
Abstract
Vascular cognitive impairment and dementia (VCID) is commonly caused by vascular injuries in cerebral large and small vessels and is a key driver of age-related cognitive decline. Severe VCID includes post-stroke dementia, subcortical ischemic vascular dementia, multi-infarct dementia, and mixed dementia. While VCID is acknowledged as the second most common form of dementia after Alzheimer’s disease (AD) accounting for 20% of dementia cases, VCID and AD frequently coexist. In VCID, cerebral small vessel disease (cSVD) often affects arterioles, capillaries, and venules, where arteriolosclerosis and cerebral amyloid angiopathy (CAA) are major pathologies. White matter hyperintensities, recent small subcortical infarcts, lacunes of presumed vascular origin, enlarged perivascular space, microbleeds, and brain atrophy are neuroimaging hallmarks of cSVD. The current primary approach to cSVD treatment is to control vascular risk factors such as hypertension, dyslipidemia, diabetes, and smoking. However, causal therapeutic strategies have not been established partly due to the heterogeneous pathogenesis of cSVD. In this review, we summarize the pathophysiology of cSVD and discuss the probable etiological pathways by focusing on hypoperfusion/hypoxia, blood–brain barriers (BBB) dysregulation, brain fluid drainage disturbances, and vascular inflammation to define potential diagnostic and therapeutic targets for cSVD.
Similar content being viewed by others


Background
Vascular cognitive impairment and dementia (VCID) is caused by various types of cerebrovascular damage such as microvascular dysfunction and large vessel stroke, impacting a large percentage of the world’s population as society ages [1]. Epidemiological studies have demonstrated that VCID is the second most common form of dementia after Alzheimer’s disease (AD), accounting for approximately 20% of dementia cases [2]. Although the clinical diagnostic criteria are somewhat vague, VCID is characterized by cognitive decline through neuropsychological testing and detection of cerebrovascular lesions through neuroimaging or clinical stroke history [3].
The Vascular Impairment of Cognition Classification Consensus Study (VICCCS) identifies four major subtypes of vascular lesions that cause dementia: 1) Post-stroke dementia, 2) Subcortical ischemic vascular dementia, 3) Multi-infarct dementia, and 4) Mixed dementia [4] (Fig. 1). Post-stroke dementia is a major consequence after large vessel strokes. Approximately 10% of patients develop dementia after their first stroke [5]. Atherothrombotic brain infarcts [6] and hemorrhagic stroke [5, 7] are associated with the higher dementia risk. Subcortical ischemic vascular dementia is caused by stenosis and occlusion of small vessels that culminate into lacunar infarct and ischemic white matter lesions. Cortical-subcortical circuit disruption often leads to impairments in information processing, complex attention, and frontal-executive function [8]. Multi-infarct dementia refers to cognitive impairment due to multiple infarcts in various cortical arteries and arterioles. Cortical symptoms such as apraxia and aphasia are often diagnosed through cognitive function tests [2]. Mixed dementia is a type of dementia with concurrent vascular and neurodegenerative pathological changes [9]. AD pathology and cerebrovascular lesions frequently coexist in autopsy cases with dementia [10]. VCID preferentially impairs attention, executive function, and sparing memory [1]. However, cognitive impairments observed in both VCID and AD cases show similar age-associated comorbidities.

Cerebral vascular lesions and vascular cognitive impairment and dementia. Vascular cognitive impairment and dementia (VCID) is a major cause of age-related cognitive decline related to cerebrovascular damages in cerebral large and small vessels. The internal carotid arteries and the vertebral arteries mediate the arterial blood entry into the brain. The blood supply to the cerebrum is mediated by anterior cerebral arteries (ACA) and middle cerebral arteries (MCA) branched from internal carotid arteries. Posterior cerebral arteries (PCA) arising from vertebral arteries are responsible for the blood supply to the brainstem, cerebellum, and occipital cortex. Leptomeningeal arteries from the cerebral arteries form a network of vessels on the pial surface, which branch into the parenchyma. Based on vascular lesions, severe VCID is generally subtyped as post-stroke dementia, subcortical ischemic vascular dementia, multi-infarct dementia, and mixed dementia. Alzheimer’s disease often coexists with cerebrovascular lesions resulting in mixed dementia
VCID is associated with heterogeneous pathological conditions in the cerebrovascular system, where cerebral small vessel disease (cSVD) is the most common pathology underlying VCID [11]. CSVD includes a heterogenous spectrum of pathological, clinical, and radiological cerebrovascular changes. In particular, vessel wall structure from leptomeningeal arteries and intraparenchymal arterioles (perforating arterioles and precapillary arterioles), capillaries, and venules often pathologically deteriorate [12, 13]. In this review, we summarize the current knowledge of cerebrovascular anatomy and cellular compositions, cSVD classification, neuroimaging characteristics, and risk factors. We also discuss probable etiology of cSVD as well as therapeutic strategies.
Basic anatomy of cerebrovascular system
Two pairs of large arteries, the internal carotid arteries and the vertebral arteries, mediate arterial blood entry into the brain. Two vertebral arteries integrate into a basilar artery, which branches out to two posterior cerebral arteries (PCA), distributing blood supply to the brainstem, cerebellum, and occipital cortex. The circle of Willis is composed of pre-communicating segments of the right and left anterior cerebral arteries (ACA), connected via the anterior communicating artery. Pre-communicating segments of the right and left PCA are connected to their corresponding internal carotid arteries via the posterior communicating arteries. An ACA and a middle cerebral artery (MCA) arise from each internal carotid artery and are responsible for blood supply to the cerebrum (Fig. 1) [14]. Second-order branches from the cerebral arteries establish a network of vessels on the pia mater in the subarachnoid space [15]. On the pial surface, leptomeningeal arteries penetrate the pia mater and glia limitans from the subarachnoid space into the brain parenchyma, ramifying into arterioles and capillaries, which end as venules that flow back into the veins [16,17,18]. Since most arterioles do not form collateral networks, arteriole damage results in hypoperfusion and hypoxia of downstream vessels and their corresponding brain regions [19]. In addition, there are cortical watershed areas in border zones between ACA, MCA, and PCA territories [20]. While perforating arterioles from MCA and ACA ascend into deep brain territories including the basal ganglia and thalamic gray matter [21], border zones between penetrating arterioles and perforating arterioles in deep subcortical white matter regions near the lateral ventricle are known as the internal watershed area [20, 22]. Low perfusion pressure in watershed areas result in hemodynamic vulnerability [18].
Cerebral small vessels including penetrating arterioles, precapillary arterioles, postcapillary venules and venules are surrounded by perivascular or paravascular space filled with cerebrospinal fluid (CSF) and/or interstitial fluid (ISF) [23]. While pia mater coats the vascular walls and brain surface, they combine into a singular layer of pia that penetrates into the brain [24]. The vascular pial sheath internally borders the perivascular space. In contrast, the glia limitans (basement membrane of astrocyte end-feet) externally borders paravascular space [25]. While perivascular space is sometimes narrowly defined as the space in basement membranes between smooth muscle cell layers, the paravascular and perivascular spaces integrate in the capillaries due to the lack of smooth muscle layers and pial sheath [23, 26] (Fig. 2). ISF is predicted to enter the periarterial space through capillaries [27, 28], flow along the vessels to leptomeningeal arteries [29], and drain into cervical lymph nodes via the wall of the internal carotid artery, referred to as the intramural periarterial drainage (IPAD) pathway [29]. On the other hand, CSF influxes from the subarachnoid space into the brain parenchyma through paravascular spaces along arterial vessels, mixed with ISF and solutes in the brain parenchyma, drained into the perivenous tracts towards the subarachnoid space, dural venous sinuses, and dural lymphatic vessels for clearance. This pathway is known as glymphatic drainage (Fig. 2). However, further studies are needed to define the specific contributions of IPAD and glymphatic drainage to ISF/CSF clearance [30,31,32,33].

Structural and cellular compositions of the cerebral small vessels. Leptomeningeal arteries penetrate the pia mater and glia limitans from the subarachnoid space into the parenchyma, ramifying into arterioles and capillaries. In leptomeningeal arteries, endothelial cells make up a single luminal layer which are covered by multiple smooth muscle cell layers. In penetrating arterioles, endothelial cells and smooth muscle cells are single layers. In capillaries, endothelial cells form the blood–brain barrier (BBB) with pericytes and basement membrane, surrounded by astrocytic end-feet. The capillaries connect to venules flowing back into veins. In contrast to arterial smooth muscle cells, venous smooth muscle cells have flattened cell bodies and multiple processes, not fully sheathing the venules and veins. Perivascular fibroblasts and macrophages are mainly localized on arterioles and venules. Subarachnoid cerebrospinal fluid (CSF) distributes into the brain parenchyma through para-arterial spaces referred as glymphatic periarterial CSF influx. Interstitial fluid (ISF) as well as CSF diffuse into the perivenous space by bulk flow, and finally efflux into the CSF-dural sinus or cervical lymph nodes. In addition, the ISF and CSF in brain parenchyma can also enter the periarterial space from the capillary level and flow countercurrent to blood flow along arterial vessels, referred as intramural periarterial drainage (IPAD) pathway
Cellular components in cerebrovascular system
The endothelial cell layer (endothelium) is continuous along cerebral vessels. Although endothelial cell is the key cell type separating blood from brain tissue, other vascular cells make up cerebral vasculature: vascular mural cells (smooth muscle cells and pericytes), astrocytes, perivascular macrophages, and perivascular fibroblasts (Fig. 2) [34, 35]. Recent single cell or single nucleus RNA-sequencing studies show cell-specific gene expression heterogeneity at each cerebrovascular segment [36], albeit some discrepancies between methodologies and species [37, 38]. These vascular and perivascular cells interactively function in maintaining cerebrovascular homeostasis.
Endothelial cells at the blood–brain barrier (BBB)
Brain capillary formation through endothelial progenitor cell angiogenesis is mediated by vascular endothelial growth factor (VEGF) and Wnt signaling [39]. By interacting with pericytes and astrocytes, the brain capillary endothelial cells mature and create the tightly sealed monolayer with high barrier integrity, referred to as the BBB [40]. During BBB maturation, the brain capillary endothelial cells exhibit distinct properties from endothelial cells in other organs: specific tight junction protein expression, selective transporter expression, suppressed transcytosis, and leukocyte adhesion molecule downregulation [39, 41]. These unique endothelial properties allow the BBB to strictly control fluid and solute exchange between blood and the parenchyma. These endothelial cells are connected by specialized tight junction proteins such as occludins and claudins, forming a high resistance paracellular barrier [42,43,44]. Both occludin and claudin are tetraspan transmembrane proteins intracellularly linked to the actin cytoskeleton through zonula occludens-1 (ZO-1) [45, 46]. In addition to tight junctions, there are adherens junctions with vascular endothelial (VE)-cadherin, platelet endothelial cell adhesion molecule-1 (PECAM-1) and neural (N)-cadherin, and connexin 43 gap junctions between endothelial cells [47]. The basement membrane is composed of extracellular matrix proteins such as collagen type IV, heparan sulfate proteoglycans, and fibronectin that surround endothelial cells and pericytes. The basement membrane is also critically involved in BBB stability [48]. BBB tight junctions, adherens junctions, gap junctions, and the basement membrane prevent passive diffusion and passive paracellular transport from blood. Selective essential nutrients and metabolites exchange such as glucose, amino acids, fatty acids, organic anions, and nucleosides across the BBB are mediated via carrier-mediated transporters [48, 49]. ATP binding cassette (ABC) transporters are also expressed at the BBB as active efflux transporters to eliminate lipids and exogenous drug from the brain [50].
Vascular mural cells in the arteries and arterioles
In cerebral arteries and arterioles, endothelial cells and the internal elastic lamina layer structure (tunica intima) are surrounded by vascular smooth muscle cell layers (tunica media) and additional layers mainly composed of collagen fibers and fibroblasts (tunica adventitia) [51]. Leptomeningeal arteries contain several smooth muscle layers, which thin into a single layer in penetrating arterioles [36]. In precapillary arterioles, pericytes are the main vascular mural cell, sharing commonalities with smooth muscle cells [52]. Vascular smooth muscle cells are contractile cells responsible for controlling cerebral blood flow. Pericyte contribution to vascular contraction and blood flow regulation is controversial [52]; however, pericytes on first-order branches from penetrating arterioles appear to predominantly regulate capillary blood flow [53]. Since systemic blood pressure substantially influences brain circulation, vascular mural cells play a critical role in cerebral autoregulation through vascular tone modulation that maintains a relatively constant baseline cerebral blood flow [54]. Vascular tone is controlled by vascular mural cell membrane polarization through K+ channels and voltage-dependent Ca2+ channels. K+ channel depolarization (suppressed K+ efflux) and voltage-dependent Ca2+ channel opening (enhanced Ca2+ influx) induce vasoconstriction, while vascular mural cell membrane hyperpolarization causes vasodilation [55]. In addition, inositol 1, 4, 5-trisphosphate receptor (IP3R)–mediated Ca2+ release [56] and RhoA/Rho-kinase activation [57] contribute to vascular smooth muscle cell contraction.
Neurovascular coupling
Vascular mural cells relax in response to nitric oxide (NO), prostaglandins, epoxyeicosatrienoic acids (EET), adenosine triphosphate (ATP), and K+ released from neurons, astrocytes, and endothelia cells depending on neuronal activity, referred to as neurovascular coupling [58]. Neurovascular coupling increases blood supply to capillaries by 84% [59]. Neuronal glutamate activates phospholipase A2 (PLA2) through metabotropic glutamate receptors (mGluRs) and promotes astrocytic prostaglandin E2 (PGE2) and EET synthesis that result in vasodilation [60]. Glutamate also triggers intracellular Ca2+ influx through N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors on the postsynaptic membrane. Cytosolic Ca2+ increases in astrocytic end-feet stimulate K+ efflux, inducing smooth muscle cell vasodilation [61]. However, excess astrocytic Ca2+ likely promotes vasoconstriction instead. NMDA and/or AMPA activation leads to neuronal NO synthase (nNOS) and cyclooxygenase 2 (COX-2) upregulation [62]. NO promotes smooth muscle cell vasodilation through cyclic guanosine monophosphate (cGMP)-dependent protein kinase (PKG) [63]. NO also inhibits vasoconstriction by impeding 20-hydroxyeicosatetraenoic acid (20-HETE) synthesis [59]. Inhibiting nNOS has been shown to reduce neurovascular response by 64% [64]. Neuronal COX facilities phospholipase I2 (PGI2) synthesis which in turn induces smooth muscle vasodilation through the cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) pathway [62, 65]. Controversially, Neuropeptide Y (NPY) released after inhibitory neuron activation likely induces vasoconstriction [66]. In the hypothalamic supraoptic nucleus (SON), vasopressin (VP) neuronal activation also causes responsive vasoconstriction upon acute salt loading challenge [67].
Angiotensin II has been shown to disrupt neurovascular coupling by increasing Ca2+ through angiotensin II type 1 (AT1) receptor in the nearby astrocytic end-feet [68]. In addition, endothelial cells are involved in NO-mediated vasodilation through endothelial NO synthase (eNOS) regulation via NMDA receptor signaling [69] or upon mechanical shear stress [70]. Hypoxia, thrombin, and inflammatory cytokines promote the production of endothelin (ET), a strong vasoconstrictor, in endothelial cells [71]. Angiotensin II also promotes peroxynitrite (ONOO−) generation through the AT1-NADPH oxidase (NOX) pathway in endothelial cells, resulting in neurovascular coupling impairment (Table 1) [72].
