
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and is the most common cause of dementia. The pathogenesis of AD still remains unclear, including two main hypotheses: amyloid cascade and tau hyperphosphorylation. The hallmark neuropathological changes of AD are extracellular deposits of amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs). Endocytosis plays an important role in a number of cellular processes including communication with the extracellular environment, nutrient uptake, and signaling by the cell surface receptors. Based on the results of genetic and biochemical studies, there is a link between neuronal endosomal function and AD pathology. Taking this into account, we can state that in the results of previous research, endolysosomal abnormality is an important cause of neuronal lesions in the brain. Endocytosis is a central pathway involved in the regulation of the degradation of amyloidogenic components. The results of the studies suggest that a correlation between alteration in the endocytosis process and associated protein expression progresses AD. In this article, we discuss the current knowledge about endosomal abnormalities in AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
According to the Alzheimer’s Association and World Health Organization (WHO), dementia affects almost 55 million people worldwide—a number that will double by 2050. Alzheimer’s disease (AD) is the most common cause of dementia and the most prevalent in the elderly population [1]. AD is a gradual and progressive neurodegenerative disorder clinically characterized by deterioration of episodic memory and successive impairment of additional cognitive domains, with behavioral changes impacting activities of daily living. Although AD has no specific symptoms, the diagnosis is currently made by meeting ICD-10 or DSM-V criteria, which include the presence of cognitive deficits in the form of memory loss without impaired consciousness, agnosia, aphasia, apraxia, impaired executive activities, a progressive disease process, deterioration in social and occupational functioning, and changes in social behavior [2]. The debilitating course, high mortality, and potential tripling of the incidence of AD by 2060 result in an increase in global economic expenditures for the healthcare sector [3]. Although most AD cases affect patients ≥ 65 years of age [4], the disease can also occur as early as the 3rd decade of life with an early diagnosis [5]. The lack of AD-specific biomarkers makes it difficult to rapidly detect the disease, especially in the early stages of AD when patients present with initial prodromal symptoms. For a very early diagnosis of AD, we need to have affordable, ultrasensitive, and selective molecular detection methods. We have till now failed to manage it effectively, with only a handful of symptomatic therapies and questionable newer disease-modifying agents [6].
In the early 1990s, the amyloid cascade hypothesis was revealed as a primary pathway that leads to AD. Currently, the major theories related to the mechanisms involved in the pathogenesis of AD are amyloid-β (Aβ) plaques, neurofibrillary tangles (NFTs) with phosphorylated tau protein (P-tau), gliosis, and neuronal loss, accompanied by cerebrovascular amyloidosis, inflammation, and major synaptic changes. The model was based on the assumption that extracellular deposits of Aβ initiate and are a direct cause of neurotoxicity, induction of tau protein pathology—NFT formation—and handicap brain vascular system, leading to neuronal death and neurodegeneration [7]. In addition, cumulating Aβ deposits in turn activate glial cells—microglia (resident immune cells of the brain, macrophages) and astrocytes—leading to an inflammatory response in the central nervous system (CNS)—neuroinflammation [8]. More recent studies suggest that astrocyte reactivity abnormality could be placed as an early biomarker model of AD progression, which may explain the validity of anti-Aβ therapy, modifying this Aβ-astrocyte-tau pathway. There is an important emphasis on a decrease in synapse density and a corresponding decrease in brain volume due to neurodegeneration [9]. The second core pathology in AD is a deposition of hyper-phosphorylated tau protein inside neurons. AD pathogenesis involves pathogenic contributions from multiple components, signaling pathways, and alterations in the behavior of various cell types. Previous studies indicated that immune response, Aβ metabolism, cholesterol/lipid dysfunction, endocytosis, and angiogenesis are strongly associated with AD. A recent large-scale genome-wide association study (GWAS) showed a number of endocytosis-related molecules in AD, and the relationship between AD and endocytosis (e.g., in Aβ aggregation) is now of great interest to the scientific community [10].
In this work, we discuss current knowledge on the role of endocytosis in AD pathophysiology and show a potential link between endocytosis and AD.
Types of endocytosis
Endocytosis is a fundamental cellular process in all eukaryotic cells, which regulate nearly every aspect of cellular physiology and are often impaired in pathological conditions. It is the cellular process of membrane vesicular transport between the plasma membrane (PM) and cytoplasmic membrane compartments, as well as within the intracellular membrane system (Fig. 1). Cells use endocytosis to take up different types of molecules that cannot otherwise pass through and is an essential mechanism for homeostasis and communication within and between cells. The surface proteins, lipids, and other macromolecules are enveloped within small membrane vesicles formed by the entrapment of the cell membrane and subsequently taken up into the cell. Endocytosis is a complex program that is strongly tied to signal transduction [9, 11] and serves as the cell’s primary communication infrastructure. Endocytosis also helps to generate cell polarity by rapidly redistributing cell surface molecules to active locations [12, 13]. Instead of planar diffusion across the plasma membrane, this fast and site-specific redistribution of membrane proteins is accomplished through a cycle of endocytosis and directed recycling. Although endocytosis is primarily associated with the function of individual cells, it also regulates the function of hundreds of assembled cells. This is interesting because it implies that the endocytosis event, which occurs at the cell level, may be synchronized across numerous cells and act in a coordinated manner.

Graphical presentation representing endocytosis. In endocytosis process, various extracellular components unbound or recognized by membrane-localized receptors become transported into plasma membrane–derived vesicles. These structures may serve to recycle the material back to the plasma membrane or dispatch it to lysosomes for future degradation
Each cargo taken up by nascent endocytotic vesicles is different. The cargo transport after endocytosis will largely depend on the combination of cargo and coat proteins involved in recognizing a specific cargo [14,15,16]. As a result, these cargoes can be employed as markers for the respective endocytotic and post-endocytotic pathways. On the other hand, it is not rare for a specific cargo to be identified in more than one endocytosis pathway. Endocytosis is usually subdivided into phagocytosis, pinocytosis, clathrin-mediated endocytosis, and caveolin-mediated endocytosis. Pinocytosis involves the internalization of small molecules, whereas phagocytosis involves the internalization of large particles.
To maintain their characteristic polar structure, neurons require the right distribution of different receptors, the perfect arrangement of proteins and organelles in dendrites and axons, continual exocytosis/endocytosis of synaptic vesicles, and the removal of defective proteins. Because synaptic vesicles must be constantly exposed to exocytosis and endocytosis, continuous membrane trafficking is essential at synapses [17,18,19]. As a result, membrane transport is involved in all aspects of neuronal function, and its malfunction is linked to neurodegeneration.
