, Bartke A, Ungvari Z. Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. Am J Physiol Heart Circ Physiol. 2008;295:H1882-1894. https://doi.org/10.1152/ajpheart.412.2008.
Higashi Y, Gautam S, Delafontaine P, Sukhanov S. IGF-1 and cardiovascular disease. Growth Horm IGF Res. 2019;45:6–16. https://doi.org/10.1016/j.ghir.2019.01.002.
Norling AM, Gerstenecker AT, Buford TW, Khan B, Oparil S, Lazar RM. The role of exercise in the reversal of IGF-1 deficiencies in microvascular rarefaction and hypertension. Geroscience. 2019. https://doi.org/10.1007/s11357-019-00139-2.
Oomen PH, Beentjes JA, Bosma E, Smit AJ, Reitsma WD, Dullaart RP. Reduced capillary permeability and capillary density in the skin of GH-deficient adults: improvement after 12 months GH replacement. Clin Endocrinol (Oxf). 2002;56:519–24. https://doi.org/10.1046/j.1365-2265.2002.01517.x.
Lin S, Zhang Q, Shao X, Zhang T, Xue C, Shi S, Zhao D, Lin Y. IGF-1 promotes angiogenesis in endothelial cells/adipose-derived stem cells co-culture system with activation of PI3K/Akt signal pathway. Cell Prolif. 2017;50 https://doi.org/10.1111/cpr.12390
Shigematsu S, Yamauchi K, Nakajima K, Iijima S, Aizawa T, Hashizume K. IGF-1 regulates migration and angiogenesis of human endothelial cells. Endocr J. 1999;46(Suppl):S59-62. https://doi.org/10.1507/endocrj.46.suppl_s59.
Wardlaw JM, Benveniste H, Williams A. Cerebral vascular dysfunctions detected in human small vessel disease and implications for preclinical studies. Annu Rev Physiol. 2022;84:409–34. https://doi.org/10.1146/annurev-physiol-060821-014521.
Vanhoutte PM. Endothelial dysfunction and atherosclerosis. Eur Heart J. 1997;18:19–29. https://doi.org/10.1016/s0195-668x(97)90005-1.
Harrison DG. Endothelial dysfunction in atherosclerosis. Basic Res Cardiol. 1994;89(Suppl 1):87–102. https://doi.org/10.1007/978-3-642-85660-0_8.
Busse R, Fleming I. Endothelial dysfunction in atherosclerosis. J Vasc Res. 1996;33:181–94. https://doi.org/10.1159/000159147.
Anderson TJ, Gerhard MD, Meredith IT, Charbonneau F, Delagrange D, Creager MA, Selwyn AP, Ganz P. Systemic nature of endothelial dysfunction in atherosclerosis. Am J Cardiol. 1995;75:71B-74B. https://doi.org/10.1016/0002-9149(95)80017-m.
Gimbrone MA Jr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–36. https://doi.org/10.1161/CIRCRESAHA.115.306301.
Tarantini S, Valcarcel-Ares MN, Toth P, Yabluchanskiy A, Tucsek Z, Kiss T, Hertelendy P, Kinter M, Ballabh P, Sule Z, et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 2019;24: 101192. https://doi.org/10.1016/j.redox.2019.101192.
Kiss T, Balasubramanian P, Valcarcel-Ares MN, Tarantini S, Yabluchanskiy A, Csipo T, Lipecz A, Reglodi D, Zhang XA, Bari F, et al. Nicotinamide mononucleotide (NMN) treatment attenuates oxidative stress and rescues angiogenic capacity in aged cerebromicrovascular endothelial cells: a potential mechanism for the prevention of vascular cognitive impairment. Geroscience. 2019;41:619–30. https://doi.org/10.1007/s11357-019-00074-2.
Csiszar A, Yabluchanskiy A, Ungvari A, Ungvari Z, Tarantini S. Overexpression of catalase targeted to mitochondria improves neurovascular coupling responses in aged mice. Geroscience. 2019;41:609–17. https://doi.org/10.1007/s11357-019-00111-0.
Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, Fulop GA, Hertelendy P, Gautam T, Farkas E, Perz A, Rabinovitch PS, Sonntag WE, et al. Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell. 2018;17 https://doi.org/10.1111/acel.12731
Toth P, Tarantini S, Tucsek Z, Ashpole NM, Sosnowska D, Gautam T, Ballabh P, Koller A, Sonntag WE, Csiszar A, Ungvari ZI. Resveratrol treatment rescues neurovascular coupling in aged mice:role of improved cerebromicrovascular endothelial function and down-regulation of NADPH oxidas. Am J Physiol Heart Circ Physiol. 2014;306:H299-308. https://doi.org/10.1152/ajpheart.00744.2013.
Csiszar A, Gautam T, Sosnowska D, Tarantini S, Banki E, Tucsek Z, Toth P, Losonczy G, Koller A, Reglodi D, et al. Caloric restriction confers persistent anti-oxidative, pro-angiogenic, and anti-inflammatory effects and promotes anti-aging miRNA expression profile in cerebromicrovascular endothelial cells of aged rats. Am J Physiol Heart Circ Physiol. 2014;307:H292-306. https://doi.org/10.1152/ajpheart.00307.2014.
Springo Z, Tarantini S, Toth P, Tucsek Z, Koller A, Sonntag WE, Csiszar A, Ungvari Z. Aging exacerbates pressure-induced mitochondrial oxidative stress in mouse cerebral arteries. J Gerontol A Biol Sci Med Sci. 2015. https://doi.org/10.1093/gerona/glu244.
Toth P, Tarantini S, Springo Z, Tucsek Z, Gautam T, Giles CB, Wren JD, Koller A, Sonntag WE, Csiszar A, Ungvari Z. Aging exacerbates hypertension-induced cerebral microhemorrhages in mice: role of resveratrol treatment in vasoprotection. Aging Cell. 2015;14:400–8. https://doi.org/10.1111/acel.12315.
Jaime Garcia D, Chagnot A, Wardlaw JM, Montagne A. A scoping review on biomarkers of endothelial dysfunction in small vessel disease: molecular insights from human studies. Int J Mol Sci. 2023;24 https://doi.org/10.3390/ijms241713114
Sleight E, Stringer MS, Clancy U, Arteaga C, Jaime Garcia D, Hewins W, Jochems ACC, Hamilton OKL, Manning C, Morgan AG, et al. Cerebrovascular reactivity in patients with small vessel disease: a cross-sectional study. Stroke. 2023;54:2776–84. https://doi.org/10.1161/STROKEAHA.123.042656.
Nyul-Toth A, Tarantini S, DelFavero J, Yan F, Balasubramanian P, Yabluchanskiy A, Ahire C, Kiss T, Csipo T, Lipecz A, et al. Demonstration of age-related blood-brain barrier disruption and cerebromicrovascular rarefaction in mice by longitudinal intravital two-photon microscopy and optical coherence tomography. Am J Physiol Heart Circ Physiol. 2021;320:H1370–92. https://doi.org/10.1152/ajpheart.00709.2020.
Galea I. The blood-brain barrier in systemic infection and inflammation. Cell Mol Immunol. 2021;18:2489–501. https://doi.org/10.1038/s41423-021-00757-x.
Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun. 2017;60:1–12. https://doi.org/10.1016/j.bbi.2016.03.010.
Evans NR, Tarkin JM, Walsh J, Chowdhury MM, Patterson AJ, Graves MJ, Rudd JHF, Warburton EA. Carotid atheroinflammation is associated with cerebral small vessel disease severity. Front Neurol. 2021;12: 690935. https://doi.org/10.3389/fneur.2021.690935.
Srinivasan V, Braidy N, Chan EK, Xu YH, Chan DK. Genetic and environmental factors in vascular dementia: an update of blood brain barrier dysfunction. Clin Exp Pharmacol Physiol. 2016;43:515–21. https://doi.org/10.1111/1440-1681.12558.
Uryga AK, Bennett MR. Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J Physiol. 2016;594:2115–24. https://doi.org/10.1113/JP270923.
Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15:973–7. https://doi.org/10.1111/acel.12458.
Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–7. https://doi.org/10.1126/science.aaf6659.
Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, Gray K, Kumar S, Clarke M, Bennett M. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132:1909–19. https://doi.org/10.1161/CIRCULATIONAHA.115.016457.
Gardner SE, Humphry M, Bennett MR, Clarke MC. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1alpha-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol. 2015;35:1963–74. https://doi.org/10.1161/ATVBAHA.115.305896.