Pericytes on the capillaries
Pericytes are localized at the abluminal side of the capillary endothelial cells and form direct synaptic-like peg-socket focal contacts with endothelium through N-cadherin and connexins [73]. Pericytes cover capillary endothelial cells with a 1:3 pericyte-to-endothelium ratio [74]. Pericytes contribute to various aspects of cerebrovascular functions including angiogenesis, BBB integrity [47, 75], and immune cell filtration [76] through the crosstalk with endothelial cells, astrocytes, neurons, and microglia [77]. Pericytes secrete VEGF to promote angiogenic sprouting and stabilization of endothelial cells [78]. Platelet-derived growth factor-BB (PDGF-BB) secreted from endothelial cells also critically mediates pericyte angiogenesis through PDGF receptor-β (PDGFRβ) [79,80,81]. Pericyte–endothelial signals, including PDGF-BB–PDGFRβ, VEGF–VEGF receptor-2 (VEGFR2), transforming growth factor-β (TGF-β)–TGF-β receptor 2 (TGFβR2), Angiopoietin (Ang)-Tie2, Notch, and major facilitator superfamily domain-containing 2a (MFSD2A), play an important roles in BBB development, maintenance of integrity, and transport [82]. Pericytes also modulate brain immune responses [76]. In vivo studies in animals have demonstrated that loss of pericytes lead to upregulation of leukocyte adhesion molecules (LAMs) on endothelial cells, exacerbating parenchymal immune cell infiltration [79, 80].
Astrocytes in the glial limitans and BBB
The glia limitans is composed of astrocytic end-feet and the basement membrane. The glia limitans constitute a continuous layer covering a large area of cerebral small vessels as external limitans of perivascular/paravascular space and mediates the ISF and CSF transport between the brain parenchyma and drainage pathways [83]. Leptomeningeal arteries and penetrating arterioles are covered by pia mater and glia limitans superficialis. In capillaries, glia limitans perivascularis surround endothelial cells and pericytes [84]. Astrocytes secrete vasculotrophic factors such as astrocyte-derived angiopoietin-1 (ANG-1), sonic hedgehog (SHH), glial-derived neurotrophic factor (GDNF), retinoic acid (RA), insulin-like growth factor-1 (IGF-1) and apolipoprotein E (APOE) involved in maintaining BBB integrity [85]. Supporting this, experiments co-culturing astrocytes and endothelial cells show upregulated expression of tight junction proteins and strengthened barrier integrity [86]. Astrocytic end-feet also express α-dystroglycan to anchor basement membrane and maintain BBB function [87]. Furthermore, water channel aquaporin 4 (AQP4) expressed on astrocytic end-feet plays an essential role in regulating cerebral water homeostasis and glymphatic drainage [88, 89].
Perivascular cells on cerebral vessels
Perivascular macrophages are yolk sac-derived immune cells detected on brain arterioles and venules [90]. Perivascular macrophages localize in perivascular or paravascular space under the glial limitans, mediating brain immune responses through phagocytosis and antigen presentation [34]. Perivascular macrophages are also involved CSF flow and glymphatic drainage regulation. A study in mice has shown that depletion of perivascular macrophages causes excess accumulation of extracellular matrix proteins in perivascular/paravascular space, thereby disturbing CSF perfusion [91].
Perivascular fibroblasts are identified as cells with flattened somata and sheet-like ruffled processes on penetrating arterioles, precapillary arterioles, and ascending venules in the brain [92]. Although physiological roles of perivascular fibroblasts remain unclear, perivascular fibroblasts appear to serve as tissue-resident mesenchymal cells [93]. It is predicted that these cells maintain the vascular basement membrane, glymphatic drainage system, and mechanosensation for neurovascular coupling [35]. Upon tissue damage, the perivascular cells are the likely major source for scar formation and fibrosis by producing extracellular matrix proteins and mediating inflammation [93].
Classifications of cSVD
Clinically cSVD shows progressive symptoms in cognitive impairment, depression, urinary disturbance, gait difficulty, dysphagia, and dysarthria [13]. While cSVD has relatively homogenous clinical features, six types of cSVD have been proposed based on etiopathogenic features: 1) Arteriolosclerosis, 2) Sporadic and hereditary cerebral amyloid angiopathy (CAA), 3) Inherited or genetic cSVD distinct from CAA, 4) Inflammatory and immunologically mediated cSVD, 5) Venous collagenosis, and 6) Other cSVD [13].
Arteriolosclerosis
Arteriolosclerosis is the most common form of cSVD, neuropathologically defined by hyaline thickening of vessel walls (< 150 μm in diameter) without association with lipid-containing cells, intramural inflammation, and amyloid or fibrinoid necrosis [94]. Smooth muscle cell loss from the tunica media and deposits of fibro-hyaline material and collagens in vessel walls are also detected in arteriolosclerosis lesions [13]. The Vascular Cognitive Impairment Neuropathology Guidelines (VCING) system has been widely used to evaluate the severity of arteriolosclerosis in a semiquantitative manner; 0 = Normal, 1 = Mid thickening of the vessel media with mid fibrosis, 2 = Partial loss of smooth muscle cells in the media with moderate hyaline fibrosis, and 3 = Complete loss of smooth muscle cells in the media with severe hyaline fibrosis and lumen stenosis [94]. Arteriolosclerosis is also exacerbated by diabetes and hypertension during aging [95]. Fibrinoid necrosis commonly accompanies arteriolosclerosis in hypertensive arteriopathy [96,97,98]. Other pathological microangiopathies include microatheroma (distal manifestations of atherosclerosis) and microaneurysms (elongated and dilated vessels) [13]. Of note, cerebral arteriolosclerosis has been reported as a predominant factor contributing to global cognitive impairments, episodic memory, working memory, perceptual speed, autonomic dysfunction, and motor symptoms [99].
Cerebral amyloid angiopathy
CAA is characterized by the progressive accumulation of amyloid-β (Aβ) in leptomeningeal arteries, penetrating arterioles, and capillaries. Aβ deposits begin at the basement membrane between smooth muscle cell layers and develop into circumferential transmural deposits [100]. Vessel integrity loss caused by Aβ deposits can lead to spontaneous lobar intracerebral hemorrhage [101]. Aβ also disrupts the vascular extracellular matrix layers, causing luminal obstruction, leading to parenchymal ischemia [102]. Population-based postmortem studies demonstrated that CAA is detected in 20–40% of elderly people without dementia and 50–60% of those with dementia [103,104,105,106]. CAA is associated with cSVD neuroimaging markers on magnetic resonance imaging (MRI), including microbleeds [107], white mater hyperintensities (WMHs) [108], and microinfarcts [109]. Of these, lobar cerebral microbleeds is strongly predictive of CAA [110]. CAA-related microbleeds are frequently identified at the gray-white matter junction of the parietal and occipital lobes [102]. CAA categorization heavily relies on intracerebral hemorrhage status to define individual cases as “definite CAA”, “probable CAA with supporting pathological evidence”, “probable CAA”, or “possible CAA” under the modified Boston criteria [111, 112] and Boston criteria version 2.0 [113]. Another non-hemorrhagic neuroimaging marker, perivascular spaces at the centrum semiovale, reflects perivascular interstitial fluid drainage impairments [114]. Subcortical WMHs [115] and posterior predominant WMHs are also observed in CAA [116]. CAA-related WMHs are likely caused by hypoperfusion associated with Aβ deposits in cortical small vessels, BBB disruption, and following increases in vascular permeability [101, 117]. WMH severity is associated with a higher risk of recurrent lobar hemorrhage, larger hematoma volume, and hematoma expansion [118, 119]. Cerebral microinfarcts are acute or subacute ischemic infarctions observed in patients with advanced CAA [109]. They appear as round or oval white colored areas that indicate high intensity regions in the subcortex and cortex on diffusion-weighted MRI [109]. CAA also causes convexity subarachnoid hemorrhage and transient focal neurological episodes (TFNEs) [120]. TFNEs are short, stereotyped episodes of somatosensory or motor disturbance, dysphasia, and visual loss, often accompanied with cortical spreading, depression, or depolarization due to the superficial hemorrhage [121].
Inherited or genetic cSVD
The Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), caused by a mutation in NOTCH3, has been known as one type of hereditary cSVD [122]. Clinical features of the mutation carriers include migraines with aura, recurrent ischemic strokes, transient ischemic attacks with cognitive impairment, and subcortical dementia [123]. Brain MRIs show hyperintense periventricular lesions and centrum semioval on T2-weighted or fluid attenuation inversion recovery (FLAIR) images [123]. This progresses to confluent leukoaraiosis with anterior temporal lobe involvement [124]. Pathological analyses show granular osmiophilic material (GOM) in the tunica media and vessel wall thickening [125]. Another form of hereditary cSVD is Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CARASIL). CARASIL is caused by mutations in the HTRA1 gene that encodes HtrA serine peptidase/protease 1 (HTRA1) [126]. Clinical features include early-onset lacunar stroke, cognitive impairment, alopecia, and lumbar spondylosis [127]. Lacunar stroke in the basal ganglia or brainstem is the most common manifestation of CARASIL, observed in approximately 50% of cases. Extensive vascular smooth muscle cells degeneration, vessel wall thickening, and lumen narrowing are histologically observed in CARASIL [125, 128]. Missense or null variants in COL4A1 and COL4A2 result in autosomal dominant cSVD [129]. These mutations are accompanied with cerebral microbleeds in the basal ganglia, centrum semiovale, and pons, and/or small deep lacunar infarcts and dilated perivascular spaces in the basal ganglia [130]. COL4A1 and COL4A2 mutations also induce other clinical manifestations in the brain (porencephaly, and intracerebral aneurysms), eyes (cataracts, retinal vascular tortuosity, and retinal hemorrhage), and kidneys (proteinuria, renal insufficiency, renal cysts, and tortuosities of retinal arteries) [131, 132]. Hereditary diffuse leukoencephalopathy with spheroids (HDLS) caused by CSF1R mutations is an early-onset dementia with brain atrophy and white matter changes [133]. HDLS is characterized by WMHs with frontal or frontoparietal predilection and asymmetric distribution, brain atrophy, and corpus callosal involvement [134]. Clinically, HDLS show symptoms related to frontal lobe syndrome such as loss of judgment, lack of social inhibition, lack of insight, and personality changes [135]. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome is a maternally inherited mitochondrial disorder with the m.3243A > G variant that results in multi-organ dysfunction [136]. The clinical MELAS manifestations are varied including stroke-like episodes, dementia, epilepsy, lactic acidosis, myopathy, hearing impairment, diabetes, headache, and short stature. Stroke-like episodes are frequently observed in occipito-temporal regions, presenting as vasogenic edema in the acute phase [137]. Fabry’s disease is an X-linked, recessive lysosomal storage disease affecting glycosphingolipid metabolism, caused by a mutation in GLA which encodes alpha-galactosidase A (α-Gal-A). The clinical symptoms include peripheral polyneuropathy, autonomic dysfunction, and posterior circulation strokes [138].
Inflammatory and immunologically mediated cSVD
This group of cSVD is characterized by excess immune cell infiltration into the vessel walls (vasculitis) due to systemic and vascular inflammation during infection, autoimmune diseases, and rare immunological diseases [13]. A community-based population study showed that high neutrophil count is associated with increased risk for enlarged perivascular spaces in the basal ganglia and lacune [139]. Plasma C-reactive protein (CRP) or interleukin 6 (IL-6) levels were also positively correlated with the presence of WMHs [140, 141].
Venous collagenosis
Venous collagenosis is noninflammatory collagenous thickening of venous walls mainly composed of collagen I and III in white matter regions along lateral ventricles. A histological study found venous collagenosis in 65% of cases in an over 60-year-old cohort [142]. Venous collagenosis is associated with leukoaraiosis severity or periventricular white matter ischemia [142]. Interestingly, increased venous collagen, but not arterial collagen, is reported as a significant predictor of higher WMH burden [143]. Since venous collagenosis also causes luminal stenosis or occlusion, venous lesions are predicted to associate with cerebral hypoperfusion, glymphatic drainage disruption, and BBB damage [144].
Other cSVD
Although radiotherapy is effective in treating cancers, cranial radiation sometimes causes irreversible cerebrovascular damage in delayed phases. It includes arteritis, intracranial aneurysm, cavernous malformation, mineralizing microangiopathy [145]. Endothelial cells and neurons are vulnerable to radiation, and cSVD is also a complication after radiation therapy. CSVD including microbleeds, microinfarcts, or white matter lesions are often observed in long term follow-up after cranial irradiation [146].
Neuroimaging hallmarks of cSVD
The STandards for Reporting and Imaging of Small Vessel Disease (STRIVE) guideline defines WMHs, recent small subcortical infarcts, lacunes, cerebral microbleeds, enlarged perivascular spaces, and brain atrophy which are common features of cSVD detected through neuroimaging [147]. These MRI hallmarks can become apparent long before symptom onset as clinically silent manifestations [13]. Such lesion accumulations subsequently lead to an increased risk of stroke [1, 21, 148], depression, and mobility disorders as well as VCID [149]. Here, we summarize MRI features of cSVD.
White matter hyperintensities (WMHs)
WMHs are frequently detected in VCID patients. A population-based study reported that WMHs are 95% pervasive in the elderly population over 60 years old [150]. WMH progression is associated with executive function, attention, and immediate/delayed memory [151]. WMHs are lesions detected as hyperintense areas on T2 or FLAIR [152]. Deep WMHs are often smaller and asymmetrically distributed in juxtacortical white matter. This distribution is suggestive of local perfusion impairments due to hypertensive arteriopathy and CAA [153]. Periventricular WMHs are located symmetrically around ventricles, suggesting occlusive periventricular venous collagenosis-related diffuse perfusion disturbances [142, 152, 154]. Pathological examination of WMHs shows varied degrees of demyelination, diffuse axonal injury, gliosis, and oligodendrocyte loss [94, 155]. In deep WMHs, hypoxia-inducible factor (HIF) levels are upregulated in cerebral capillary endothelial cells, supporting ischemic associations [156]. Furthermore, BBB disruption characterized by activated astrocytes and fibrinogen positivity is also associated with both deep and periventricular WMHs [157].
Recent small subcortical infarcts
Recent small subcortical infarcts, commonly called lacunar infarction, account for about 25% of all ischemic strokes [158]. Lacunar infarcts are defined by neuroimaging evidence of recent infarction around a single perforating arteriole with a diameter of less than 20 mm in axial section. “Recent” refers to symptoms or imaging features formed during the hyperacute phase and the first few weeks before diagnostic imaging [147].
Lacunes of presumed vascular origin
Lacune is used to describe round or ovoid, subcortical, fluid-filled cavities with a diameter of 3–15 mm, formed during healing from lacunar infarcts or small hemorrhages. Lacune prevalence range from 8–28% (mean age: 50–75 years) [159]. Increased lacune counts are associated with a higher risk of cognitive impairment and stroke [159, 160].
Enlarged perivascular space
In axial MRI imaging, enlarged perivascular spaces are observed as hyperintense round lesions surrounding perforating arteries and arterioles in the basal ganglia and linear in the centrum semiovale on T2-weighted images. Mechanisms underlying enlarged perivascular spaces is not well understood. However, it is hypothesized to represent perivascular fluid stagnation due to lymphatic drainage blockage [161]. Larger numbers of enlarged perivascular spaces are associated with worsened cognitive function or dementia [162].