Phagocytosis
Phagocytosis is a kind of clathrin-independent endocytosis (CIE) in which membrane-derived vesicles known as phagosomes selectively detect and internalize particles with sizes greater than 500 nm. Phagocytosis is divided into four stages: target recognition, signal transduction that initiates the internalization process, phagosome formation, and phagolysosome maturation. The cytoskeleton must be involved in membrane modification during this process. Phagocytosis is a complicated process that is necessary for growth and tissue homeostasis and removing pathogens and apoptotic cells [20]. It is also the first line of defense involving innate immune cells against infections, and immune system phagocytes such as neutrophils, macrophages, monocytes, and dendritic cells use phagocytosis to consume invading organisms, pathogens, dead cells, and debris [21]. Surface receptors recognize phagocytosis targets and are divided into two types: opsonin receptors and non-opsonin receptors. Opsonin receptors detect foreign particles by binding to opsonin produced by the host. Non-opsonin receptors recognize a collection of chemicals on the surface of the phagocytosed item [21, 22]. When these receptors bind to target particles, they activate the protein tyrosine kinase Syk, which generates phosphoinositide second messengers and initiates an intracellular signaling cascade that recruits activated Rho GTPases. Small GTPases (which contain Ras, Rho, Rab, Ran, and Arf proteins) are central regulators of a wide variety of signal transduction pathways (regulate actin reorganization, influence cell polarity, microtubule dynamics, membrane transport pathways, and transcription factor activity) in all eukaryotic cells. Rho GTPases reorganize actin polymerization in phagocytic cups to form pseudopods, which seal their ends and transform into phagosomes after surrounding the target particle [23]. In the final step of phagocytosis, phagosomes continually merge and split with endocytic vesicles before fusing with lysosomes to form phagolysosomes. This process involves a continuous decrease in pH and the acquisition of digestive enzymes, which leads to target digestion and antigen retrieval for presentation on the phagocytic surface [24].
Macropinocytosis
Pinocytosis is a type of nonspecific endocytosis in which dissolved objects are integrated into vesicles of arbitrary size. Macropinocytosis is an actine-mediated bulk type of endocytosis, a large-volume endocytic pathway that produces vesicles (macropinosomes) with diameters ranging from 200 nm to 5 µm [25]. Under the cell membrane, actin polymerization rings (circular ruffles) can form, forming a cup-like entry opening. When this cup closes, it transforms into a macropinosome. Because the cargo taken up by macropinocytosis can range from liquids to particles and can be taken up in large quantities, this pathway has piqued the interest of drug delivery researchers. Unlike phagocytosis, amiloride specifically inhibits macropinosomes by blocking the Na + /H + exchanger in the cell membrane. Pathogens have evolved sophisticated macropinocytosis mechanisms, which allow for metabolic adaptation and survival in nutrient-deficient environments [26]. Micropinocytosis is regulated by a group of small GTPases. Rab family proteins, which are small GTPases, govern cytoskeleton movement and membrane retention. A small GTPase is a molecular switch that has two states: inactive (bound to GDP) and active (attached to GTP). The activation/inactivation cycle is made up of a guanine exchange factor (GEF) that activates the GTPase by promoting GTP binding, a GTPase activating protein (GAP) that inactivates the GTPase by hydrolyzing GTP, and a guanine nucleotide that prevents GDP dissociation from the GTPase and keeps the GTPase inactive [27]. Ras GTPases activate phosphatidylinositol 3-kinase, causing the cell membrane to form a PIP3-rich membrane domain. This membrane domain functions as a docking site for Rho GTPases and promotes membrane ruffling through actin remodeling. The maturation process is mediated by small Rab GTPases and phosphoinositides once the macropinosome is closed at the plasma membrane. The macropinosome fuses with the early endosome initially. Rab5 and Rab34 facilitate fusion, and Rab5 changes to Rab7 during macropinosome maturation, encouraging fusion to the late endosome/lysosome compartment [28,29,30].
The dysregulation of endocytosis in AD
Evidence from both genetic and biochemical studies supports the involvement of endosomal abnormalities in the pathogenesis of AD. Many endosomal pathways play a role in the amyloidogenesis process. The results are not always unambiguous. In addition, it remains essential to answer the question of whether the changes are the result of the cause of developing AD.
Caveolin-mediated endocytosis
The caveolar pathway involves caveolae, which are bulb-shaped, 50–60-nm plasma membrane invaginations. Caveolae as cholesterol-rich microinvaginations in the cell membrane are associated with the production of Aβ peptides and may play a role in the pathogenesis of AD [31]. However, the results obtained are inconsistent. A positive immunoreactivity to caveolin-1 (CAV-1) is commonly observed in these endocytic structures. In aging brain tissues, CAV-1 was overexpressed, the distribution of which was particularly pronounced in the neurons of the II-VI cortical layers, mainly in pyramidal cells and in the pyramidal cell layer of the CA1 region of the hippocampus, and its subcellular localization at the ultrastructural level was observed in the 50–200-nm-sized vesicle-like caveolae structures. Moreover, the increased level of CAV-1 had a negative effect on the expression of alpha-secretase-released APP ectodomain (sαAPP), probably by blocking the phosphorylation of protein kinase C (PKC) [32]. Contrary to these results, in the post-mortem brain tissue from AD cases, CAV-1 expression was not significantly higher than in control tissues; however, the abundant accumulation of Aβ in the walls of cerebral vessels was associated with loss of CAV-1 and caveolin-2 (CAV-2) expression [56]. There were also changes in the distribution and phenotype of these structures associated with the AD pathological signs. In post-mortem studies of the human brain tissue, accumulation of the voltage-dependent anion channel (VDAC) was observed in caveolae, the distribution of which was particularly pronounced in vanishing neurites of senile plaques, and estrogen receptor alpha (ERα) expression was noted mainly in astrocytes surrounding Aβ plaques [33]. Caveolae may play an important role in the enhanced capture of extracellular Aβ peptides. The inducing of the caveolae-mediated endocytosis allows to bypass the lysosomal degradation of the transferred load, which increases the efficiency of the endocytic cargo transfer mechanism [34]. Changes in the expression level of caveolae markers and AD-related pathological molecules are also influenced by certain risk factors for this dementia. High glucose levels induced increased activation of mTOR signaling, which led to inhibition of CAV-1 expression and hyperphosphorylation of the tau protein [35]. The hypothetical importance of caveolae in the pathogenesis of AD has been demonstrated (Fig. 2).