Hayashi T, Kotani H, Yamaguchi T, Taguchi K, Iida M, Ina K, Maeda M, Kuzuya M, Hattori Y, Ignarro LJ. Endothelial cellular senescence is inhibited by liver X receptor activation with an additional mechanism for its atheroprotection in diabetes. Proc Natl Acad Sci U S A. 2014;111:1168–73. https://doi.org/10.1073/pnas.1322153111.
Shi Q, Hornsby PJ, Meng Q, Vandeberg JF, Vandeberg JL. Longitudinal analysis of short-term high-fat diet on endothelial senescence in baboons. Am J Cardiovasc Dis. 2013;3:107–19.
Gorenne I, Kumar S, Gray K, Figg N, Yu H, Mercer J, Bennett M. Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation. 2013;127:386–96. https://doi.org/10.1161/CIRCULATIONAHA.112.124404.
Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, De Meyer GRY. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. 2018;114:622–34. https://doi.org/10.1093/cvr/cvy007.
Hayashi T, Matsui-Hirai H, Miyazaki-Akita A, Fukatsu A, Funami J, Ding QF, Kamalanathan S, Hattori Y, Ignarro LJ, Iguchi A. Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci USA. 2006;103:17018–23.
Marfella R, Di Filippo C, Laieta MT, Vestini R, Barbieri M, Sangiulo P, Crescenzi B, Ferraraccio F, Rossi F, D’Amico M, Paolisso G. Effects of ubiquitin-proteasome system deregulation on the vascular senescence and atherosclerosis process in elderly patients. J Gerontol A Biol Sci Med Sci. 2008;63:200–3. https://doi.org/10.1093/gerona/63.2.200.
Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, Bennett M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99:156–64. https://doi.org/10.1161/01.RES.0000233315.38086.bc.
Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15–26.
Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–4.
Minamino T, Yoshida T, Tateno K, Miyauchi H, Zou Y, Toko H, Komuro I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation. 2003;108:2264–9.
Ota H, Eto M, Ogawa S, Iijima K, Akishita M, Ouchi Y. SIRT1/eNOS axis as a potential target against vascular senescence, dysfunction and atherosclerosis. J Atheroscler Thromb. 2010;17:431–5.
Faakye J, Nyul-Toth A, Muranyi M, Gulej R, Csik B, Shanmugarama S, Tarantini S, Negri S, Prodan C, Mukli P, et al. Preventing spontaneous cerebral microhemorrhages in aging mice: a novel approach targeting cellular senescence with ABT263/navitoclax. Geroscience. 2023. https://doi.org/10.1007/s11357-023-01024-9.
Kiss T, Nyul-Toth A, Balasubramanian P, Tarantini S, Ahire C, DelFavero J, Yabluchanskiy A, Csipo T, Farkas E, Wiley G, et al. Single-cell RNA sequencing identifies senescent cerebromicrovascular endothelial cells in the aged mouse brain. Geroscience. 2020. https://doi.org/10.1007/s11357-020-00177-1.
Tarantini S, Balasubramanian P, Delfavero J, Csipo T, Yabluchanskiy A, Kiss T, Nyul-Toth A, Mukli P, Toth P, Ahire C, et al. Treatment with the BCL-2/BCL-xL inhibitor senolytic drug ABT263/Navitoclax improves functional hyperemia in aged mice. Geroscience. 2021;43:2427–40. https://doi.org/10.1007/s11357-021-00440-z.
Ungvari Z, Podlutsky A, Sosnowska D, Tucsek Z, Toth P, Deak F, Gautam T, Csiszar A, Sonntag WE. Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J Gerontol A Biol Sci Med Sci. 2013;68:1443–57. https://doi.org/10.1093/gerona/glt057glt057[pii].
Kiss T, Nyul-Toth A, DelFavero J, Balasubramanian P, Tarantini S, Faakye J, Gulej R, Ahire C, Ungvari A, Yabluchanskiy A, et al. Spatial transcriptomic analysis reveals inflammatory foci defined by senescent cells in the white matter, hippocampi and cortical grey matter in the aged mouse brain. Geroscience. 2022;44:661–81. https://doi.org/10.1007/s11357-022-00521-7.
Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to disease and back. Physiol Rev. 2019;99:21–78. https://doi.org/10.1152/physrev.00050.2017.
Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270–6. https://doi.org/10.1038/s41591-018-0297-y.
Verheggen ICM, de Jong JJA, van Boxtel MPJ, Postma AA, Jansen JFA, Verhey FRJ, Backes WH. Imaging the role of blood-brain barrier disruption in normal cognitive ageing. Geroscience. 2020. https://doi.org/10.1007/s11357-020-00282-1.
Verheggen ICM, de Jong JJA, van Boxtel MPJ, Gronenschild E, Palm WM, Postma AA, Jansen JFA, Verhey FRJ, Backes WH. Increase in blood-brain barrier leakage in healthy, older adults. Geroscience. 2020;42:1183–93. https://doi.org/10.1007/s11357-020-00211-2.
Malkiewicz MA, Szarmach A, Sabisz A, Cubala WJ, Szurowska E, Winklewski PJ. Blood-brain barrier permeability and physical exercise. J Neuroinflammation. 2019;16:15. https://doi.org/10.1186/s12974-019-1403-x.
Buttler L, Jordao MT, Fragas MG, Ruggeri A, Ceroni A, Michelini LC. Maintenance of blood-brain barrier integrity in hypertension: a novel benefit of exercise training for autonomic control. Front Physiol. 2017;8:1048. https://doi.org/10.3389/fphys.2017.01048.
Funding
Open access funding provided by Semmelweis University. This work was supported by grants from the American Heart Association (RG: AHA916225, ANT: AHA834339, and ST: AHA CDA941290), the Oklahoma Center for the Advancement of Science and Technology, the National Institute on Aging (RF1AG072295, R01AG055395, R01AG068295, R01AG070915, K01AG073614, K01AG073613, and R03AG070479), the National Institute of Neurological Disorders and Stroke (R01NS100782), the National Cancer Institute (R01CA255840), the Oklahoma Shared Clinical and Translational Resources (U54GM104938) with an Institutional Development Award (IDeA) from NIGMS, the Presbyterian Health Foundation, the Reynolds Foundation, the Oklahoma Nathan Shock Center (P30AG050911), and the Cellular and Molecular GeroScience CoBRE (P20GM125528), the NCI Cancer Center Support Grant (P30 CA225520) and the Oklahoma Tobacco Settlement Endowment Trust. ANT and AU were supported by TKP2021-NKTA-47, implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-NKTA funding scheme; by funding through the National Cardiovascular Laboratory Program (RRF-2.3.1–21-2022–00003) provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund; Project no. 135784 implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the K20 funding scheme and the European University for Well-Being (EUniWell) program (grant agreement number: 101004093/EUniWell/EAC-A02-2019/EAC-A02-2019–1). The 4.0 version of ChatGPT, developed by OpenAI, was used as a language tool to refine our writing, enhancing the clarity of our work.
Author information
Authors and Affiliations
Contributions
The study’s conception is from ANT, AU, and ZU. Literature summary and data collection have been done by ANT, RP, and AU. The initial draft of the manuscript was jointly composed by ANT, AU, AC, and ZU. Subsequent revisions to the manuscript were conducted by all authors, who also collectively reviewed and provided their approval for the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Dr. Anna Csiszar serves as the Associate Editor for The Journal of Gerontology, Series A: Biological Sciences and Medical Sciences and GeroScience. Dr. Zoltan Ungvari serves as the Editor-in-Chief for GeroScience and has personal relationships with individuals involved in the submission of this paper. Dr. Stefano Tarantini, Dr. Calin Prodan, Dr. Eric Liotta, Dr. Ádám Nyúl-Tóth, Dr. Péter Mukli, Dr. Farzaneh A. Sorond, and Dr. Andriy Yabluchanskiy serve as the Associate Editors for GeroScience.
Disclaimer
The funding sources had no role in the study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the American Heart Association, or the Presbyterian Health Foundation.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Nyúl-Tóth, Á., Patai, R., Csiszar, A. et al. Linking peripheral atherosclerosis to blood–brain barrier disruption: elucidating its role as a manifestation of cerebral small vessel disease in vascular cognitive impairment. GeroScience (2024). https://doi.org/10.1007/s11357-024-01194-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11357-024-01194-0