Microbleeds
The prevalence of cerebral microbleeds is between 11.1–15.3%, increasing in an age-dependent manner [163, 164]. Cerebral microbleeds are MRI-visible small hypointense oval or round lesions with a diameter of 2–10 mm detected through T2* weighted gradient imaging [165] or magnetic susceptibility weighted images [165]. Cerebral microbleeds are perivascular hemosiderin deposits that reflect previous subtle hemorrhages from small vessels involved in arteriolosclerosis or CAA [147, 166]. Lobar cerebral microbleeds in cortico-subcortical areas are associated with cognitive impairment [167,168,169] and lobar intracranial hemorrhage [170]. Deep/infratentorial cerebral microbleeds are detected as hypertensive vasculopathy in deep gray or white matter of the cerebral hemispheres, brainstem, and cerebellum [171].
Brain atrophy
Brain atrophy manifests as general or focal, and symmetrical or asymmetrical decrease of gray or white matter volumes. Brain atrophy frequently occurs with increased ventricular volumes, enlarged superficial sulci, and WMHs. Some studies show that increased WMHs further aggravate brain atrophy [172, 173].
Risk factors for cSVD
Collective evidence suggests that age, hypertension, diabetes, hyperlipidemia, smoking, and obstructive sleep apnea substantially impact cSVD pathogenesis [174,175,176]. Among them, aging and hypertension are the predominant factors associated with cSVD risk [177]. Epidemiological studies show that cSVD prevalence is higher in cases with longstanding hypertension in middle age [178, 179]. In addition, recent reports indicate that a subset of COVID-19 patients have neuroimaging features of cSVD [180, 181]. In this section, we summarize the pathogenic conditions that influence cSVD risk and genetic risk factors.
Aging
Aging is involved in various pathogenic conditions, including hypertension, hyperlipidemia, diabetes, cardiovascular diseases, and dementia [182]. A meta-analysis study found that BBB permeability increases with age in both healthy and demented individuals [183]. A neuropathological study also showed that age exacerbates cSVD score in AD brains [184]. While WMH burden increases with age, age-dependent effects are accelerated in the presence of hypertension, abnormal body mass index (BMI), and diabetes mellitus after 50 years of age [185]. Overall prevalence of cerebral microbleeds was high and increased with age from 17.8% in persons aged 60–69 years to 38.3% in those over 80 years [171]. Thus, aging contributes to cSVD risk as a predominant factor. Although various pathogenic mechanisms such as oxidative stress, mitochondrial dysfunction, and chronic inflammation contribute to age-related vascular dysfunction [182], vascular senescence is also a critical cause compromising cerebrovascular function [186].
Hypertension
Hypertension is a leading risk factor for cSVD [13, 147] and VCID [187], which critically contributes to disease pathogenesis. WMH severity has a positive linear correlation with blood pressure [188]. A population-based study found that premorbid systolic blood pressure preceding 20 years before neuroimaging is more predominantly associated with cSVD burden than current systolic blood pressure [189]. Hypertension is also the most consistent predictor of cerebral microhemorrhage in healthy adult individuals and stroke patients [165]. While blood pressure variability has been shown to correlate with cardiovascular disease risk [190], it is also involved in cSVD development. Larger variation in systolic blood pressure in midlife is related to WMH development and ventricular atrophy later in life [191]. Hypertension damages cerebral vessels through multiple mechanisms: suppression of NO production, induction of reactive oxygen species (ROS), and extracellular matrix remodeling [192, 193].
Diabetes mellitus
Diabetes mellitus is a well-established risk factor for developing ischemic, hemorrhagic stroke, dementia, and cardiovascular diseases [194, 195]. Type 2 diabetes mellitus is associated with a higher risk of lacunar occurrence, although its impact on WMHs is controversial [196,197,198]. Since increased cSVD burden is detected in type 2 diabetes patients with retinopathy compared with those without retinopathy, small vessels are likely damaged in multiple organs during diabetes [199]. Hyperglycemia, insulin resistance, and altered fatty acid metabolism accompanied with diabetes mellitus have been known to induce oxidate stress and activation of the PKC pathway and receptors for advanced glycation endproducts (RAGE). These factors lead to suppression of NO production, inflammation, and thrombosis activation, resulting in substantial damages in endothelial and vascular mural cells [200].
Smoking
Smoking is known to cause deleterious effects on the vascular system, resulting in coronary heart disease, hypertension, arteriosclerosis, and stroke [201]. A meta-analysis showed that stroke morbidity and mortality are significantly higher in ever smoker groups with OR 1.45 and OR 1.44, and in current smoker groups with OR 1.90 and OR 1.70, compared to non-smoker groups [202]. A dose-dependent negative association between cigarette smoking and cortical thickness has also been reported [203]. Smoking is a strong factor associated with increased cSVD burden [204]. While smoking exacerbates WMHs [205] and white matter microstructural integrity [206], the effects on microbleeds, lacunes, and perivascular space enlargement are controversial [159, 207]. Toxic effects of cigarette smoking on endothelial cells are mainly induced by oxidative stress initiated by ROS, reactive nitrogen species, and other oxidant constituents [208]. Smoking also activates immune cells and induces vascular inflammation including leukocyte infiltration, matrix metalloproteinase (MMP) upregulation, and platelet/coagulation activation [208].
Obstructive sleep apnea
Obstructive sleep apnea are recurrent breathing interruptions during sleep [209] and is a strong risk factor for vascular diseases including hypertension, atherosclerosis, cardiovascular disease, and stroke [210,211,212]. As acute sleep deprivation decreases regional cerebral blood flow in healthy individuals [213], sleep fragmentation is related to the severity of cSVD neuroimaging markers [214]. Consistently, several studies show a significant association of obstructive sleep apnea with WMHs, but not microhemorrhage [215]. Obstructive sleep apnea causes ischemia/reperfusion injury [216, 217], where increased ROS and pro-inflammatory molecules cause brain damage [218]. Furthermore, accumulating evidence indicates that sleep disturbances compromise glymphatic drainage [219]. The severity of obstructive sleep apnea is associated with enlarged perivascular space in the basal ganglia and centrum semiovale [215].
Hyperlipidemia
Healthy individuals with dyslipidemia (total triglyceride > 150 mg/dL and/or high-density lipoprotein [HDL] < 40 mg/dL) are associated with subcortical WMHs [220]. Another study also demonstrated that total triglyceride levels, but not low-density lipoprotein (LDL) or HDL, were associated with larger WMH volume and lacune [221]. Controversially, there is a report showing that ischemic stroke patients with a history of hyperlipidemia (total cholesterol > 220 mg/dL or total triglyceride > 150 mg/dL, and prescription of statin) have less severe WMHs [222]. Higher total cholesterol (200–225 mg/dL) is significantly associated with a lower cSVD risk such as lacunar infarctions and WMHs as detected by MRI in a middle-aged population who visited a hospital for a brain checkup [223]. Although a meta-analysis showed the positive correlation between hyperlipidemia and cSVD risk [174], it remains controversial. Thus, hypertriglyceridemia but not hypercholesterolemia may be associated with increased cSVD risk as hypertriglyceridemia compromises endothelial function by causing oxidative stress [224]. Higher blood LDL is also associated with exacerbated AD neuropathology [225].
COVID-19
COVID-19 due to SARS-CoV-2 infection has substantially impacted the population health worldwide since December in 2019. In severe COVID-19 cases, there is respiratory failure and systemic inflammation. COVID-19 patients frequently show neurological symptoms including encephalopathy, encephalitis, and ischemic stroke [226]. Of note, a prospective study enrolling 60 recovered COVID-19 patients demonstrated that neurological symptoms are present in 55% of cases. Neuroimaging found micro-structural and functional brain integrity disruption during COVID-19 recovery [227]. Several case reports also indicate an association between COVID-19 and cSVD [228, 229]. While ischemic stroke occurs during the acute phase of COVID-19, cSVD phenotypes such as microinfarctions and vessel wall contrast enhancement are detected in the later phase [230]. In addition, COVID-19 has been reported to cause cerebrovascular endothelial loss, increasing the number of thin collagen IV-positive vessels lacking endothelial cells [231]. When SARS-CoV-2 infects endothelial cells, a viral protease (Mpro) appears to reduce nuclear factor (NF)-κB essential modulator (NEMO) and suppress the receptor-interacting protein kinase 3 (RIPK3) pathway, leading to endothelial apoptosis and BBB breakdown [231]. Although further studies in larger cohorts are required, COVID-19 is likely associated with a higher risk for cSVD compared to other infective diseases.
Socioeconomic disparities
Several studies demonstrate that socioeconomic disparities are associated with stroke incidence, outcomes, and recurrence [232,233,234]. Other factors associated with lower socioeconomic status such as substance dependence, mental illness, and infectious diseases may contribute to pathogenesis. Consistently, socially marginalized individuals have shown higher prevalence (32%) for cSVD and often at a younger age (median 44.7 years old) [235]. Higher prevalence of WMHs is also associated with alcohol consumption [236], nonprescribed drug usage [237, 238], and nutritional deficiencies [239].
Genetic factors
Recent genome-wide association studies (GWAS) identified 31 loci associated with cSVD-related imaging traits including WMHs, mean diffusivity, and fractional anisotropy in 42,310 individuals. CSVD risk loci include gene coding proteins related to AD (APOE and MAPT), immune system (HLA-B and HLA-S), and extracellular matrix (COL4A2 and VCAN) [240]. Although APOE-ε2 is protective against AD, both APOE-ε2 and ε4 have been known to increase CAA and CAA-related hemorrhagic risk [241, 242]. APOE-ε2/ε4 carriers are prone to developing CAA at an early age [243]. APOE-ε2 and ε4 are associated with CAA in arteries/arterioles and capillaries, respectively [244]. In addition to CAA, a meta-analysis implies that APOE-ε2 and ε4 are associated with increased WMH burden. While APOE-ε4 is correlated with lobar microbleeds, APOE-ε2 increases the risk of brain infarct [245].
Probable etiology of cSVD
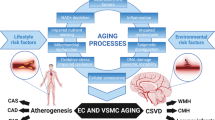
The etiological mechanisms of cSVD can be summarized in the following four pathways: 1) hypoperfusion/hypoxia, 2) BBB dysregulation, 3) ISF/CSF drainage disturbances, and 4) vascular inflammation (Fig. 3) [246]. Each of them is predicted to contribute independently and interactively to cSVD pathogenesis.

Risk factors and pathogenic mechanisms of cerebral small vessel disease. The cerebral small vessel disease (cSVD) can be classified into six groups including arteriolosclerosis, sporadic and hereditary cerebral amyloid angiopathy (CAA), inherited or genetic cSVD distinct from CAA, inflammatory and immunologically mediated cSVD, venous collagenosis, and others. Neuroimaging hallmarks of cSVD include white matter hyperintensities (WMHs), microbleeds, subcortical infarcts, lacunes, and enlarged perivascular space. While various molecular mechanisms are involved in cSVD pathogenesis, hypoperfusion/hypoxia, blood–brain barrier (BBB) dysregulation, interstitial fluid (ISF)/cerebrospinal fluid (CSF) drainage disturbances, and vascular inflammation are likely the major etiological pathways. Hypertension, smoking, diabetes, and sleep apnea are strongly associated with the risk of cSVD, where aging, lifestyle, and genetic factors also contribute to the pathogenic pathways as modifiers
Hypoperfusion/hypoxia
The brain needs a constant supply of oxygen and nutrition from blood to maintain the cellular and functional homeostasis. Arteriosclerosis, CAA, venous collagenosis, and other pathological changes detected in cSVD might cause not only luminal narrowing but also dysregulation of cerebral autoregulation, resulting in the reduction of cerebral blood supply. Capillary endothelial cells are vulnerable to elevated shear stress and hypoperfusion [247]. Chronic cerebral hypoperfusion and subsequent intermittent hypoxia provoke oxidative stress, mitochondrial dysfunction, inflammation, and proteinopathy, leading to neurodegeneration [248]. Particularly, white matter is susceptible to hypoperfusion/hypoxia due to poor collateral flow [249]. Cross-sectional studies demonstrated that lower cerebral blood flow is associated with higher WMH burden [250]. It is possible that brain atrophy due to white matter damage cause cerebral blood flow reduction [251]. However, a population-based study showed that lower cerebral perfusion at the baseline is associated with accelerated cognitive decline during follow-up. Animal studies also demonstrated that chronic hypoperfusion leads to white matter injury, lacunar infarcts, hemorrhages, and cognitive impairment, further exacerbated by APOE-ε4 [252]. Consistently, carotid revascularization has proved to improve cognitive function in patients with severe carotid stenosis by ameliorating cerebral hypoperfusion [253]. While the cascade to cSVD might vary depending on subtype, cerebrovascular hypoperfusion is predicted to predominantly trigger the etiological pathway [254, 255].
BBB dysregulation
Altered paracellular and transcellular transport, decreased tight junction proteins, basement membrane abnormality, and pericyte dysfunction characterize BBB dysregulation, which lead to aggravated plasma protein leakage and leukocyte infiltration into the brain parenchyma resulting in glial activation, demyelination, and neurodegeneration [50]. Neuroimaging studies have demonstrated greater BBB leakage in regions with WMH than normal-appearing white matter, which positively correlates with WMH severity, age, and hypertension [256, 257]. BBB leakage in WMH likely proceeds cognitive decline. As such, BBB dysregulation is causatively involved in cSVD symptoms. Notably, chronic cerebral hypoperfusion or hypoxia is a major factor causing BBB damage. For example, HIF-1 upregulates VEGF in pericytes and astrocytes during hypoxic conditions [258,259,260]. Excessive VEGF exacerbates BBB leakage through altering the distribution of tight junction proteins [261], despite VEGF having beneficial effects including collateral vessel formation, reparative angiogenesis, and neuroprotection after ischemic stroke [262]. Cerebral hypoperfusion also reduces capillary pericyte coverage, disrupting BBB integrity [78]. CSVD risk factors such as hypertension, diabetes, and smoking induce oxidative stress. That oxidative stress subsequently damages the BBB through mitochondrial dysfunction and ROS production, followed by excitotoxicity, altered iron metabolism, inflammatory responses, pyroptosis, and necroptosis in the neurovascular unit [263]. Inflammation is also associated with BBB dysregulation through pathways mediated by inflammatory cytokines and lipid inflammatory mediators. In severe inflammation, parenchymal infiltration of peripheral immune cells and activation of MMPs lead to BBB structural damage [264]. Accumulating evidence demonstrates that APOE-ε4 leads to BBB dysfunction [265] where the mechanism is likely mediated by excess activation of cyclophilin A-MMP9 pathway in pericytes [266].