Hypothesized model of endocytosis pathway in Alzheimer’s disease (AD). In AD, endocytosis resulted in forming increased deposition of Aβ peptides by omitting lysosomal digestion of processed endosomes. Increased activation of mTOR signaling leads to inhibition of CAV-1 expression and hyperphosphorylation of the tau protein degradation of CAV-1-rich caveolae resulting in decreased level of this marker in AD-affected brain tissue
Clathrin-mediated endocytosis (CME)
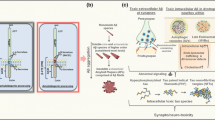
Clathrin-mediated endocytosis (CME) is the major endocytic pathway for the internalization of numerous cargos. In a mouse AD model, the cortical overexpression was confirmed for clathrin-mediated endocytic proteins such as clathrin, dynamin II, and some phosphatidylinositol-binding clathrin assembly protein (PICAM) variants [36]. PICALM belongs to endocytic-related proteins with important regulatory action in the process of endocytosis. Silencing the expression of PICALM isoforms decreased the levels of intracellular amyloid precursor protein (APP), intracellular β-C-terminal fragment (β-CTF), and soluble peptide APPβ (sAPPβ) in H4 cells but had no effect on Aβ40. PICALM and clathrin depletion had a negative effect on the endocytosis process and decreased the expression of beta-secretase (BACE1) at the mRNA level; however, only silencing PICALM expression inhibited BACE1 at the protein level and negatively influenced the cytoplasmic level of clathrin [37]. PICALM as the key clathrin adapter protein was an important mediator of endocytosis for the CME pathway of which the expression at the protein level was decreased in AD brain homogenates, but its immunoreactivity showed a clear cellular differentiation between AD cases and control tissues, showing a clearly higher level of expression in microglia and in neurons in late-onset AD. Moreover, the expression of PICALM in AD was characterized by co-localization with NFT and P-tau protein, which was especially crucial for vanishing neurites and the vicinity of Aβ plaques [38]. PICALM was responsible for endocytosis in the CME pathway associated with the low-density lipoprotein receptor–related protein-1 (LRP1), which regulated Aβ trafficking to Rab5 and Rab11 ensuring proper amyloid clearance. In AD, the reduction of PICALM expression in endothelial cells limited transcytosis and Aβ clearance [39]. PICALM protein is a key molecule in the regulation of the CME mechanism, which in turn increases the distribution of clathrins and adaptor protein 2 (AP-2) in the regions of cell membranes, which facilitates the formation of clathrin-coated vesicles (CCVs). In the AD brain, PICALM was expressed mainly in endothelial cells, which may affect the removal of Aβ through the brain vascular walls [40] (Fig. 3).

Regulation of clathrin-mediated endocytosis (CME) by PICALM. (A) In a healthy brain, PICALM interacts with adaptor protein (AP) and clathrin at the plasma membrane. PICALM participates in CME and facilitates the formation of clathrin-coated vesicles (CCVs). This provides Aβ clearance. (B) PICALM and clathrin depletion in AD brain limited or stopped Aβ clearance. This increases the distribution of Aβ, intensifying neurotoxicity to neurons. In AD, abnormally cleaved PICALM was also associated with neurofibrillary tangles, co-localizing with conformationally abnormal and hyper-phosphorylated tau (P-tau) which contributes to endocytic dysfunction in AD
AP-2 as one of the clathrin’s adaptor proteins may have an influence on the phenomenon of APP endocytosis in the AD brain [41]. Overexpression of AP-2 occurring under the influence of a high-fat diet increased the formation of BACE1/AP-2/clathrin complexes on the surface of cell membranes and induced the redistribution of the resulting complex into the cytoplasm, which promoted APP cleavage [42] and may increase the distribution of Aβ, intensifying neurotoxicity to nervous tissue. The phenomenon of Aβ transcytosis via the brain endothelial cells was caused, among others, by the ABC transporter P-glycoprotein (ABCB1/P-gp) and LRP1 which, in a mechanism dependent on PICALM, regulated the outflow of Aβ through the blood–brain barrier (BBB) [43]. CME via Ca2+ signaling may induce axonal damage resulting from the increased distribution of Aβ peptides, while inhibition of CME in both the in vivo and cellular culture models inhibited axon loss and improved cognitive functioning in the mouse AD model [44]. The amyloidogenic process of APP, which produces Aβ peptide, occurs in an intracellular compartment requiring endocytic trafficking [45,46,47]. APP is processed by BACE1 in Rab5-positive early endosomes under physiological conditions, resulting in a β-CTF [48, 49]. This β-CTF is converted to an Aβ peptide at late endosomes and the Golgi [50]. This emphasizes the relevance of GTPases and membrane trafficking in the etiology of AD. Furthermore, numerous genes associated with endocytic trafficking are linked to the likelihood of acquiring AD [51,52,53,54,55,56].
During cellular senescence in neurons, APP endocytosis was potentiated and Aβ secretion was increased, due to the higher distribution of F actin and clathrin. Other changes observed at the cellular level included the increased formation of early endosomes, APP co-localization to nascent endosomes, and degeneration of synaptic connections associated with the endocytic generation of Aβ progressing with age [57]. Endocytosis in the CME pathway mediated by γ-secretase internalized by clathrin assembly lymphoid myeloid leukemia protein (CALM) through endosome formation regulated Aβ41 production is trafficking by retrograde transport via the endosome-to-trans-Golgi network (TGN) pathway [58]. The loss of CALM disrupted γ-secretase-mediated APP processing leading to reduced release of Aβ42 by HeLa cells [59]. Another possibility related to the increased Aβ distribution in the AD brain is autophagy. The expression of proteins that regulate autophagy was changed during cellular senescence. LC-3-associated endocytosis is an essential process for the processing of Aβ receptors, including TREM2, which in effect ensured protection against the formation of Aβ plaques and the formation of cognitive deficits in an AD animal model [60]. One of the potential causes of the cognitive deficit observed in AD patients was the CME pathway–related loss of AMPA receptors (AMPARs) in the postsynaptic regions, induced by the intensification of endocytosis due to Aβ plaques [61]. AMPA-type glutamate receptors (AMPARs) are a group of receptor protein complexes that regulate neuroplasticity and synaptic transmission related to the learning and memory processes. The endocytic adapter CALM and AP180 N-terminal homology (ANTH) domain–containing proteins HIP1 and HIP1R regulated Ca2+-permeable GluA1 AMPAR homomer endocytosis by affecting the membrane expression of these receptors in the postsynaptic region of neurons through membrane remodeling by a clathrin mechanism independent of clathrin [62].
Microglia acting in the AD-affected brain via phagocytosis and clathrin-independent pathway
Phagocytosis is one of the subtypes of endocytosis in which cargo assimilation can occur regarding the receptor-mediated or non-receptor-mediated pathway [63]. Aβ deposits in the brain activate the local immune response, which leads to the migration of astrocytes and microglial cells into the region of plaque formation, whose biological task is to phagocytose the formed lesions. Nevertheless, amyloid peptides that accumulate in the AD brain may impair the phagocytic capacity of astroglial cells to remove the produced Aβ oligomers [64]. Phagocytosis of Aβ plaques by microglia changes over time the gene expression profiles of late-onset AD and modulates the activity of these cells toward increased phagocytosis of synaptic components located in the vicinity of the plaques [65]. The generation of Aβ peptides was characterized by high heterogeneity in proteolytic cleavages and in the relevant post-translational modification processes, which may translate into the rate of amyloid aggregate formation, their neurotoxicity, and the effect of plaques against phagocytic cells (Fig. 4).