ISF/CSF drainage disturbances
Diffusion tensor imaging-based analysis along the perivascular space (DTI-ALPS) index has been used to evaluate the glymphatic clearance function [267]. The ALPS index is associated with cSVD neuroimaging markers including WMHs, lacunas, microbleeds, and enlarged perivascular spaces [268]. However, the ALPS index is correlated with cognitive function independent of other factors [269]. Furthermore, lower ALPS index is associated with lower Aβ42 levels in CSF [270]. Glymphatic impairment is predicted to stagnate ISF/CSF drainage and exacerbate brain accumulations of deleterious protein/cell debris, which eventually leads to cSVD-related cognitive impairment. IPAD disturbance also contributes to perivascular space enlargement in the white matter and CAA formation [271]. Thus, ISF/CSF drainage dysregulation through the glymphatic system and IPAD pathways should also be a key etiological mechanism of cSVD. Although CAA is mainly detected between smooth muscle cell layers of leptomeningeal arteries and penetrating arterioles in the IPAD pathway [272], Aβ deposition is sometimes detected in capillaries, venules, and veins [273]. While arterial CAA is fivefold more frequent compared to venous CAA in AD cases [274], Aβ deposition in veins are observed in 78% of severe CAA cases with cerebral hemorrhage [275]. An animal study using a TgF344-AD rat model showed that arteriolar Aβ accumulation precedes venular Aβ accumulation [276]. These observations suggest that IPAD and glymphatic dysfunction are connected. Continuous IPAD dysfunction causes arterial CAA and subsequentially compromises the glymphatic pathway resulting in Aβ deposition on venous vessels in severe cases. Cerebrovascular pulsatility has been identified as the driving force of ISF/CSF bulk flow along cerebral vessels. Thus, altered vascular wall compliance and reactivity due to cerebrovascular damages might disturb the homeostasis of IPAD and glymphatic system in cSVD [277]. Ultrafast magnetic resonance encephalography (MREG) shows that cardiac pulsations drive fluid drainage along periarterial spaces, whereas respiratory pulsations mediate perivenous fluid flow [278]. Hence, altered cardiovascular or respiratory systems also impact the IPAD and glymphatic drainage pathways. In addition, astrocytic AQP4 plays an essential role in regulating brain water homeostasis and glymphatic clearance system [88]; mislocalization or reduction of AQP4 is detected in white matter with cSVD [279].
Vascular inflammation
Vascular inflammation is often characterized by increases of homocysteine, ICAM-1 (intercellular adhesion molecule 1), VCAM-1 (vascular cell adhesion molecule 1), lipoprotein-associated phospholipase A2 (Lp-PLA2), VEGF, E-selectin, P-selectin, MMP9, neopterin, or CD40 [280]. Vascular inflammation is causatively or consequently involved in oxidative stress, vascular endothelial dysfunction [281], BBB damage [282], atherosclerotic plaque formation [283], narrowing of the lumen [284], and hemodynamic impairment [285], all of which eventually culminate in the development of cSVD (Fig. 3). A meta-analysis indicated that vascular inflammation associates with cSVD development in the brain regions supplied by deep perforating arteries such as basal ganglia [280]. The increase of macrophage-derived proinflammatory enzyme Lp-PLA2 has been shown as an risk factor for WMHs [285] as well as cardiovascular disease and stroke [286]. In addition, higher baseline levels of systemic inflammatory markers likely predict cSVD severity and progression [280]. Neutrophil count is also suggestively correlated with increased cSVD burden and prevalence [139]. Animal studies have demonstrated robust association between inflammation and cSVD, providing strong evidence that inflammation could be a major etiological factor of cSVD [287, 288]. Indeed, cSVD risk factors such as aging [289, 290] and hypertension [291, 292] have been known to cause both systemic and vascular inflammation.
Preventative and therapeutic strategy for cSVD
Since cSVD is often a secondary phenotype of a metabolic syndrome, pharmacological approaches to ameliorate hypertension and atherosclerosis are the current standard to treat cSVD [293]. A meta-analysis reported that patients treated with intensive anti-hypertensive drugs have significantly slower WMH progression compared with non-treated groups [294]. While statins are 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors used to treat hyperlipidemia, a randomized controlled trial showed that a low dose of rosuvastatin treatment over 5 years suppresses WMH progression and reduces the risk for microbleeds in aged hypertension patients [295]. Pre-stroke statin medication also slowed post-stroke WMH progression during 2-year follow-up [296]. However, another study found that statins may exacerbate CAA-related lobar hemorrhage risk in APOE-ε4/ε4 and APOE-ε2/ε4 carriers [297]. Other reports failed to detect preventive effects of anti-hypertensive drugs and statins on WMH progression [298,299,300,301].
Antiplatelet therapies using aspirin, clopidogrel, dipyridamole, cilostazol, and ticagrelor are a main strategy for secondary stroke prevention as platelet activation predominantly causes vessel occlusion [302]. Antiplatelet therapy in secondary stroke prevention after lacunar stroke has been reported to reduce recurrence of any stroke and ischemic stroke with superior effects in dual-antiplatelet therapy compared to single-antiplatelet therapy [303]. However, long-term dual antiplatelet therapy with clopidogrel and aspirin resulted in increased rate of major bleeding and all-cause mortality than aspirin alone [304]. Prolonged antiplatelet therapy needs careful consideration when used for cSVD patients especially with CAA. As cilostazol administration showed the lower incidence of hemorrhagic stroke in lacunar stroke patients than aspirin, cilostazol appears to be safer treatment for cSVD [305]. Cilostazol has also been known to ameliorate cognitive decline and gliovascular damage through endothelial stabilization [306].
Although aducanumab and lecanemab have been approved by FDA to treat patients with early AD by reducing brain Aβ amyloids, it is not recommended for cases with diagnosed CAA [307]. Amyloid-related imaging abnormalities (ARIA) were reported in significant number of patients receiving anti-amyloid immunotherapy for AD, in particular in individuals carrying APOE4 gene allele in a gene dose-dependent manner [308, 309]. CAA is likely involved in the pathogenic mechanism of ARIA as APOE4 also increases the prevalence of CAA [310]. ARIA-E is characterized by vasogenic parenchymal edema or leptomeninges/sulci sulcal effusions. ARIA-H exhibits microhemorrhages or superficial siderosis hemosiderin deposits. Antibodies used in Aβ immunotherapy may directly attack vascular amyloid deposition, causing ARIA. Aβ-targeted immunotherapy could also increase perivascular Aβ accumulation, further exacerbating CAA and ARIA. ARIA is generally managed through temporary treatment suspension or dosage reduction, pulsed steroid therapy may ameliorate it [311].
Behavioral metrics (smoking, BMI, physical activity, and diet) are associated with WMH and lacunes risk [312]. Thus, lifestyle interventions are promising approaches for cSVD therapy. While smoking cessation is essential in current smokers with cSVD, multidomain intervention (diet, exercise, cognitive training, vascular risk monitoring) could improve or maintain cognitive functioning in the elderly [313]. Since exercise [314] and the Mediterranean diet improve endothelial function. [315], they could be beneficial in treating cSVD patients. Furthermore, the association between low serum vitamin B12 levels and increased white matter volume was identified [316]. A study reported that B-vitamin (folate, vitamins B12 and B6) supplementation lowers plasma homocysteine levels and reduces WMH burden in patients with severe cSVD intervention [317]. Vitamin E tocotrienols were also found to be beneficial in the attenuation of WMHs among cognitively unimpaired individuals [318].
There are also several emerging new therapeutic strategies for cSVD based on animal studies. For example, an angiotensin II receptor blocker, candesartan, attenuates vascular distensibility and cerebral blood flow by modulating pathological extracellular matrix accumulation in CARASIL model mice [319]. In addition, several active or passive immunotherapy strategies targeting NOTCH3 have been reported to be effective for CADASIL in mouse models [320,321,322]. Since nicotinamide mononucleotide supplementation could restore cerebrovascular endothelial dysfunction and improve cognitive function in aged wild-type mice, nicotinamide mononucleotide may pose protective effects age-related VCID [323]. Furthermore, G-CSF administration has been shown to restore white matter damage and improve non-spatial cognitive function in spontaneously hypertensive rats [324]. Minocycline could also reduce white matter damage, improved behavior, and prolonged life in spontaneously hypertensive/stroke prone rats [325].
Conclusions
As contributions of VCID to age-related cognitive decline have been increasingly recognized, greater understanding of cSVD pathophysiology and etiology is desired to develop novel diagnostic and therapeutic strategies for the disease. Given that hypertension, smoking, and diabetes are strong risk factors for cSVD, it is reasonable to consider cSVD as a primary phenotype of metabolic syndrome in the brain. Although how these risk factors relate to each aspect of cSVD pathogenesis remains to be elucidated, lifestyle interventions focusing on vascular health might be the most effective approach to reduce cSVD risk, cardiovascular diseases, and stroke. In most of cSVD cases, clinical symptoms silently progress for many years before symptoms become evident. Although further studies are needed to define the adequate therapeutic window, earlier intervention at the pre-symptomatic stage should be beneficial to treat cSVD. Current advancements in neuroimaging enables precise cSVD diagnosis. However, there is a difficulty in predicting the onset and progression of cSVD. Early accurate diagnosis or prediction of cSVD may be accelerated through easily identifiable fluid biomarkers. Accumulating evidence indicates etiological pathways such as hypoperfusion/hypoxia, BBB dysregulation, ISF/CSF drainage disturbances, and vascular inflammation in cSVD, where a variety of cell types are involved in the pathogenesis. Since these different aspects of cerebrovascular damages are likely associated with one another, it might be critical to target multiple pathways to establish effective cSVD therapies.
In addition, cSVD might be causatively and consequently involved in AD pathogenesis as CAA is a common type of cSVD. While cSVD and AD frequently coexist in the elderly, future studies should further define how ameliorating cSVD phenotypes influence the onset and development of AD.
Availability of data and materials
Not applicable.
Abbreviations
- VCID:
-
Vascular cognitive impairment and dementia
- AD:
-
Alzheimer’s disease
- CSVD:
-
Cerebral small vessel disease
- ACA:
-
Anterior cerebral artery
- MCA:
-
Middle cerebral artery
- PCA:
-
Posterior cerebral arteries
- CSF:
-
Cerebrospinal fluid
- ISF:
-
Interstitial fluid
- IPAD:
-
Intramural periarterial drainage
- BBB:
-
Blood–brain barrier
- VEGF:
-
Vascular endothelial growth factor
- ZO-1:
-
Zonula occludens-1
- VE-cadherin:
-
Vascular endothelial-cadherin
- PECAM-1:
-
Platelet endothelial cell adhesion molecule-1
- Neural (N)-cadherin:
-
N-cadherin
- ABC:
-
ATP binding cassette
- IP3R:
-
Inositol 1, 4, 5-trisphosphate receptor
- NO:
-
Nitric oxide
- EET:
-
Epoxyeicosatrienoic acids
- ATP:
-
Adenosine triphosphate
- PLA2:
-
Phospholipase A2
- mGluR:
-
Metabotropic glutamate receptor
- PGE2 :
-
Prostaglandin E2
- NMDA:
-
N-methyl-D-aspartate receptor
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- nNOS:
-
Neuronal NO synthase
- COX:
-
Cyclooxygenase
- cGMP:
-
Cyclic guanosine monophosphate
- PKG:
-
Protein kinase G
- 20-HETE:
-
20-Hydroxyeicosatetraenoic acid
- PGI2 :
-
Prostaglandin I2
- cAMP:
-
Cyclic adenosine monophosphate
- PKA:
-
Protein kinase A
- NPY:
-
Neuropeptide Y
- SON:
-
Supraoptic nucleus
- VP:
-
Vasopressin
- AT1:
-
Angiotensin II type 1
- eNOS:
-
Endothelial NO synthase
- ET:
-
Endothelin
- ONOO-:
-
Peroxynitrite
- NOX:
-
NADPH oxidase
- PDGF:
-
Platelet-derived growth factor
- PDGFRβ:
-
PDGF receptor-β
- VEGFR2:
-
Vascular endothelial growth factor receptor-2
- TGF-β:
-
Transforming growth factor-β
- TGFβR2:
-
TGF-β receptor-2
- Ang:
-
Angiopoietin
- MFSD2A:
-
Major facilitator superfamily domain-containing 2a
- LAMs:
-
Leukocyte adhesion molecules
- ANG-1:
-
Angiopoietin-1
- SHH:
-
Sonic hedgehog
- GDNF:
-
Glial-derived neurotrophic factor
- RA:
-
Retinoic acid
- IGF-1:
-
Insulin-like growth factor-1
- APOE:
-
Apolipoprotein E
- AQP4:
-
Aquaporin 4
- CAA:
-
Cerebral amyloid angiopathy
- Aβ:
-
Amyloid-β
- MRI:
-
Magnetic resonance imaging
- WMH:
-
White mater hyperintensity
- TFNEs:
-
Transient focal neurological episodes
- CADASIL:
-
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- FLAIR:
-
Fluid attenuation inversion recovery
- GOM:
-
Granular osmiophilic material
- CARASIL:
-
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy
- HTRA1:
-
HtrA serine peptidase/protease 1
- HDLS:
-
Hereditary diffuse leukoencephalopathy with spheroids
- MELAS:
-
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes
- α-Gal-A:
-
Alpha-galactosidase A
- CRP:
-
C-reactive protein
- IL-6:
-
Interleukin 6
- HIF:
-
Hypoxia-inducible factor
- BMI:
-
Body mass index
- ROS:
-
Reactive oxygen species
- RAGE:
-
Receptors for advanced glycation endproducts
- MMP:
-
Matrix metalloproteinase
- HDL:
-
High-density lipoprotein
- LDL:
-
Low-density lipoprotein
- NF-κB:
-
Nuclear factor (NF)-κB
- NEMO:
-
NF-κB essential modulator
- RIPK3:
-
Receptor-interacting protein kinase 3
- GWAS:
-
Genome-wide association studies
- DTI-ALPS:
-
Diffusion tensor imaging-based analysis along the perivascular space
- MREG:
-
Magnetic resonance encephalography
- ICAM-1:
-
Intercellular adhesion molecule
- VCAM-1:
-
Vascular cell adhesion molecule
- Lp-PLA2:
-
Lipoprotein-associated phospholipase A2
- HMG-CoA:
-
3-hydroxy-3-methylglutaryl coenzyme A
- ARIA:
-
Amyloid-related imaging abnormalities
References
O’Brien JT, Erkinjuntti T, Reisberg B, Roman G, Sawada T, Pantoni L, Bowler JV, Ballard C, DeCarli C, Gorelick PB, et al. Vascular cognitive impairment. Lancet Neurol. 2003;2:89–98.
Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–66.
Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42:2672–713.
Skrobot OA, O’Brien J, Black S, Chen C, DeCarli C, Erkinjuntti T, Ford GA, Kalaria RN, Pantoni L, Pasquier F, et al. The Vascular Impairment of Cognition Classification Consensus Study. Alzheimers Dement. 2017;13:624–33.
Pendlebury ST, Rothwell PM. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 2009;8:1006–18.
Ivan CS, Seshadri S, Beiser A, Au R, Kase CS, Kelly-Hayes M, Wolf PA. Dementia after stroke: the Framingham Study. Stroke. 2004;35:1264–8.
Barba R, Martinez-Espinosa S, Rodriguez-Garcia E, Pondal M, Vivancos J, Del Ser T. Poststroke dementia : clinical features and risk factors. Stroke. 2000;31:1494–501.
Roman GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC. Subcortical ischaemic vascular dementia. Lancet Neurol. 2002;1:426–36.
Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–204.
Jorgensen IF, Aguayo-Orozco A, Lademann M, Brunak S. Age-stratified longitudinal study of Alzheimer’s and vascular dementia patients. Alzheimers Dement. 2020;16:908–17.
Erkinjuntti T, Gauthier S. The concept of vascular cognitive impairment. Front Neurol Neurosci. 2009;24:79–85.
Bullmore E, Sporns O. The economy of brain network organization. Nat Rev Neurosci. 2012;13:336–49.
Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701.
Campbell BCV, De Silva DA, Macleod MR, Coutts SB, Schwamm LH, Davis SM, Donnan GA. Ischaemic stroke Nat Rev Dis Primers. 2019;5:70.