Graphical presentation representing the theoretical basis of Aβ accumulation in the brain tissue. Aβ deposits in the brain cause the migration of astrocytes and microglial cells into the region of plaque formation. Biological task of these cells is to phagocytose the formed lesions. Heterogeneity in proteolytic cleavages during Aβ generation and the relevant post-translational modification processes of Aβ may translate into the rate of amyloid aggregate formation, their neurotoxicity, and the effect of plaques against phagocytic cells. The symbols of the stop sign and the plus sign refer to the intensity of a given process or to the effect of increased or decreased endocytosis in relation to selected types of cells making up the nervous tissue
Post-translational modifications of Aβ peptides related to their phosphorylation seem to hinder phagocytosis of amyloid deposits by microglial cells in a TREM2-dependent mechanism [66]. TREM2 as a membrane-associated receptor is located not only in microglia cells, but also in macrophages and dendritic cells, while its influence on phagocytosis seems to depend on the level of its expression and the time of exposure to microglia. Decreased TREM2 expression in a mouse model led to the deposition of Aβ in late-stage AD, a decline in plaque-associated microglia, and caused the downstream of tau phosphorylation [67]. The phenotype of microglia may be altered in the pathogenesis of AD, which also plays a role in the activity of these cells. The dietary antioxidants such as cyani-din-3-O-glucoside (C3G) can change the direction of microglia polarization from the pro-inflammatory M1 phenotype to the M2 phenotype by overexpression and activation of PPARγ and subsequent phagocytic removal of Aβ42 peptides associated with TREM2 overexpression [68]. The phagocytic activity of microglia toward tau was also dependent on some dietary components, such as α-linolenic acid (ALA), which intensified the phagocytosis process. Microglia also showed the potential for phagocytosis of the extracellular tau protein; however, the rate of this phenomenon depended on the ability of these cells to migrate and induce the repolarization of the microtubule-organizing center [69]. Microglial activity in a mouse animal model of AD was focused in the vicinity of Aβ plaques and was associated with increased expression of phagocytic regulating factors such as TREM2 and CTSD, which improved the Aβ clearance and thus reduced neurotoxicity to neural cells [70]. Ren and colleagues showed that in a mouse animal model, the deficit of microglial cells led to a decrease in Aβ phagocytosis, increased distribution of Aβ peptides, increased activity of glial cells, and intensified dystrophic changes in neurites, which was related to the acceleration of cognitive impairment [71]. Moreover, microglial cells in AD clean the parenchyma of brain tissue by phagocytic removal of fibrillary Aβ and elimination of soluble Aβ (sAβ) peptides by internalizing these molecules from the extracellular space through macropinocytosis [72].
Phosphatidylinositol-4,5-bisphosphate (PIP2) and AD endocytosis
Aβ deposits may impair synaptic transmission leading to cognitive deficits in AD by hydrolysis of PIP2 [73]. Familial AD (FAD)-associated presenilin mutations affected PIP2 metabolism. The cellular level of PIP2 had a positive effect on the functional potential of the transient receptor potential melastatin 7 (TRPM7)-associated Mg2+-inhibited cation (MIC) channels and is inversely correlated with the level of Aβ42 peptides [74]. Mutations in the PLCG2 gene were related to PIP2 depletion and decreased level of this marker, which in vivo showed a relationship with reduced phagocytosis and increased endocytosis expressed by acceleration of the clearance of Aβ1–42 oligomers [75]. Changes in PIP2 expression may also depend on the exogenous dietary factors. In an animal model, cerebral overexpression of phospholipase C (PLβC1) induced by high dietary cholesterol levels led to decreased PIP2 expression [76]. High cholesterol levels affect the excitability of neural cells through a potential affinity for the PIP2 transmembrane domain that stabilizes its interaction with G protein–activated inward rectifier potassium channel 2 (GIRK2) [77]. PIP2 through its effect on capillary endothelial Kir2.1 channels positively influenced the cerebral blood flow, which often remains impaired in AD brain [78].
Regulation of endosome secretion and AD pathogenesis
The mechanism of the formation of Aβ aggregates in AD was dependent on the endocytosis of the APP through YTSI, the motif that promotes early endosome formation, which positively regulated this process by binding to APP tail 1a (PAT1a) and subsequent overexpression of RME-6, which was an activator of Rab5 [79]. GTPase Rab5-positive APP-carrying endocytic vesicles have been described in a rat brain model. In vesicular organelles, the highest frequency of Rab5-positive structures was found in the small synaptic vesicles (SSVs), large bilamellar vesicles (LLVs), and the lowest in multivesicular bodies (MVBs) [80]. Changes in the expression level of proteins of the Rab GTPases family were noted in the AD brain tissues. Examination of post-mortem human brain tissue homogenates revealed elevated levels of Rab5 and Rab7 proteins in the basal forebrain, frontal cortex, and hippocampus regions in AD patients compared to normal brain tissues [81]. The number and size of the formed Rab5-positive endosomes depended on the exogenous expression of APP (V642I) and the APP-binding protein APP–BP1 and increased the level of Rab5 and early endosomal antigen 1 (EEA1) proteins, which also were elevated and carried in early endosomes [82]. Moreover, the Rab5 inhibition increased the release of Aβ40 and Aβ42 peptides in N2a cell cultures [83]. Other endosome-related proteins may also mediate the pathogenesis of AD. Overexpression of Ras and Rab interactor 3 (RIN3) led to the accumulation of APP carboxyl-terminal fragments (CTFs) and increased tau protein phosphorylation, which in the AD model led to enlarged early endosome secretion [84]. The increased level of β-CTF induced the recruitment of a protein adapter containing pleckstrin homology domain, phosphotyrosine-binding domain, and leucine zipper motif (APPL1) to Rab5 endosomes, which resulted in intensification of the endocytosis phenomenon, including impaired axonal transport and endosome edema [85]. The retromer complex plays a key role in regulating endosome sorting processes with respect to the endosome-to-Golgi retrieval pathway and the endosome-to-cell-surface recycling pathway, and increased Rab7a activity dependent on TBC1D5 protein inhibition promoted recruitment of retromer cargo selective complex (CSC) to endosomes [86].
Sorting nexins (SNXs) and AD-associated endocytosis
Sorting nexins (SNXs), which function as adaptor proteins, sort a variety of protein cargoes via the endolysosomal system to promote intracellular protein transport and signaling [87,88,89,90,91,92]. SNXs are made up of “membrane-bound” sorting dimers (SNX1, SNX2, SNX5, SNX6) and vacuolar protein sorting trimers with a membrane curvature detection domain (BAR domain) (Vps26, Vps29, Vps35) [21,22,23]. In a yeast-2 hybrid screen, SNX1 was found to interact with the epidermal growth factor receptor (EGFR), making it the first SNX found in mammals [93, 94]. SNX belongs to a protein family and features a conserved phosphoinositol-binding domain (PX domain) [95]. When the PX domain connects to PIP, SNX can bind to PIP-rich regions of the endocytosis network [96]. Abnormalities in SNX family proteins are associated with pathologies of the central nervous system. In particular, aberrant expression of SNX and mutations in autosomal recessive genes cause cerebellar ataxia, intellectual disability syndrome, AD, PD, and Down syndrome (DS) [91, 97,98,99]. Interestingly, in the DS mouse model, restoring hippocampal levels of SNX27 restores synaptic and cognitive deficits [100, 101]. SNX dysfunction has also been seen in epilepsy and schizophrenia [102]. Furthermore, changing SNX protein expression is linked to endocytosis, the basis of neural function and synaptic plasticity, and modulates complex behaviors such as learning and memory [103]. As a result, an improved understanding of SNX’s role in neuropsychiatric and neurodegenerative illnesses necessitates a reconsideration of how SNX maintains and inhibits normal brain function. SNX1 shares similarities with Vps5, a recognized yeast retromer complex component [104]. Retromer complexes are “hetero-pentameric” complexes that enable cargo transit and recovery from the endolysosomal system to the plasma membrane and trans-Golgi network (TGN), implying that SNX participates in cargo recovery from the degradation route [105,106,107,108,109].
SNXs may regulate distinct signaling pathways with divergent effects on the pathogenesis of AD. Markers modulating endocytosis may be related to APP shedding related to the pathogenesis of AD. SNX33 is one of the endocytic proteins that binds to dynamin through the SH3 domain. This resulted in the inhibition of endocytosis expressed by an increase in α-secretase cleavage and inhibition of both transferrin uptake and APP endocytosis [110]. SNX3 overexpression inhibited the APP uptake in the cell culture model, which led to an increase in the level of the precursor on the cell membrane and led to an increased cellular distribution of the full-length APP. SNX3 overexpression decreased the production of Aβ40, Aβ42, and sAPPβ [111]. Sorting nexin-4 (SNX4) overexpression led to increased levels of BACE1 and Aβ, which were presumed to be associated with an increase in BACE1 half-life time and with an enhanced Aβ release. The expression of SNX4 was impaired in the AD brain, emphasizing the dynamic distribution of the protein depending on the duration of the disease. In the early stages, the increased expression of this nexin was noted, and in the late stage of AD, its level significantly decreased [111]. SNX4 expression was common in nervous tissue, and its neural localization at early and recycling endosomes as well as in the synaptic regions was related to the regulation of neurotransmitter production and the release and transport of synaptic vesicles [112]. The biological importance of SNX4 complexes was related to the recognition and sorting of the charge on endosomal regions of cell membranes and was one of the pathways responsible for the proper functioning of the endosome [36]. Knockdown of sorting nexin 6 (SNX6) increased the levels of BACE1, sAPPβ, and CTFAβ in SK-N-SH and HEK293 cells [113]. Sorting nexin 12 (SNX12) was also expressed in nervous tissue, its localization at the ultrastructural level of the cell concerned mainly early endosomes, and overexpression of this nexin significantly decreased the levels of Aβ, soluble APPβ, and APPβ-carboxyl-terminal fragments. In the AD brain, a decreased level of SNX12 was noted, the interaction of which with BACE1 affected Aβ clearance [114].
Conclusions and future perspectives
The causes of AD remain unknown. The etiology is based on two main hypotheses: amyloid plaques and neurofibrillary tangles. Additionally, several risk factors such as increasing age, genetic factors, head injuries, vascular diseases, infections, and environmental factors play a role in the pathogenesis of the disease. Besides the well-established AD pathogenesis processes, there is an increasing role of endocytosis in AD pathophysiology. Endocytosis is a pathway involved in the production, trafficking, and clearance of Aβ. Endolysosomal abnormalities occur within neurons in AD, and they are linked to both Aβ and tau pathologies. Upregulation in the endocytosis process with other factors could predispose to develop AD by increasing the internalization of APP and thus its subsequent metabolism to generate Aβ deposits. Hyperphosphorylation of tau protein may accelerate endocytic dysregulation. Microglia cells are activated by Aβ and secrete neurotoxic molecules. In contrast, microglia use phagocytosis to reduce neurotoxicity and improve Aβ internalization and the clearance of Aβ.
Aging and genetic risk factors for Alzheimer’s also influence endocytosis. Experimental data suggest that during cell aging, the endocytosis of APP increases [115]. Revving up of APP processing leads to Aβ over-production. If APP endocytosis is increased with neuronal aging, it should be emphasized to develop an inhibitor that does not interfere with synaptic vesicle endocytosis. Potential drugs should prevent amyloid accumulation and synaptic decline with aging. Thus, regulating APP trafficking may be a therapeutic strategy to prevent AD [115]. Understanding endocytosis processes (neuron-specific functions of endocytosis and also autophagy) and their regulatory systems can help to recognize the pathophysiology of AD. Focusing on how exactly genetic alterations in specific endocytosis protein genes interfere with neuronal survival should also be investigated. Therefore, as we presented, the endocytosis may be a target for therapeutic interventions aimed at stopping or slowing the pathogenesis of AD.
References
World Health Organization. Global action plan on the public health response to dementia 2017 - 2025 http://apps.who.int/iris/bitstream/10665/259615/1/9789241513487-eng.pdf. Accessed 29 Aug 2023
Apostolova LG. Alzheimer disease. Continuum (Minneap Minn). 2016;22(2 Dementia):419–34.
Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, Cummings J, van der Flier WM. Alzheimer’s disease. Lancet. 2021;397:1577–90.
TahamiMonfared AA, Byrnes MJ, White LA, Zhang Q. Alzheimer’s disease: epidemiology and clinical progression. Neurol Ther. 2022;11:553–69.
Chen KL, Li PX, Sun YM, Chen SF, Zuo CT, Wang J, Dong Q, Cui M, Yu JT. Very early-onset Alzheimer’s disease in the third decade of life with de novo PSEN1 mutations. J Alzheimers Dis. 2022;85:65–71.
Monterey MD, Wei H, Wu X, Wu JQ. The many faces of astrocytes in Alzheimer’s disease. Front Neurol. 2021;6:9625.
Castellani RJ, Smith MA. Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is “too big to fail.” J Pathol. 2011;224(2):147–52.
Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405.
Brunt L, Scholpp S. The function of endocytosis in Wnt signaling. Cell Mol Life Sci. 2018;75(5):785–95.