Mastorakos P, McGavern D. The anatomy and immunology of vasculature in the central nervous system. Sci Immunol. 2019;4:eaav0492.
Duvernoy HM, Delon S, Vannson JL. Cortical blood vessels of the human brain. Brain Res Bull. 1981;7:519–79.
Cannistraro RJ, Badi M, Eidelman BH, Dickson DW, Middlebrooks EH, Meschia JF. CNS small vessel disease: A clinical review. Neurology. 2019;92:1146–56.
Iadecola C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron. 2017;96:17–42.
Nishimura N, Schaffer CB, Friedman B, Lyden PD, Kleinfeld D. Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc Natl Acad Sci U S A. 2007;104:365–70.
Momjian-Mayor I, Baron JC. The pathophysiology of watershed infarction in internal carotid artery disease: review of cerebral perfusion studies. Stroke. 2005;36:567–77.
Greenfield JG, Love S, Louis DN, Ellison D. Greenfield’s neuropathology. 8th ed. London: Hodder Arnold; 2008.
Pantoni L, Garcia JH. Pathogenesis of leukoaraiosis: a review. Stroke. 1997;28:652–9.
Wardlaw JM, Benveniste H, Nedergaard M, Zlokovic BV, Mestre H, Lee H, Doubal FN, Brown R, Ramirez J, MacIntosh BJ, et al. Perivascular spaces in the brain: anatomy, physiology and pathology. Nat Rev Neurol. 2020;16:137–53.
Engelhardt B, Carare RO, Bechmann I, Flugel A, Laman JD, Weller RO. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016;132:317–38.
Bakker ENTP, Bacskai BJ, Arbel-Ornath M, Aldea R, Bedussi B, Morris AWJ, Weller RO, Carare RO. Lymphatic Clearance of the Brain: Perivascular, Paravascular and Significance for Neurodegenerative Diseases. Cell Mol Neurobiol. 2016;36:181–94.
Bacyinski A, Xu M, Wang W, Hu J. The Paravascular Pathway for Brain Waste Clearance: Current Understanding. Significance Controversy Front Neuroanat. 2017;11:101.
Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, Weller RO. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34:131–44.
Albargothy NJ, Johnston DA, MacGregor-Sharp M, Weller RO, Verma A, Hawkes CA, Carare RO. Convective influx/glymphatic system: tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018;136:139–52.
Morris AW, Sharp MM, Albargothy NJ, Fernandes R, Hawkes CA, Verma A, Weller RO, Carare RO. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 2016;131:725–36.
Agarwal N, Carare RO. Cerebral Vessels: An Overview of Anatomy, Physiology, and Role in the Drainage of Fluids and Solutes. Front Neurol. 2020;11: 611485.
Benveniste H, Liu X, Koundal S, Sanggaard S, Lee H, Wardlaw J. The Glymphatic System and Waste Clearance with Brain Aging: A Review. Gerontology. 2019;65:106–19.
Hladky SB, Barrand MA. Elimination of substances from the brain parenchyma: efflux via perivascular pathways and via the blood-brain barrier. Fluids Barriers CNS. 2018;15:30.
Mestre H, Mori Y, Nedergaard M. The Brain’s Glymphatic System: Current Controversies. Trends Neurosci. 2020;43:458–66.
Yang Q, Wei X, Deng B, Chang Z, Jin D, Huang Y, Zhang JH, Yenari MA, Jin K, Wang Q. Cerebral small vessel disease alters neurovascular unit regulation of microcirculation integrity involved in vascular cognitive impairment. Neurobiol Dis. 2022;170: 105750.
Dorrier CE, Jones HE, Pintaric L, Siegenthaler JA, Daneman R. Emerging roles for CNS fibroblasts in health, injury and disease. Nat Rev Neurosci. 2022;23:23–34.
Schaeffer S, Iadecola C. Revisiting the neurovascular unit. Nat Neurosci. 2021;24:1198–209.
Garcia FJ, Sun N, Lee H, Godlewski B, Mathys H, Galani K, Zhou B, Jiang X, Ng AP, Mantero J, et al. Single-cell dissection of the human brain vasculature. Nature. 2022;603:893–9.
Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, Losada PM, Chen MB, Schaum N, Khoury N, et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature. 2022;603:885–92.
Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–96.
Abbott NJ, Romero IA. Transporting therapeutics across the blood-brain barrier. Mol Med Today. 1996;2:106–13.
Potente M, Mäkinen T. Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol. 2017;18:477–94.
Wong AD, Ye M, Levy AF, Rothstein JD, Bergles DE, Searson PC. The blood-brain barrier: an engineering perspective. Front Neuroeng. 2013;6:7.
Hannocks MJ, Pizzo ME, Huppert J, Deshpande T, Abbott NJ, Thorne RG, Sorokin L. Molecular characterization of perivascular drainage pathways in the murine brain. J Cereb Blood Flow Metab. 2018;38:669–86.
Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol. 2010;2: a002907.
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–88.
Gunzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev. 2013;93:525–69.
Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–405.
Yamazaki Y, Kanekiyo T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer's Disease. Int J Mol Sci. 2017;18:1965.
Ohtsuki S, Terasaki T. Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm Res. 2007;24:1745–58.
Knox EG, Aburto MR, Clarke G, Cryan JF, O’Driscoll CM. The blood-brain barrier in aging and neurodegeneration. Mol Psychiatry. 2022;27:2659–73.
Cipolla MJ. In The Cerebral Circulation. San Rafael: Integrated Systems Physiology: From Molecule to Function; 2009.
Uemura MT, Maki T, Ihara M, Lee VMY, Trojanowski JQ. Brain Microvascular Pericytes in Vascular Cognitive Impairment and Dementia. Front Aging Neurosci. 2020;12:80.
Zambach SA, Cai C, Helms HCC, Hald BO, Dong Y, Fordsmann JC, Nielsen RM, Hu J, Lonstrup M, Brodin B, Lauritzen MJ: Precapillary sphincters and pericytes at first-order capillaries as key regulators for brain capillary perfusion. Proc Natl Acad Sci U S A. 2021;118:e2023749118.
Aries MJ, Elting JW, De Keyser J, Kremer BP, Vroomen PC. Cerebral autoregulation in stroke: a review of transcranial Doppler studies. Stroke. 2010;41:2697–704.
Dunn KM, Nelson MT. Potassium channels and neurovascular coupling. Circ J. 2010;74:608–16.
Lin Q, Zhao G, Fang X, Peng X, Tang H, Wang H, Jing R, Liu J, Lederer WJ, Chen J, Ouyang K. IP(3) receptors regulate vascular smooth muscle contractility and hypertension. JCI Insight. 2016;1: e89402.
Shimokawa H, Sunamura S, Satoh K. RhoA/Rho-Kinase in the Cardiovascular System. Circ Res. 2016;118:352–66.
Zhu WM, Neuhaus A, Beard DJ, Sutherland BA, DeLuca GC. Neurovascular coupling mechanisms in health and neurovascular uncoupling in Alzheimer’s disease. Brain. 2022;145:2276–92.
Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60.
Mishra A. Binaural blood flow control by astrocytes: listening to synapses and the vasculature. J Physiol. 2017;595:1885–902.
Longden TA, Nelson MT. Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation. 2015;22:183–96.
Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–43.
Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1994;91:7583–7.
Hosford PS, Gourine AV. What is the key mediator of the neurovascular coupling response? Neurosci Biobehav Rev. 2019;96:174–81.
Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–95.
Uhlirova H, Kilic K, Tian P, Thunemann M, Desjardins M, Saisan PA, Sakadzic S, Ness TV, Mateo C, Cheng Q, et al. Cell type specificity of neurovascular coupling in cerebral cortex. Elife. 2016;5:e14315.
Roy RK, Althammer F, Seymour AJ, Du W, Biancardi VC, Hamm JP, Filosa JA, Brown CH, Stern JE. Inverse neurovascular coupling contributes to positive feedback excitation of vasopressin neurons during a systemic homeostatic challenge. Cell Rep. 2021;37: 109925.
Boily M, Li L, Vallerand D, Girouard H. Angiotensin II Disrupts Neurovascular Coupling by Potentiating Calcium Increases in Astrocytic Endfeet. J Am Heart Assoc. 2021;10: e020608.
Lourenco CF, Laranjinha J. Nitric Oxide Pathways in Neurovascular Coupling Under Normal and Stress Conditions in the Brain: Strategies to Rescue Aberrant Coupling and Improve Cerebral Blood Flow. Front Physiol. 2021;12: 729201.
Sriram K, Laughlin JG, Rangamani P, Tartakovsky DM. Shear-Induced Nitric Oxide Production by Endothelial Cells. Biophys J. 2016;111:208–21.
Koyama Y. Endothelin systems in the brain: involvement in pathophysiological responses of damaged nerve tissues. Biomol Concepts. 2013;4:335–47.
Bloch S, Obari D, Girouard H. Angiotensin and neurovascular coupling: beyond hypertension. Microcirculation. 2015;22:159–67.
Sims DE. The pericyte–a review. Tissue Cell. 1986;18:153–74.
von Tell D, Armulik A, Betsholtz C. Pericytes and vascular stability. Exp Cell Res. 2006;312:623–9.
Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215.
Torok O, Schreiner B, Schaffenrath J, Tsai HC, Maheshwari U, Stifter SA, Welsh C, Amorim A, Sridhar S, Utz SG, et al. Pericytes regulate vascular immune homeostasis in the CNS. Proc Natl Acad Sci U S A. 2021;118:e2016587118.
Nikolakopoulou AM, Montagne A, Kisler K, Dai Z, Wang Y, Huuskonen MT, Sagare AP, Lazic D, Sweeney MD, Kong P, et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci. 2019;22:1089–98.
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–27.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–6.
Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–61.
Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–5.
Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. 2016;19:771–83.
Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–76.
Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008;67:1113–21.
Michinaga S, Koyama Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int J Mol Sci. 2019;20:571.
Siddharthan V, Kim YV, Liu S, Kim KS. Human astrocytes/astrocyte-conditioned medium and shear stress enhance the barrier properties of human brain microvascular endothelial cells. Brain Res. 2007;1147:39–50.
Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, Sorokin LM. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. 2006;203:1007–19.
Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14:265–77.
Ghersi-Egea JF, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, Engelhardt B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol. 2018;135:337–61.
Stremmel C, Schuchert R, Wagner F, Thaler R, Weinberger T, Pick R, Mass E, Ishikawa-Ankerhold HC, Margraf A, Hutter S, et al. Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat Commun. 2018;9:75.
Drieu A, Du S, Storck SE, Rustenhoven J, Papadopoulos Z, Dykstra T, Zhong F, Kim K, Blackburn S, Mamuladze T, et al. Parenchymal border macrophages regulate the flow dynamics of the cerebrospinal fluid. Nature. 2022;611:585–93.
Bonney SK, Sullivan LT, Cherry TJ, Daneman R, Shih AY. Distinct features of brain perivascular fibroblasts and mural cells revealed by in vivo two-photon imaging. J Cereb Blood Flow Metab. 2022;42:966–78.
Di Carlo SE, Peduto L. The perivascular origin of pathological fibroblasts. J Clin Invest. 2018;128:54–63.
Skrobot OA, Attems J, Esiri M, Hortobagyi T, Ironside JW, Kalaria RN, King A, Lammie GA, Mann D, Neal J, et al. Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain. 2016;139:2957–69.
Furuta A, Ishii N, Nishihara Y, Horie A. Medullary arteries in aging and dementia. Stroke. 1991;22:442–6.
Cole FM, Yates P. Intracerebral microaneurysms and small cerebrovascular lesions. Brain. 1967;90:759–68.
Feigin I, Prose P. Hypertensive fibrinoid arteritis of the brain and gross cerebral hemorrhage: a form of “hyalinosis.” Arch Neurol. 1959;1:98–110.
Rosenblum WI. Cerebral hemorrhage produced by ruptured dissecting aneurysm in miliary aneurysm. Ann Neurol. 2003;54:376–8.
Blevins BL, Vinters HV, Love S, Wilcock DM, Grinberg LT, Schneider JA, Kalaria RN, Katsumata Y, Gold BT, Wang DJJ, et al. Brain arteriolosclerosis. Acta Neuropathol. 2021;141:1–24.
Yamaguchi H, Yamazaki T, Lemere CA, Frosch MP, Selkoe DJ. Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer’s disease. An immunoelectron microscopic study. Am J Pathol. 1992;141:249–59.
Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol. 2011;70:871–80.
Gilbert JJ, Vinters HV. Cerebral amyloid angiopathy: incidence and complications in the aging brain. I Cerebral hemorrhage Stroke. 1983;14:915–23.
Keage HA, Carare RO, Friedland RP, Ince PG, Love S, Nicoll JA, Wharton SB, Weller RO, Brayne C. Population studies of sporadic cerebral amyloid angiopathy and dementia: a systematic review. BMC Neurol. 2009;9:3.
Xuereb JH, Brayne C, Dufouil C, Gertz H, Wischik C, Harrington C, Mukaetova-Ladinska E, McGee MA, O’Sullivan A, O’Connor D, et al. Neuropathological findings in the very old. Results from the first 101 brains of a population-based longitudinal study of dementing disorders. Ann N Y Acad Sci. 2000;903:490–6.
Neuropathology Group. Medical Research Council Cognitive F, Aging S: Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet. 2001;357:169–75.
Tanskanen M, Lindsberg PJ, Tienari PJ, Polvikoski T, Sulkava R, Verkkoniemi A, Rastas S, Paetau A, Kiuru-Enari S. Cerebral amyloid angiopathy in a 95+ cohort: complement activation and apolipoprotein E (ApoE) genotype. Neuropathol Appl Neurobiol. 2005;31:589–99.
Koennecke HC. Cerebral microbleeds on MRI: prevalence, associations, and potential clinical implications. Neurology. 2006;66:165–71.
Imaoka K, Kobayashi S, Fujihara S, Shimode K, Nagasaki M. Leukoencephalopathy with cerebral amyloid angiopathy: a semiquantitative and morphometric study. J Neurol. 1999;246:661–6.
Kimberly WT, Gilson A, Rost NS, Rosand J, Viswanathan A, Smith EE, Greenberg SM. Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology. 2009;72:1230–5.
Martinez-Ramirez S, Romero JR, Shoamanesh A, McKee AC, Van Etten E, Pontes-Neto O, Macklin EA, Ayres A, Auriel E, Himali JJ, et al. Diagnostic value of lobar microbleeds in individuals without intracerebral hemorrhage. Alzheimers Dement. 2015;11:1480–8.
Linn J, Halpin A, Demaerel P, Ruhland J, Giese AD, Dichgans M, van Buchem MA, Bruckmann H, Greenberg SM. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology. 2010;74:1346–50.
Charidimou A, Linn J, Vernooij MW, Opherk C, Akoudad S, Baron JC, Greenberg SM, Jager HR, Werring DJ. Cortical superficial siderosis: detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain. 2015;138:2126–39.
Charidimou A, Boulouis G, Frosch MP, Baron JC, Pasi M, Albucher JF, Banerjee G, Barbato C, Bonneville F, Brandner S, et al. The Boston criteria version 2.0 for cerebral amyloid angiopathy: a multicentre, retrospective MRI-neuropathology diagnostic accuracy study. Lancet Neurol. 2022;21:714–25.
Charidimou A, Boulouis G, Pasi M, Auriel E, van Etten ES, Haley K, Ayres A, Schwab KM, Martinez-Ramirez S, Goldstein JN, et al. MRI-visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology. 2017;88:1157–64.