Tey HJ, Ng CH. Computational analysis of functional SNPs in Alzheimer’s disease-associated endocytosis genes. PeerJ. 2019;7:e7667.
Schmid SL. Reciprocal regulation of signaling and endocytosis: implications for the evolving cancer cell. J Cell Biol. 2017;216(9):2623–32.
Shafaq-Zadah M, Dransart E, Johannes L. Clathrin-independent endocytosis, retrograde trafficking, and cell polarity. Curr Opin Cell Biol. 2020;65:112–21.
Peterman E, Prekeris R. The postmitotic midbody: regulating polarity, stemness, and proliferation. J Cell Biol. 2019;218(12):3903–11.
Briant K, Redlingshöfer L, Brodsky FM. Clathrin’s life beyond 40: connecting biochemistry with physiology and disease. Curr Opin Cell Biol. 2020;65:141–9.
Tu Y, Seaman MNJ. Navigating the controversies of retromer-mediated endosomal protein sorting. Front Cell Dev Biol. 2021;9:658741.
Dell’Angelica EC, Bonifacino JS. Coatopathies: genetic disorders of protein coats. Annu Rev Cell Dev Biol. 2019;35:131–68.
Wu LG, Hamid E, Shin W, Chiang HC. Exocytosis and endocytosis: modes, functions, and coupling mechanisms. Annu Rev Physiol. 2014;76:301–31.
Morgan JR, Comstra HS, Cohen M, Faundez V. Presynaptic membrane retrieval and endosome biology: defining molecularly heterogeneous synaptic vesicles. Cold Spring Harb Perspect Biol. 2013;5(10):a016915.
Saheki Y, De Camilli P. Synaptic vesicle endocytosis. Cold Spring Harb Perspect Biol. 2012;4(9):a005645.
El-Sayed A, Harashima H. Endocytosis of gene delivery vectors: from clathrin-dependent to lipid raft-mediated endocytosis. Mol Ther. 2013;21(6):1118–30.
Uribe-Querol E, Rosales C. Phagocytosis: our current understanding of a universal biological process. Front Immunol. 2020;11:1066.
Rosales C, Uribe-Querol E. Phagocytosis: a fundamental process in immunity. Biomed Res Int. 2017;2017:9042851.
Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35.
Nguyen JA, Yates RM. Better together: current insights into phagosome-lysosome fusion. Front Immunol. 2021;25(12):636078.
Li YX, Pang HB. Macropinocytosis as a cell entry route for peptide-functionalized and bystander nanoparticles. J Control Release. 2021;10(329):1222–30.
Lin XP, Mintern JD, Gleeson PA. Macropinocytosis in different cell types: similarities and differences. Membranes (Basel). 2020;10(8):177.
Li G, Marlin MC. Rab family of GTPases. Methods Mol Biol. 2015;1298:1–15.
Tu H, Wang Z, Yuan Y, Miao X, Li D, Guo H, et al. The PripA-TbcrA complex-centered Rab GAP cascade facilitates macropinosome maturation in Dictyostelium. Nat Commun. 2022;13(1):1787.
Morishita S, Wada N, Fukuda M, Nakamura T. Rab5 activation on macropinosomes requires ALS2, and subsequent Rab5 inactivation through ALS2 detachment requires active Rab7. FEBS Lett. 2019;593(2):230–41.
Condon ND, Heddleston JM, Chew TL, Luo L, McPherson PS, Ioannou MS, et al. Macropinosome formation by tent pole ruffling in macrophages. J Cell Biol. 2018;217(11):3873–85.
van Helmond ZK, Miners JS, Bednall E, Chalmers KA, Zhang Y, Wilcock GK, et al. Caveolin-1 and -2 and their relationship to cerebral amyloid angiopathy in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2007;33(3):317–27.
Kang MJ, Chung YH, Hwang CI, Murata M, Fujimoto T, Mook-Jung IH, et al. Caveolin-1 upregulation in senescent neurons alters amyloid precursor protein processing. Exp Mol Med. 2006;38(2):126–33.
Ramírez CM, González M, Díaz M, Alonso R, Ferrer I, Santpere G, et al. VDAC and ERalpha interaction in caveolae from human cortex is altered in Alzheimer’s disease. Mol Cell Neurosci. 2009;42(3):172–83.
Park TE, Singh B, Li H, Lee JY, Kang SK, Choi YJ, et al. Enhanced BBB permeability of osmotically active poly(mannitol-co-PEI) modified with rabies virus glycoprotein via selective stimulation of caveolar endocytosis for RNAi therapeutics in Alzheimer’s disease. Biomaterials. 2015;38:61–71.
Wu J, Zhou SL, Pi LH, Shi XJ, Ma LR, Chen Z, et al. High glucose induces formation of tau hyperphosphorylation via Cav-1-mTOR pathway: a potential molecular mechanism for diabetes-induced cognitive dysfunction. Oncotarget. 2017;8(25):40843–56.
Thomas RS, Lelos MJ, Good MA, Kidd EJ. Clathrin-mediated endocytic proteins are upregulated in the cortex of the Tg2576 mouse model of Alzheimer’s disease-like amyloid pathology. Biochem Biophys Res Commun. 2011;415(4):656–61.
Thomas RS, Henson A, Gerrish A, Jones L, Williams J, Kidd EJ. Decreasing the expression of PICALM reduces endocytosis and the activity of β-secretase: implications for Alzheimer’s disease. BMC Neurosci. 2016;17(1):50.
Ando K, Brion JP, Stygelbout V, Suain V, Authelet M, Dedecker R, et al. Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer’s brains. Acta Neuropathol. 2013;125(6):861–78.
Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat Neurosci. 2015;18(7):978–87.
Baig S, Joseph SA, Tayler H, Abraham R, Owen MJ, Williams J, Kehoe PG, Love S. Distribution and expression of picalm in Alzheimer disease. J Neuropathol Exp Neurol. 2010;69(10):1071–7.
Poulsen ET, Larsen A, Zollo A, Jørgensen AL, Sanggaard KW, Enghild JJ, et al. New insights to clathrin and adaptor protein 2 for the design and development of therapeutic strategies. Int J Mol Sci. 2015;16(12):29446–53.
Maesako M, Uemura M, Tashiro Y, Sasaki K, Watanabe K, Noda Y, et al. High fat diet enhances β-site cleavage of amyloid precursor protein (APP) via promoting β-site APP cleaving enzyme 1/adaptor protein 2/clathrin complex formation. PLoS One. 2015;10(9):e0131199.
Storck SE, Hartz AMS, Bernard J, Wolf A, Kachlmeier A, Mahringer A, et al. The concerted amyloid-beta clearance of LRP1 and ABCB1/P-gp across the blood-brain barrier is linked by PICALM. Brain Behav Immun. 2018;73:21–33.