Charidimou A, Boulouis G, Haley K, Auriel E, van Etten ES, Fotiadis P, Reijmer Y, Ayres A, Vashkevich A, Dipucchio ZY, et al. White matter hyperintensity patterns in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology. 2016;86:505–11.
Thanprasertsuk S, Martinez-Ramirez S, Pontes-Neto OM, Ni J, Ayres A, Reed A, Swords K, Gurol ME, Greenberg SM, Viswanathan A. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology. 2014;83:794–800.
Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry. 2012;83:124–37.
Smith EE, Gurol ME, Eng JA, Engel CR, Nguyen TN, Rosand J, Greenberg SM. White matter lesions, cognition, and recurrent hemorrhage in lobar intracerebral hemorrhage. Neurology. 2004;63:1606–12.
Lou M, Al-Hazzani A, Goddeau RP Jr, Novak V, Selim M. Relationship between white-matter hyperintensities and hematoma volume and growth in patients with intracerebral hemorrhage. Stroke. 2010;41:34–40.
Banerjee G, Carare R, Cordonnier C, Greenberg SM, Schneider JA, Smith EE, Buchem MV, Grond JV, Verbeek MM, Werring DJ. The increasing impact of cerebral amyloid angiopathy: essential new insights for clinical practice. J Neurol Neurosurg Psychiatry. 2017;88:982–94.
Charidimou A, Peeters A, Fox Z, Gregoire SM, Vandermeeren Y, Laloux P, Jager HR, Baron JC, Werring DJ. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke. 2012;43:2324–30.
Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–10.
Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil. Lancet Neurol. 2009;8:643–53.
Markus HS, Martin RJ, Simpson MA, Dong YB, Ali N, Crosby AH, Powell JF. Diagnostic strategies in CADASIL. Neurology. 2002;59:1134–8.
Tikka S, Baumann M, Siitonen M, Pasanen P, Poyhonen M, Myllykangas L, Viitanen M, Fukutake T, Cognat E, Joutel A, Kalimo H. CADASIL and CARASIL. Brain Pathol. 2014;24:525–44.
Hara K, Shiga A, Fukutake T, Nozaki H, Miyashita A, Yokoseki A, Kawata H, Koyama A, Arima K, Takahashi T, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. 2009;360:1729–39.
Nozaki H, Nishizawa M, Onodera O. Features of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke. 2014;45:3447–53.
Oide T, Nakayama H, Yanagawa S, Ito N, Ikeda S, Arima K. Extensive loss of arterial medial smooth muscle cells and mural extracellular matrix in cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). Neuropathology. 2008;28:132–42.
Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, Bousser MG, Heutink P, Miner JH, Tournier-Lasserve E, John SW. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354:1489–96.
Vahedi K, Alamowitch S. Clinical spectrum of type IV collagen (COL4A1) mutations: a novel genetic multisystem disease. Curr Opin Neurol. 2011;24:63–8.
Gould DB, Marchant JK, Savinova OV, Smith RS, John SW. Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum Mol Genet. 2007;16:798–807.
Plaisier E, Gribouval O, Alamowitch S, Mougenot B, Prost C, Verpont MC, Marro B, Desmettre T, Cohen SY, Roullet E, et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med. 2007;357:2687–95.
Sundal C, Baker M, Karrenbauer V, Gustavsen M, Bedri S, Glaser A, Myhr KM, Haugarvoll K, Zetterberg H, Harbo H, et al. Hereditary diffuse leukoencephalopathy with spheroids with phenotype of primary progressive multiple sclerosis. Eur J Neurol. 2015;22:328–33.
Codjia P, Ayrignac X, Mochel F, Mouzat K, Carra-Dalliere C, Castelnovo G, Ellie E, Etcharry-Bouyx F, Verny C, Belliard S, et al. Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia: An MRI Study of 16 French Cases. AJNR Am J Neuroradiol. 2018;39:1657–61.
Freeman SH, Hyman BT, Sims KB, Hedley-Whyte ET, Vossough A, Frosch MP, Schmahmann JD. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol. 2009;19:39–47.
Kraya T, Neumann L, Paelecke-Habermann Y, Deschauer M, Stoevesandt D, Zierz S, Watzke S. Cognitive impairment, clinical severity and MRI changes in MELAS syndrome. Mitochondrion. 2019;44:53–7.
Ng YS, Bindoff LA, Gorman GS, Horvath R, Klopstock T, Mancuso M, Martikainen MH, McFarland R, Nesbitt V, Pitceathly RDS, et al. Consensus-based statements for the management of mitochondrial stroke-like episodes. Wellcome Open Res. 2019;4:201.
Viana-Baptista M. Stroke and Fabry disease. J Neurol. 2012;259:1019–28.
Jiang L, Cai X, Yao D, Jing J, Mei L, Yang Y, Li S, Jin A, Meng X, Li H, et al. Association of inflammatory markers with cerebral small vessel disease in community-based population. J Neuroinflammation. 2022;19:106.
van Dijk EJ, Prins ND, Vermeer SE, Vrooman HA, Hofman A, Koudstaal PJ, Breteler MM. C-reactive protein and cerebral small-vessel disease: the Rotterdam Scan Study. Circulation. 2005;112:900–5.
Noz MP, Ter Telgte A, Wiegertjes K, Joosten LAB, Netea MG, de Leeuw FE, Riksen NP. Trained immunity characteristics are associated with progressive cerebral small vessel disease. Stroke. 2018;49:2910–7.
Moody DM, Brown WR, Challa VR, Anderson RL. Periventricular venous collagenosis: association with leukoaraiosis. Radiology. 1995;194:469–76.
Lahna D, Schwartz DL, Woltjer R, Black SE, Roese N, Dodge H, Boespflug EL, Keith J, Gao F, Ramirez J, Silbert LC. Venous collagenosis as pathogenesis of white matter hyperintensity. Ann Neurol. 2022;92:992–1000.
Nan D, Cheng Y, Feng L, Zhao M, Ma D, Feng J. Potential mechanism of venous system for leukoaraiosis: from post-mortem to in vivo research. Neurodegener Dis. 2019;19:101–8.
Katsura M, Sato J, Akahane M, Furuta T, Mori H, Abe O. Recognizing radiation-induced changes in the central nervous system: where to look and what to look for. Radiographics. 2021;41:224–48.
Miura M, Nakajima M, Fujimoto A, Kaku Y, Kawano T, Watanabe M, Kuratsu JI, Ando Y. High prevalence of small vessel disease long after cranial irradiation. J Clin Neurosci. 2017;46:129–35.
Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, Lindley RI, O’Brien JT, Barkhof F, Benavente OR, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12:822–38.
Vermeer SE, Hollander M, van Dijk EJ, Hofman A, Koudstaal PJ, Breteler MM, Rotterdam Scan S. Silent brain infarcts and white matter lesions increase stroke risk in the general population: the Rotterdam scan study. Stroke. 2003;34:1126–9.
Debette S, Schilling S, Duperron MG, Larsson SC, Markus HS. Clinical significance of magnetic resonance imaging markers of vascular brain injury: a systematic review and meta-analysis. JAMA Neurol. 2019;76:81–94.
de Leeuw FE, de Groot JC, Achten E, Oudkerk M, Ramos LM, Heijboer R, Hofman A, Jolles J, van Gijn J, Breteler MM. Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study. The Rotterdam Scan Study. J Neurol Neurosurg Psychiatry. 2001;70:9–14.
Kloppenborg RP, Nederkoorn PJ, Geerlings MI, van den Berg E. Presence and progression of white matter hyperintensities and cognition: a meta-analysis. Neurology. 2014;82:2127–38.
Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1 5 T in Alzheimer’s dementia and normal aging. AJR Am J Roentgenol. 1987;149:351–6.
Park JH, Kwon HM, Lee J, Kim DS, Ovbiagele B. Association of intracranial atherosclerotic stenosis with severity of white matter hyperintensities. Eur J Neurol. 2015;22(44–52):e42–43.
Brant-Zawadzki M, Fein G, Van Dyke C, Kiernan R, Davenport L, de Groot J. MR imaging of the aging brain: patchy white-matter lesions and dementia. AJNR Am J Neuroradiol. 1985;6:675–82.
Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, Geurts JJ. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry. 2011;82:126–35.
Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R, Kalaria RN, Forster G, Esteves F, Wharton SB, et al. White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke. 2006;37:1391–8.
Sahlas DJ, Bilbao JM, Swartz RH, Black SE. Clasmatodendrosis correlating with periventricular hyperintensity in mixed dementia. Ann Neurol. 2002;52:378–81.
Sudlow CL, Warlow CP. Comparable studies of the incidence of stroke and its pathological types: results from an international collaboration. International Stroke Incidence Collaboration Stroke. 1997;28:491–9.
Vermeer SE, Longstreth WT Jr, Koudstaal PJ. Silent brain infarcts: a systematic review. Lancet Neurol. 2007;6:611–9.
Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med. 2003;348:1215–22.
Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117:1–14.
Maclullich AM, Wardlaw JM, Ferguson KJ, Starr JM, Seckl JR, Deary IJ. Enlarged perivascular spaces are associated with cognitive function in healthy elderly men. J Neurol Neurosurg Psychiatry. 2004;75:1519–23.
Sveinbjornsdottir S, Sigurdsson S, Aspelund T, Kjartansson O, Eiriksdottir G, Valtysdottir B, Lopez OL, van Buchem MA, Jonsson PV, Gudnason V, Launer LJ. Cerebral microbleeds in the population based AGES-Reykjavik study: prevalence and location. J Neurol Neurosurg Psychiatry. 2008;79:1002–6.
Poels MM, Ikram MA, van der Lugt A, Hofman A, Krestin GP, Breteler MM, Vernooij MW. Incidence of cerebral microbleeds in the general population: the Rotterdam Scan Study. Stroke. 2011;42:656–61.
Cordonnier C, Al-Shahi Salman R, Wardlaw J. Spontaneous brain microbleeds: systematic review, subgroup analyses and standards for study design and reporting. Brain. 2007;130:1988–2003.
Shi Y, Wardlaw JM. Update on cerebral small vessel disease: a dynamic whole-brain disease. Stroke Vasc Neurol. 2016;1:83–92.
Poels MM, Ikram MA, van der Lugt A, Hofman A, Niessen WJ, Krestin GP, Breteler MM, Vernooij MW. Cerebral microbleeds are associated with worse cognitive function: the Rotterdam Scan Study. Neurology. 2012;78:326–33.
Chung CP, Chou KH, Chen WT, Liu LK, Lee WJ, Chen LK, Lin CP, Wang PN. Strictly lobar cerebral microbleeds are associated with cognitive impairment. Stroke. 2016;47:2497–502.
van Norden AG, van den Berg HA, de Laat KF, Gons RA, van Dijk EJ, de Leeuw FE. Frontal and temporal microbleeds are related to cognitive function: the Radboud University Nijmegen Diffusion Tensor and Magnetic Resonance Cohort (RUN DMC) Study. Stroke. 2011;42:3382–6.
Roob G, Lechner A, Schmidt R, Flooh E, Hartung HP, Fazekas F. Frequency and location of microbleeds in patients with primary intracerebral hemorrhage. Stroke. 2000;31:2665–9.
Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Niessen WJ, Hofman A, Krestin GP, Breteler MM. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology. 2008;70:1208–14.
Muller M, Appelman AP, van der Graaf Y, Vincken KL, Mali WP, Geerlings MI. Brain atrophy and cognition: interaction with cerebrovascular pathology? Neurobiol Aging. 2011;32:885–93.
Mok V, Wong KK, Xiong Y, Wong A, Schmidt R, Chu W, Hu X, Leung EY, Chen S, Chen Y, et al. Cortical and frontal atrophy are associated with cognitive impairment in age-related confluent white-matter lesion. J Neurol Neurosurg Psychiatry. 2011;82:52–7.
Wang Z, Chen Q, Chen J, Yang N, Zheng K. Risk factors of cerebral small vessel disease: a systematic review and meta-analysis. Medicine (Baltimore). 2021;100: e28229.
Ferini-Strambi L, Lombardi GE, Marelli S, Galbiati A. Neurological deficits in obstructive sleep apnea. Curr Treat Options Neurol. 2017;19:16.
Hakim AM. A proposed hypothesis on dementia: inflammation, small vessel disease, and hypoperfusion is the sequence that links all harmful lifestyles to cognitive impairment. Front Aging Neurosci. 2021;13: 679837.
Gyanwali B, Shaik MA, Tan BY, Venketasubramanian N, Chen C, Hilal S. Risk factors for and clinical relevance of incident and progression of cerebral small vessel disease markers in an asian memory clinic population. J Alzheimers Dis. 2019;67:1209–19.
Jeerakathil T, Wolf PA, Beiser A, Massaro J, Seshadri S, D’Agostino RB, DeCarli C. Stroke risk profile predicts white matter hyperintensity volume: the framingham study. Stroke. 2004;35:1857–61.
White WB, Wolfson L, Wakefield DB, Hall CB, Campbell P, Moscufo N, Schmidt J, Kaplan RF, Pearlson G, Guttmann CR. Average daily blood pressure, not office blood pressure, is associated with progression of cerebrovascular disease and cognitive decline in older people. Circulation. 2011;124:2312–9.
Sachs JR, Gibbs KW, Swor DE, Sweeney AP, Williams DW, Burdette JH, West TG, Geer CP. COVID-19-associated Leukoencephalopathy. Radiology. 2020;296:E184–5.
Radmanesh A, Derman A, Lui YW, Raz E, Loh JP, Hagiwara M, Borja MJ, Zan E, Fatterpekar GM. COVID-19-associated diffuse leukoencephalopathy and microhemorrhages. Radiology. 2020;297:E223–7.
Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of vascular aging. Circ Res. 2018;123:849–67.
Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease–systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–52.
Esiri MM, Joachim C, Sloan C, Christie S, Agacinski G, Bridges LR, Wilcock GK, Smith AD. Cerebral subcortical small vessel disease in subjects with pathologically confirmed Alzheimer disease: a clinicopathologic study in the Oxford Project to Investigate Memory and Ageing (OPTIMA). Alzheimer Dis Assoc Disord. 2014;28:30–5.
King KS, Peshock RM, Rossetti HC, McColl RW, Ayers CR, Hulsey KM, Das SR. Effect of normal aging versus hypertension, abnormal body mass index, and diabetes mellitus on white matter hyperintensity volume. Stroke. 2014;45:255–7.
Yamazaki Y, Baker DJ, Tachibana M, Liu CC, van Deursen JM, Brott TG, Bu G, Kanekiyo T. Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke. 2016;47:1068–77.
Kennelly SP, Lawlor BA, Kenny RA. Blood pressure and dementia - a comprehensive review. Ther Adv Neurol Disord. 2009;2:241–60.
Dufouil C, de Kersaint-Gilly A, Besancon V, Levy C, Auffray E, Brunnereau L, Alperovitch A, Tzourio C. Longitudinal study of blood pressure and white matter hyperintensities: the EVA MRI Cohort. Neurology. 2001;56:921–6.
Lau KK, Li L, Simoni M, Mehta Z, Kuker W, Rothwell PM, Oxford Vascular S. Long-term premorbid blood pressure and cerebral small vessel disease burden on imaging in transient ischemic attack and ischemic stroke. Stroke. 2018;49:2053–60.
Stevens SL, Wood S, Koshiaris C, Law K, Glasziou P, Stevens RJ, McManus RJ. Blood pressure variability and cardiovascular disease: systematic review and meta-analysis. BMJ. 2016;354: i4098.