Kuboyama T, Lee YA, Nishiko H, Tohda C. Inhibition of clathrin-mediated endocytosis prevents amyloid β-induced axonal damage. Neurobiol Aging. 2015;36(5):1808–19.
Angelopoulou E, Paudel YN, Shaikh MF, Piperi C. Flotillin: a promising biomarker for Alzheimer’s disease. J Pers Med. 2020;10(2):E20.
Tiwari S, Atluri V, Kaushik A, Yndart A, Nair M. Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomedicine. 2019;14:5541–54.
Kimura N, Yanagisawa K. Traffic jam hypothesis: relationship between endocytic dysfunction and Alzheimer’s disease. Neurochem Int. 2018;119:35–41.
Zhang QY, Tan MS, Yu JT, Tan L. The role of retromer in Alzheimer’s disease. Mol Neurobiol. 2016;53(6):4201–9.
Kwart D, Gregg A, Scheckel C, Murphy EA, Paquet D, Duffield M, et al. A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, not Aβ. Neuron. 2019;104(2):256-270.e5.
Tan JZA, Gleeson PA. The trans-Golgi network is a major site for α-secretase processing of amyloid precursor protein in primary neurons. J Biol Chem. 2019;294(5):1618–31.
Goldstein LSB, Das U. The cellular machinery of post-endocytic APP trafficking in Alzheimer’s disease: a future target for therapeutic intervention? Prog Mol Biol Transl Sci. 2021;177:109–22.
Chae CW, Lee HJ, Choi GE, Jung YH, Kim JS, Lim JR, et al. High glucose-mediated PICALM and mTORC1 modulate processing of amyloid precursor protein via endosomal abnormalities. Br J Pharmacol. 2020;177(16):3828–47.
Perdigão C, Barata MA, Araújo MN, Mirfakhar FS, Castanheira J, Guimas AC. Intracellular trafficking mechanisms of synaptic dysfunction in Alzheimer’s disease. Front Cell Neurosci. 2020;14:72.
Merthan L, Haller A, Thal DR, von Einem B, von Arnim CAF. The role of PTB domain containing adaptor proteins on PICALM-mediated APP endocytosis and localization. Biochem J. 2019;476(14):2093–109.
Xu W, Tan L, Yu JT. The role of PICALM in Alzheimer’s disease. Mol Neurobiol. 2015;52(1):399–413.
Tian Y, Chang JC, Fan EY, Flajolet M, Greengard P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci U S A. 2013;110(42):17071–6.
Burrinha T, Martinsson I, Gomes R, Terrasso AP, Gouras GK, Almeida CG. Upregulation of APP endocytosis by neuronal aging drives amyloid-dependent synapse loss. J Cell Sci. 2021;134(9):jcs255752.
Kanatsu K, Hori Y, Ebinuma I, Chiu YW, Tomita T. Retrograde transport of γ-secretase from endosomes to the trans-Golgi network regulates Aβ42 production. J Neurochem. 2018;147(1):110–23.
Kanatsu K, Morohashi Y, Suzuki M, Kuroda H, Watanabe T, Tomita T, et al. Decreased CALM expression reduces Aβ42 to total Aβ ratio through clathrin-mediated endocytosis of γ-secretase. Nat Commun. 2014;5:3386.
Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, et al. LC3-associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell. 2019;178(3):536-551.e14.
Zhang J, Yin Y, Ji Z, Cai Z, Zhao B, Li J, et al. Endophilin2 interacts with GluA1 to mediate AMPA receptor endocytosis induced by oligomeric amyloid-β. Neural Plast. 2017;2017:8197085.
Azarnia Tehran D, Kochlamazashvili G, Pampaloni NP, Sposini S, Shergill JK, Lehmann M, et al. Selective endocytosis of Ca2+-permeable AMPARs by the Alzheimer’s disease risk factor CALM bidirectionally controls synaptic plasticity. Sci Adv. 2022;8(21):eabl5032.
Carraway KL, Carraway CAC. Signaling components and pathways. In: Carraway KL, Carraway CAC, Carraway KL (red). Signaling and the Cytoskeleton. Berlin, Heidelberg: Springer; 1998 s. 41–95. Available at: https://doi.org/10.1007/978-3-662-12993-7_2.
Sanchez-Mico MV, Jimenez S, Gomez-Arboledas A, Muñoz-Castro C, Romero-Molina C, Navarro V, et al. Amyloid-β impairs the phagocytosis of dystrophic synapses by astrocytes in Alzheimer’s disease. Glia. 2021;69(4):997–1011.
Grubman A, Choo XY, Chew G, Ouyang JF, Sun G, Croft NP, et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat Commun. 2021;12(1):3015.
Joshi P, Riffel F, Satoh K, Enomoto M, Qamar S, Scheiblich H, et al. Differential interaction with TREM2 modulates microglial uptake of modified Aβ species. Glia. 2021;69(12):2917–32.
Schoch KM, Ezerskiy LA, Morhaus MM, Bannon RN, Sauerbeck AD, Shabsovich M, et al. Acute Trem2 reduction triggers increased microglial phagocytosis, slowing amyloid deposition in mice. Proc Natl Acad Sci U S A. 2021;118(27):e2100356118.
Sanjay, Shin JH, Park M, Lee HJ. Cyanidin-3-O-glucoside regulates the M1/M2 polarization of microglia via PPARγ and Aβ42 phagocytosis through TREM2 in an Alzheimer’s disease model. Mol Neurobiol. 2022;59(8):5135–48.
Desale SE, Chinnathambi S. α- Linolenic acid modulates phagocytosis and endosomal pathways of extracellular Tau in microglia. Cell Adh Migr. 2021;15(1):84–100.
Ruiz-Pérez G, de Martín Esteban SR, Marqués S, Aparicio N, Grande MT, Benito-Cuesta I, et al. Potentiation of amyloid beta phagocytosis and amelioration of synaptic dysfunction upon FAAH deletion in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2021;18(1):223.
Ren X, Yao L, Wang Y, Mei L, Xiong WC. Microglial VPS35 deficiency impairs Aβ phagocytosis and Aβ-induced disease-associated microglia, and enhances Aβ associated pathology. J Neuroinflammation. 2022;19(1):61.
Mandrekar S, Jiang Q, Lee CYD, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J Neurosci. 2009;29(13):4252–62.
He Y, Wei M, Wu Y, Qin H, Li W, Ma X, et al. Amyloid β oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4,5-bisphosphate. Nat Commun. 2019;10(1):1193.
Landman N, Jeong SY, Shin SY, Voronov SV, Serban G, Kang MS, et al. Presenilin mutations linked to familial Alzheimer’s disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc Natl Acad Sci U S A. 2006;103(51):19524–9.
Maguire E, Menzies GE, Phillips T, Sasner M, Williams HM, Czubala MA, et al. PIP2 depletion and altered endocytosis caused by expression of Alzheimer’s disease-protective variant PLCγ2 R522. EMBO J. 2021;40(17):e105603.