Havlik RJ, Foley DJ, Sayer B, Masaki K, White L, Launer LJ. Variability in midlife systolic blood pressure is related to late-life brain white matter lesions: the Honolulu-Asia Aging study. Stroke. 2002;33:26–30.
Higashi Y, Kihara Y, Noma K. Endothelial dysfunction and hypertension in aging. Hypertens Res. 2012;35:1039–47.
Ungvari Z, Toth P, Tarantini S, Prodan CI, Sorond F, Merkely B, Csiszar A. Hypertension-induced cognitive impairment: from pathophysiology to public health. Nat Rev Nephrol. 2021;17:639–54.
Emerging Risk Factors C, Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio E, Ingelsson E, Lawlor DA, Selvin E, et al: Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375:2215–2222.
Biessels GJ, Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol. 2018;14:591–604.
Gerstein HC, Smith EE, Ramasundarahettige C, Desai D, Awadalla P, Broet P, Black S, Dummer TJB, Hicks J, Moody A, et al. Diabetes, brain infarcts, cognition, and small vessels in the canadian alliance for healthy hearts and minds study. J Clin Endocrinol Metab. 2021;106:e891–8.
Liu J, Rutten-Jacobs L, Liu M, Markus HS, Traylor M. Causal impact of Type 2 diabetes mellitus on cerebral small vessel disease: a mendelian randomization analysis. Stroke. 2018;49:1325–31.
Biessels GJ, Reijmer YD. Brain changes underlying cognitive dysfunction in diabetes: what can we learn from MRI? Diabetes. 2014;63:2244–52.
Sanahuja J, Alonso N, Diez J, Ortega E, Rubinat E, Traveset A, Alcubierre N, Betriu A, Castelblanco E, Hernandez M, et al. Increased burden of cerebral small vessel disease in patients with Type 2 diabetes and retinopathy. Diabetes Care. 2016;39:1614–20.
Creager MA, Luscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation. 2003;108:1527–32.
Ockene IS, Miller NH. Cigarette smoking, cardiovascular disease, and stroke: a statement for healthcare professionals from the American Heart Association. American Heart Association Task Force on Risk Reduction. Circulation. 1997;96:3243–7.
Luo J, Tang X, Li F, Wen H, Wang L, Ge S, Tang C, Xu N, Lu L. Cigarette smoking and risk of different pathologic types of stroke: a systematic review and dose-response meta-analysis. Front Neurol. 2021;12: 772373.
Karama S, Ducharme S, Corley J, Chouinard-Decorte F, Starr JM, Wardlaw JM, Bastin ME, Deary IJ. Cigarette smoking and thinning of the brain’s cortex. Mol Psychiatry. 2015;20:778–85.
Staals J, Makin SD, Doubal FN, Dennis MS, Wardlaw JM. Stroke subtype, vascular risk factors, and total MRI brain small-vessel disease burden. Neurology. 2014;83:1228–34.
van Dijk EJ, Prins ND, Vrooman HA, Hofman A, Koudstaal PJ, Breteler MM. Progression of cerebral small vessel disease in relation to risk factors and cognitive consequences: Rotterdam Scan study. Stroke. 2008;39:2712–9.
Gons RA, van Norden AG, de Laat KF, van Oudheusden LJ, van Uden IW, Zwiers MP, Norris DG, de Leeuw FE. Cigarette smoking is associated with reduced microstructural integrity of cerebral white matter. Brain. 2011;134:2116–24.
Doubal FN, MacLullich AM, Ferguson KJ, Dennis MS, Wardlaw JM. Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke. 2010;41:450–4.
Csordas A, Bernhard D. The biology behind the atherothrombotic effects of cigarette smoke. Nat Rev Cardiol. 2013;10:219–30.
Dopp JM, Reichmuth KJ, Morgan BJ. Obstructive sleep apnea and hypertension: mechanisms, evaluation, and management. Curr Hypertens Rep. 2007;9:529–34.
Drager LF, McEvoy RD, Barbe F, Lorenzi-Filho G, Redline S, Initiative I. Sleep apnea and cardiovascular disease: lessons from recent trials and need for team science. Circulation. 2017;136:1840–50.
Li M, Hou WS, Zhang XW, Tang ZY. Obstructive sleep apnea and risk of stroke: a meta-analysis of prospective studies. Int J Cardiol. 2014;172:466–9.
Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;353:2034–41.
Zhou F, Huang M, Gu L, Hong S, Jiang J, Zeng X, Gong H. Regional cerebral hypoperfusion after acute sleep deprivation: A STROBE-compliant study of arterial spin labeling fMRI. Medicine (Baltimore). 2019;98: e14008.
Wang J, Chen X, Liao J, Zhou L, Han H, Tao J, Lu Z. Non breathing-related sleep fragmentation and imaging markers in patients with atherosclerotic cerebral small vessel disease (CSVD): a cross-sectional case-control study. BMC Neurol. 2020;20:98.
Huang S, Wang D, Zhou H, Chen Z, Wang H, Li Y, Yin S. Neuroimaging consequences of cerebral small vessel disease in patients with obstructive sleep apnea-hypopnea syndrome. Brain Behav. 2019;9: e01364.
Alvarez-Sabin J, Romero O, Delgado P, Quintana M, Santamarina E, Ferre A, Maisterra O, Riba-Llena I, Montaner J, Sampol G. Obstructive sleep apnea and silent cerebral infarction in hypertensive individuals. J Sleep Res. 2018;27:232–9.
Mohsenin V. Sleep-related breathing disorders and risk of stroke. Stroke. 2001;32:1271–8.
Calvin AD, Albuquerque FN, Lopez-Jimenez F, Somers VK. Obstructive sleep apnea, inflammation, and the metabolic syndrome. Metab Syndr Relat Disord. 2009;7:271–8.
Chong PLH, Garic D, Shen MD, Lundgaard I, Schwichtenberg AJ. Sleep, cerebrospinal fluid, and the glymphatic system: a systematic review. Sleep Med Rev. 2022;61: 101572.
Bokura H, Yamaguchi S, Iijima K, Nagai A, Oguro H. Metabolic syndrome is associated with silent ischemic brain lesions. Stroke. 2008;39:1607–9.
Schilling S, Tzourio C, Dufouil C, Zhu Y, Berr C, Alperovitch A, Crivello F, Mazoyer B, Debette S. Plasma lipids and cerebral small vessel disease. Neurology. 2014;83:1844–52.
Jimenez-Conde J, Biffi A, Rahman R, Kanakis A, Butler C, Sonni S, Massasa E, Cloonan L, Gilson A, Capozzo K, et al. Hyperlipidemia and reduced white matter hyperintensity volume in patients with ischemic stroke. Stroke. 2010;41:437–42.
Ohwaki K, Yano E, Tamura A, Inoue T, Saito I. Hypercholesterolemia is associated with a lower risk of cerebral ischemic small vessel disease detected on brain checkups. Clin Neurol Neurosurg. 2013;115:669–72.
Bae JH, Bassenge E, Kim KB, Kim YN, Kim KS, Lee HJ, Moon KC, Lee MS, Park KY, Schwemmer M. Postprandial hypertriglyceridemia impairs endothelial function by enhanced oxidant stress. Atherosclerosis. 2001;155:517–23.
Wingo AP, Vattathil SM, Liu J, Fan W, Cutler DJ, Levey AI, Schneider JA, Bennett DA, Wingo TS. LDL cholesterol is associated with higher AD neuropathology burden independent of APOE. J Neurol Neurosurg Psychiatry. 2022;93:930–8.
Iadecola C, Anrather J, Kamel H. Effects of COVID-19 on the nervous system. Cell. 2020;183(16–27): e11.
Lu Y, Li X, Geng D, Mei N, Wu PY, Huang CC, Jia T, Zhao Y, Wang D, Xiao A, Yin B. Cerebral Micro-Structural Changes in COVID-19 Patients - An MRI-based 3-month Follow-up Study. EClinicalMedicine. 2020;25: 100484.
Hanafi R, Roger PA, Perin B, Kuchcinski G, Deleval N, Dallery F, Michel D, Hacein-Bey L, Pruvo JP, Outteryck O, Constans JM. COVID-19 Neurologic Complication with CNS vasculitis-like pattern. AJNR Am J Neuroradiol. 2020;41:1384–7.
Brun G, Hak JF, Coze S, Kaphan E, Carvelli J, Girard N, Stellmann JP. COVID-19-White matter and globus pallidum lesions: Demyelination or small-vessel vasculitis? Neurol Neuroimmunol Neuroinflamm. 2020;7:e777.
Keller E, Brandi G, Winklhofer S, Imbach LL, Kirschenbaum D, Frontzek K, Steiger P, Dietler S, Haeberlin M, Willms J, et al. Large and small cerebral vessel involvement in severe COVID-19: detailed clinical workup of a case series. Stroke. 2020;51:3719–22.
Wenzel J, Lampe J, Muller-Fielitz H, Schuster R, Zille M, Muller K, Krohn M, Korbelin J, Zhang L, Ozorhan U, et al. The SARS-CoV-2 main protease M(pro) causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat Neurosci. 2021;24:1522–33.
Addo J, Ayerbe L, Mohan KM, Crichton S, Sheldenkar A, Chen R, Wolfe CD, McKevitt C. Socioeconomic status and stroke: an updated review. Stroke. 2012;43:1186–91.
Pennlert J, Asplund K, Glader EL, Norrving B, Eriksson M. Socioeconomic status and the risk of stroke recurrence: persisting gaps observed in a nationwide swedish study 2001 to 2012. Stroke. 2017;48:1518–23.
Marshall IJ, Wang Y, Crichton S, McKevitt C, Rudd AG, Wolfe CD. The effects of socioeconomic status on stroke risk and outcomes. Lancet Neurol. 2015;14:1206–18.
Zhou LW, Panenka WJ, Al-Momen G, Gicas KM, Thornton AE, Jones AA, Woodward M, Heran MKS, Vertinsky AT, Su W, et al. Cerebral small vessel disease, risk factors, and cognition in tenants of precarious housing. Stroke. 2020;51:3271–8.
Pflugrad H, Bronzlik P, Raab P, Tryc AB, Goldbecker A, Barg-Hock H, Strassburg CP, Ding XQ, Lanfermann H, Weissenborn K. Cerebral white matter lesions in patients with cirrhosis - causative for hepatic encephalopathy or bystanders? Liver Int. 2015;35:1816–23.
Rojas R, Riascos R, Vargas D, Cuellar H, Borne J. Neuroimaging in drug and substance abuse part I: cocaine, cannabis, and ecstasy. Top Magn Reson Imaging. 2005;16:231–8.
Borne J, Riascos R, Cuellar H, Vargas D, Rojas R. Neuroimaging in drug and substance abuse part II: opioids and solvents. Top Magn Reson Imaging. 2005;16:239–45.
Gu Y, Scarmeas N. Diet and neuroimaging markers of cerebrovascular disease. Curr Nutr Rep. 2013;2:81–9.
Persyn E, Hanscombe KB, Howson JMM, Lewis CM, Traylor M, Markus HS. Genome-wide association study of MRI markers of cerebral small vessel disease in 42,310 participants. Nat Commun. 2020;11:2175.
Biffi A, Sonni A, Anderson CD, Kissela B, Jagiella JM, Schmidt H, Jimenez-Conde J, Hansen BM, Fernandez-Cadenas I, Cortellini L, et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann Neurol. 2010;68:934–43.
Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38:254–9.
O’Donnell HC, Rosand J, Knudsen KA, Furie KL, Segal AZ, Chiu RI, Ikeda D, Greenberg SM. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med. 2000;342:240–5.
Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Del Tredici K, Braak H. Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2002;61:282–93.
Schilling S, DeStefano AL, Sachdev PS, Choi SH, Mather KA, DeCarli CD, Wen W, Hogh P, Raz N, Au R, et al. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta-analysis. Neurology. 2013;81:292–300.
Kang P, Ying C, Chen Y, Ford AL, An H, Lee JM. Oxygen metabolic stress and white matter injury in patients with cerebral small vessel disease. Stroke. 2022;53:1570–9.
Humphrey JD. Mechanisms of arterial remodeling in hypertension: coupled roles of wall shear and intramural stress. Hypertension. 2008;52:195–200.
Burtscher J, Mallet RT, Burtscher M, Millet GP. Hypoxia and brain aging: neurodegeneration or neuroprotection? Ageing Res Rev. 2021;68: 101343.
Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–63.
Shi Y, Thrippleton MJ, Makin SD, Marshall I, Geerlings MI, de Craen AJM, van Buchem MA, Wardlaw JM. Cerebral blood flow in small vessel disease: a systematic review and meta-analysis. J Cereb Blood Flow Metab. 2016;36:1653–67.
van der Veen PH, Muller M, Vincken KL, Hendrikse J, Mali WP, van der Graaf Y, Geerlings MI, Group SS. Longitudinal relationship between cerebral small-vessel disease and cerebral blood flow: the second manifestations of arterial disease-magnetic resonance study. Stroke. 2015;46:1233–8.
Duncombe J, Kitamura A, Hase Y, Ihara M, Kalaria RN, Horsburgh K. Chronic cerebral hypoperfusion: a key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin Sci (Lond). 2017;131:2451–68.
De Rango P, Caso V, Leys D, Paciaroni M, Lenti M, Cao P. The role of carotid artery stenting and carotid endarterectomy in cognitive performance: a systematic review. Stroke. 2008;39:3116–27.
Markus HS, Lythgoe DJ, Ostegaard L, O’Sullivan M, Williams SC. Reduced cerebral blood flow in white matter in ischaemic leukoaraiosis demonstrated using quantitative exogenous contrast based perfusion MRI. J Neurol Neurosurg Psychiatry. 2000;69:48–53.
O’Sullivan M, Lythgoe DJ, Pereira AC, Summers PE, Jarosz JM, Williams SC, Markus HS. Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology. 2002;59:321–6.
Wardlaw JM, Makin SJ, Valdés Hernández MC, Armitage PA, Heye AK, Chappell FM, Muñoz-Maniega S, Sakka E, Shuler K, Dennis MS, Thrippleton MJ. Blood-brain barrier failure as a core mechanism in cerebral small vessel disease and dementia: evidence from a cohort study. Alzheimers Dement. 2017;13:634–43.
Munoz Maniega S, Chappell FM, Valdes Hernandez MC, Armitage PA, Makin SD, Heye AK, Thrippleton MJ, Sakka E, Shuler K, Dennis MS, Wardlaw JM. Integrity of normal-appearing white matter: Influence of age, visible lesion burden and hypertension in patients with small-vessel disease. J Cereb Blood Flow Metab. 2017;37:644–56.
Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–13.
Bai Y, Zhu X, Chao J, Zhang Y, Qian C, Li P, Liu D, Han B, Zhao L, Zhang J, et al. Pericytes contribute to the disruption of the cerebral endothelial barrier via increasing VEGF expression: implications for stroke. PLoS ONE. 2015;10: e0124362.
Li YN, Pan R, Qin XJ, Yang WL, Qi Z, Liu W, Liu KJ. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J Neurochem. 2014;129:120–9.
Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125:2549–57.
Lange C, Storkebaum E, de Almodovar CR, Dewerchin M, Carmeliet P. Vascular endothelial growth factor: a neurovascular target in neurological diseases. Nat Rev Neurol. 2016;12:439–54.
Song K, Li Y, Zhang H, An N, Wei Y, Wang L, Tian C, Yuan M, Sun Y, Xing Y, Gao Y. Oxidative stress-mediated Blood-Brain Barrier (BBB) disruption in neurological diseases. Oxid Med Cell Longev. 2020;2020:4356386.
Galea I. The blood-brain barrier in systemic infection and inflammation. Cell Mol Immunol. 2021;18:2489–501.
Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501–18.
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–6.
Taoka T, Masutani Y, Kawai H, Nakane T, Matsuoka K, Yasuno F, Kishimoto T, Naganawa S. Evaluation of glymphatic system activity with the diffusion MR technique: diffusion tensor image analysis along the perivascular space (DTI-ALPS) in Alzheimer’s disease cases. Jpn J Radiol. 2017;35:172–8.
Zhang W, Zhou Y, Wang J, Gong X, Chen Z, Zhang X, Cai J, Chen S, Fang L, Sun J, Lou M. Glymphatic clearance function in patients with cerebral small vessel disease. Neuroimage. 2021;238: 118257.
Tang J, Zhang M, Liu N, Xue Y, Ren X, Huang Q, Shi L, Fu J. The association between glymphatic system dysfunction and cognitive impairment in cerebral small vessel disease. Front Aging Neurosci. 2022;14:916633.
Kamagata K, Andica C, Takabayashi K, Saito Y, Taoka T, Nozaki H, Kikuta J, Fujita S, Hagiwara A, Kamiya K, et al. Association of MRI Indices of Glymphatic System With Amyloid Deposition and Cognition in Mild Cognitive Impairment and Alzheimer Disease. Neurology. 2022;99:e2648–60.
Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO. White matter changes in dementia: role of impaired drainage of interstitial fluid. Brain Pathol. 2015;25:63–78.
Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain. 2017;140:1829–50.
Morrone CD, Bishay J, McLaurin J. Potential Role of Venular Amyloid in Alzheimer's Disease Pathogenesis. Int J Mol Sci. 2020;21:1985.
Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol. 1998;153:725–33.
Mendel T, Wierzba-Bobrowicz T, Stepien T, Szpak GM. beta-amyloid deposits in veins in patients with cerebral amyloid angiopathy and intracerebral haemorrhage. Folia Neuropathol. 2013;51:120–6.
Bishay J, Beckett TL, Lai AY, Hill ME, McMahon D, McLaurin J. Venular amyloid accumulation in transgenic Fischer 344 Alzheimer’s disease rats. Sci Rep. 2022;12:15287.
Mestre H, Kostrikov S, Mehta RI, Nedergaard M. Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin Sci (Lond). 2017;131:2257–74.
Kiviniemi V, Wang X, Korhonen V, Keinanen T, Tuovinen T, Autio J, LeVan P, Keilholz S, Zang YF, Hennig J, Nedergaard M. Ultra-fast magnetic resonance encephalography of physiological brain activity - Glymphatic pulsation mechanisms? J Cereb Blood Flow Metab. 2016;36:1033–45.
Owasil R, O'Neill R, Keable A, Nimmo J, MacGregor Sharp M, Kelly L, Saito S, Simpson JE, Weller RO, Smith C, et al. The Pattern of AQP4 Expression in the Ageing Human Brain and in Cerebral Amyloid Angiopathy. Int J Mol Sci. 2020;21:1225.
Low A, Mak E, Rowe JB, Markus HS, O’Brien JT. Inflammation and cerebral small vessel disease: a systematic review. Ageing Res Rev. 2019;53: 100916.
Mohamed IN, Ishrat T, Fagan SC, El-Remessy AB. Role of inflammasome activation in the pathophysiology of vascular diseases of the neurovascular unit. Antioxid Redox Signal. 2015;22:1188–206.
Muramatsu R, Kuroda M, Matoba K, Lin H, Takahashi C, Koyama Y, Yamashita T. Prostacyclin prevents pericyte loss and demyelination induced by lysophosphatidylcholine in the central nervous system. J Biol Chem. 2015;290:11515–25.
Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74.
Douglas G, Channon KM. The pathogenesis of atherosclerosis Medicine. 2014;42:480–4.
Huang CJ, Zhou X, Yuan X, Zhang W, Li MX, You MZ, Zhu XQ, Sun ZW. Contribution of inflammation and hypoperfusion to white matter hyperintensities-related cognitive impairment. Front Neurol. 2021;12: 786840.
Tellis CC, Tselepis AD. Pathophysiological role and clinical significance of lipoprotein-associated phospholipase A(2) (Lp-PLA(2)) bound to LDL and HDL. Curr Pharm Des. 2014;20:6256–69.
Kaiser D, Weise G, Moller K, Scheibe J, Posel C, Baasch S, Gawlitza M, Lobsien D, Diederich K, Minnerup J, et al. Spontaneous white matter damage, cognitive decline and neuroinflammation in middle-aged hypertensive rats: an animal model of early-stage cerebral small vessel disease. Acta Neuropathol Commun. 2014;2:169.
Rajani RM, Quick S, Ruigrok SR, Graham D, Harris SE, Verhaaren BFJ, Fornage M, Seshadri S, Atanur SS, Dominiczak AF, et al. Reversal of endothelial dysfunction reduces white matter vulnerability in cerebral small vessel disease in rats. Sci Transl Med. 2018;10:eaam9507.
Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54.
Vitale G, Salvioli S, Franceschi C. Oxidative stress and the ageing endocrine system. Nat Rev Endocrinol. 2013;9:228–40.
Petrie JR, Guzik TJ, Touyz RM. Diabetes, hypertension, and cardiovascular disease: clinical insights and vascular mechanisms. Can J Cardiol. 2018;34:575–84.
Evans LE, Taylor JL, Smith CJ, Pritchard HAT, Greenstein AS, Allan SM. Cardiovascular comorbidities, inflammation, and cerebral small vessel disease. Cardiovasc Res. 2021;117:2575–88.
Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 2019;18:684–96.
van Middelaar T, Argillander TE, Schreuder F, Deinum J, Richard E, Klijn CJM. Effect of antihypertensive medication on cerebral small vessel disease: a systematic review and meta-analysis. Stroke. 2018;49:1531–3.
Ji T, Zhao Y, Wang J, Cui Y, Duan D, Chai Q, Zhang H, Liu Z. Effect of low-dose statins and apolipoprotein e genotype on cerebral small vessel disease in older hypertensive patients: a subgroup analysis of a randomized clinical trial. J Am Med Dir Assoc. 2018;19(995–1002): e1004.
Xiong Y, Wong A, Cavalieri M, Schmidt R, Chu WW, Liu X, Wong KS, Mok V. Prestroke statins, progression of white matter hyperintensities, and cognitive decline in stroke patients with confluent white matter hyperintensities. Neurotherapeutics. 2014;11:606–11.
Woo D, Deka R, Falcone GJ, Flaherty ML, Haverbusch M, Martini SR, Greenberg SM, Ayres AM, Sauerbeck L, Kissela BM, et al. Apolipoprotein E, statins, and risk of intracerebral hemorrhage. Stroke. 2013;44:3013–7.
Arima H, Tzourio C, Anderson C, Woodward M, Bousser MG, MacMahon S, Neal B, Chalmers J, Group PC. Effects of perindopril-based lowering of blood pressure on intracerebral hemorrhage related to amyloid angiopathy: the PROGRESS trial. Stroke. 2010;41:394–6.
Weber R, Weimar C, Blatchford J, Hermansson K, Wanke I, Moller-Hartmann C, Gizewski ER, Forsting M, Demchuk AM, Sacco RL, et al. Telmisartan on top of antihypertensive treatment does not prevent progression of cerebral white matter lesions in the prevention regimen for effectively avoiding second strokes (PRoFESS) MRI substudy. Stroke. 2012;43:2336–42.
ten Dam VH, van den Heuvel DM, van Buchem MA, Westendorp RG, Bollen EL, Ford I, de Craen AJ, Blauw GJ, Group PS: Effect of pravastatin on cerebral infarcts and white matter lesions. Neurology 2005, 64:1807-1809
Mok VC, Lam WW, Fan YH, Wong A, Ng PW, Tsoi TH, Yeung V, Wong KS. Effects of statins on the progression of cerebral white matter lesion: Post hoc analysis of the ROCAS (Regression of Cerebral Artery Stenosis) study. J Neurol. 2009;256:750–7.
Shah J, Liu S, Yu W. Contemporary antiplatelet therapy for secondary stroke prevention: a narrative review of current literature and guidelines. Stroke Vasc Neurol. 2022;7:406–14.
Kwok CS, Shoamanesh A, Copley HC, Myint PK, Loke YK, Benavente OR. Efficacy of antiplatelet therapy in secondary prevention following lacunar stroke: pooled analysis of randomized trials. Stroke. 2015;46:1014–23.
Group SPSS, Benavente OR, Coffey CS, Conwit R, Hart RG, McClure LA, Pearce LA, Pergola PE, Szychowski JM. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet. 2013;382:507–15.
Uchiyama S, Shinohara Y, Katayama Y, Yamaguchi T, Handa S, Matsuoka K, Ohashi Y, Tanahashi N, Yamamoto H, Genka C, et al. Benefit of cilostazol in patients with high risk of bleeding: subanalysis of cilostazol stroke prevention study 2. Cerebrovasc Dis. 2014;37:296–303.
Kitamura A, Manso Y, Duncombe J, Searcy J, Koudelka J, Binnie M, Webster S, Lennen R, Jansen M, Marshall I, et al. Long-term cilostazol treatment reduces gliovascular damage and memory impairment in a mouse model of chronic cerebral hypoperfusion. Sci Rep. 2017;7:4299.
Greenberg SM, Cordonnier C, Schneider JA, Smith EE, van Buchem MA, van Veluw SJ, Verbeek MM, Viswanathan A, Werring DJ. Off-label use of aducanumab for cerebral amyloid angiopathy. Lancet Neurol. 2021;20:596–7.
Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061–70.
Sperling RA, Jack CR Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367–85.
Sveikata L, Charidimou A, Viswanathan A. Vessels Sing Their ARIAs: the role of vascular amyloid in the age of aducanumab. Stroke. 2022;53:298–302.
VandeVrede L, Gibbs DM, Koestler M, La Joie R, Ljubenkov PA, Provost K, Soleimani-Meigooni D, Strom A, Tsoy E, Rabinovici GD, Boxer AL. Symptomatic amyloid-related imaging abnormalities in an APOE epsilon4/epsilon4 patient treated with aducanumab. Alzheimers Dement (Amst). 2020;12: e12101.
Liu D, Cai X, Yang Y, Wang S, Yao D, Mei L, Jing J, Li S, Yan H, Meng X, et al. Associations of life’s simple 7 with cerebral small vessel disease. Stroke. 2022;53:2859–67.
Ngandu T, Lehtisalo J, Solomon A, Levalahti E, Ahtiluoto S, Antikainen R, Backman L, Hanninen T, Jula A, Laatikainen T, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385:2255–63.
Pedralli ML, Marschner RA, Kollet DP, Neto SG, Eibel B, Tanaka H, Lehnen AM. Different exercise training modalities produce similar endothelial function improvements in individuals with prehypertension or hypertension: a randomized clinical trial Exercise, endothelium and blood pressure. Sci Rep. 2020;10:7628.
Yubero-Serrano EM, Fernandez-Gandara C, Garcia-Rios A, Rangel-Zuniga OA, Gutierrez-Mariscal FM, Torres-Pena JD, Marin C, Lopez-Moreno J, Castano JP, Delgado-Lista J, et al. Mediterranean diet and endothelial function in patients with coronary heart disease: an analysis of the CORDIOPREV randomized controlled trial. PLoS Med. 2020;17: e1003282.
Pieters B, Staals J, Knottnerus I, Rouhl R, Menheere P, Kessels A, Lodder J. Periventricular white matter lucencies relate to low vitamin B12 levels in patients with small vessel stroke. Stroke. 2009;40:1623–6.
Cavalieri M, Schmidt R, Chen C, Mok V, de Freitas GR, Song S, Yi Q, Ropele S, Grazer A, Homayoon N, et al. B vitamins and magnetic resonance imaging-detected ischemic brain lesions in patients with recent transient ischemic attack or stroke: the VITAmins TO Prevent Stroke (VITATOPS) MRI-substudy. Stroke. 2012;43:3266–70.
Gopalan Y, Shuaib IL, Magosso E, Ansari MA, Abu Bakar MR, Wong JW, Khan NA, Liong WC, Sundram K, Ng BH, et al. Clinical investigation of the protective effects of palm vitamin E tocotrienols on brain white matter. Stroke. 2014;45:1422–8.
Kato T, Manabe RI, Igarashi H, Kametani F, Hirokawa S, Sekine Y, Fujita N, Saito S, Kawashima Y, Hatano Y, et al. Candesartan prevents arteriopathy progression in cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy model. J Clin Invest. 2021;131:e140555.
Oliveira DV, Coupland KG, Shao W, Jin S, Del Gaudio F, Wang S, Fox R, Rutten JW, Sandin J, Zetterberg H, et al. Active immunotherapy reduces NOTCH3 deposition in brain capillaries in a CADASIL mouse model. EMBO Mol Med. 2023;15: e16556.
Machuca-Parra AI, Bigger-Allen AA, Sanchez AV, Boutabla A, Cardona-Velez J, Amarnani D, Saint-Geniez M, Siebel CW, Kim LA, D’Amore PA, Arboleda-Velasquez JF. Therapeutic antibody targeting of Notch3 signaling prevents mural cell loss in CADASIL. J Exp Med. 2017;214:2271–82.
Ghezali L, Capone C, Baron-Menguy C, Ratelade J, Christensen S, Ostergaard Pedersen L, Domenga-Denier V, Pedersen JT, Joutel A. Notch3(ECD) immunotherapy improves cerebrovascular responses in CADASIL mice. Ann Neurol. 2018;84:246–59.
Tarantini S, Valcarcel-Ares MN, Toth P, Yabluchanskiy A, Tucsek Z, Kiss T, Hertelendy P, Kinter M, Ballabh P, Sule Z, et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 2019;24: 101192.
Tuo M, Xiao Y, Xu Y, Wang L, Wei X, Zhang L. Role of Granulocyte-colony stimulating factor in the protection of cerebral vascular endothelium, white matter, and cognition. Curr Neurovasc Res. 2019;16:425–32.
Jalal FY, Yang Y, Thompson JF, Roitbak T, Rosenberg GA. Hypoxia-induced neuroinflammatory white-matter injury reduced by minocycline in SHR/SP. J Cereb Blood Flow Metab. 2015;35:1145–53.
Acknowledgements
We thank Ms. Danielle Feathers for proofreading of the manuscript.
Funding
This work was supported by NIH grants U19AG069701, RF1AG071226, RF1AG068034, and RF1AG057181 (to T.K.), a Cure Alzheimer’s Fund grant (to T.K.), and Florida Department of Health Ed and Ethel Moore Alzheimer’s Disease Research Program 22A08 (to Y.I.).
Author information
Authors and Affiliations
Contributions
YI led the writing of the manuscript and prepared the figures. SF contributed to the editing of the manuscript. GB and TK supervised the writing and co-edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors agreed to publish.
Competing interests
G.B. is an employee of SciNeuro Pharmaceuticals. Other authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Inoue, Y., Shue, F., Bu, G. et al. Pathophysiology and probable etiology of cerebral small vessel disease in vascular dementia and Alzheimer’s disease. Mol Neurodegeneration 18, 46 (2023). https://doi.org/10.1186/s13024-023-00640-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-023-00640-5