Chun YS, Chung S. High-cholesterol diet decreases the level of phosphatidylinositol 4,5-bisphosphate by enhancing the expression of phospholipase C (PLCβ1) in rat brain. Int J Mol Sci. 2020;21(3):1161.
Mathiharan YK, Glaaser IW, Zhao Y, Robertson MJ, Skiniotis G, Slesinger PA. Structural insights into GIRK2 channel modulation by cholesterol and PIP2. Cell Rep. 2021;36(8):109619.
Mughal A, Harraz OF, Gonzales AL, Hill-Eubanks D, Nelson MT. PIP2 improves cerebral blood flow in a mouse model of Alzheimer’s disease. Function (Oxf). 2021;2(2):zqab010.
Eggert S, Gruebl T, Rajender R, Rupp C, Sander B, Heesch A, et al. The Rab5 activator RME-6 is required for amyloid precursor protein endocytosis depending on the YTSI motif. Cell Mol Life Sci. 2020;77(24):5223–42.
Ikin AF, Annaert WG, Takei K, De Camilli P, Jahn R, Greengard P, et al. Alzheimer amyloid protein precursor is localized in nerve terminal preparations to Rab5-containing vesicular organelles distinct from those implicated in the synaptic vesicle pathway. J Biol Chem. 1996;271(50):31783–6.
Ginsberg SD, Mufson EJ, Counts SE, Wuu J, Alldred MJ, Nixon RA, et al. Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2010;22(2):631–9.
Laifenfeld D, Patzek LJ, McPhie DL, Chen Y, Levites Y, Cataldo AM, et al. Rab5 mediates an amyloid precursor protein signaling pathway that leads to apoptosis. J Neurosci. 2007;27(27):7141–53.
Caudano F, Montalto G, Passalacqua M, Pronzato MA, Fedele E, Ricciarelli R. cGMP favors the interaction between APP and BACE1 by inhibiting Rab5 GTPase activity. Sci Rep. 2020;10(1):1358.
Shen R, Zhao X, He L, Ding Y, Xu W, Lin S, et al. Upregulation of RIN3 induces endosomal dysfunction in Alzheimer’s disease. Transl Neurodegener. 2020;9(1):26.
Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini A, Rigoglioso A, et al. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol Psychiatry. 2016;21(5):707–16.
Seaman MNJ, Mukadam AS, Breusegem SY. Inhibition of TBC1D5 activates Rab7a and can enhance the function of the retromer cargo-selective complex. J Cell Sci. 2018;131(12):jcs217398.
Granger E, McNee G, Allan V, Woodman P. The role of the cytoskeleton and molecular motors in endosomal dynamics. Semin Cell Dev Biol. 2014;31:20–9.
Yong X, Mao L, Seaman MNJ, Jia D. An evolving understanding of sorting signals for endosomal retrieval. iScience. 2022;25(5):104254.
Amatya B, Lee H, Asico LD, Konkalmatt P, Armando I, Felder RA, et al. SNX-PXA-RGS-PXC subfamily of SNXs in the regulation of receptor-mediated signaling and membrane trafficking. Int J Mol Sci. 2021;22(5):2319.
Yang L, Tan W, Yang X, You Y, Wang J, Wen G et al. Sorting nexins: a novel promising therapy target for cancerous/neoplastic diseases. J Cell Physiol. 2020;9. https://doi.org/10.1002/jcp.30093.
Reitz C. Retromer dysfunction and neurodegenerative disease. Curr Genomics. 2018;19(4):279–88.
Gallon M, Cullen PJ. Retromer and sorting nexins in endosomal sorting. Biochem Soc Trans. 2015;43(1):33–47.
Nishimura Y, Takiguchi S, Ito S, Itoh K. EGF-stimulated AKT activation is mediated by EGFR recycling via an early endocytic pathway in a gefitinib-resistant human lung cancer cell line. Int J Oncol. 2015;46(4):1721–9.
Zhang Y, Pang X, Li J, Xu J, Hsu VW, Sun F. Structural insights into membrane remodeling by SNX1. Proc Natl Acad Sci U S A. 2021;118(10):e2022614118.
Chandra M, Collins BM. The phox homology (PX) domain. Adv Exp Med Biol. 2019;1111:1–17.
Vieira N, Rito T, Correia-Neves M, Sousa N. Sorting out sorting nexins functions in the nervous system in health and disease. Mol Neurobiol. 2021;58(8):4070–106.
Sanderson LE, Lanko K, Alsagob M, Almass R, Al-Ahmadi N, Najafi M, et al. Bi-allelic variants in HOPS complex subunit VPS41 cause cerebellar ataxia and abnormal membrane trafficking. Brain. 2021;144(3):769–80.
Tao X, Che Y, Li C, Ruan W, Xu J, Yu Y, et al. Novel SNX13 frameshift variant in an individual with developmental delay. Cytogenet Genome Res. 2021;161(10–11):514–9.
Kim NY, Cho MH, Won SH, Kang HJ, Yoon SY, Kim DH. Sorting nexin-4 regulates β-amyloid production by modulating β-site-activating cleavage enzyme-1. Alzheimers Res Ther. 2017;9(1):4.
Wang X, Zhou Y, Wang J, Tseng IC, Huang T, Zhao Y, et al. SNX27 deletion causes hydrocephalus by impairing ependymal cell differentiation and ciliogenesis. J Neurosci. 2016;36(50):12586–97.
Wang X, Huang T, Zhao Y, Zheng Q, Thompson RC, Bu G, et al. Sorting nexin 27 regulates Aβ production through modulating γ-secretase activity. Cell Rep. 2014;9(3):1023–33.
Vieira N, Rito T, Correia-Neves M, et al. Sorting out sorting nexins functions in the nervous system in health and disease. Mol Neurobiol. 2021;58:4070–106.
Pasek JG, Wang X, Colbran RJ. Differential CaMKII regulation by voltage-gated calcium channels in the striatum. Mol Cell Neurosci. 2015;68:234–43.
Trousdale C, Kim K. Retromer: structure, function, and roles in mammalian disease. Eur J Cell Biol. 2015;94(11):513–21.
Simonetti B, Guo Q, Giménez-Andrés M, Chen KE, Moody ERR, Evans AJ, et al. SNX27-retromer directly binds ESCPE-1 to transfer cargo proteins during endosomal recycling. PLoS Biol. 2022;20(4):e3001601.
Hariri H, Henne WM. Filling in the gaps: SNX-RGS proteins as multiorganelle tethers. J Cell Biol. 2022;221(5):e202203061.
Suzuki SW, Oishi A, Nikulin N, Jorgensen JR, Baile MG, Emr SD. A PX-BAR protein Mvp1/SNX8 and a dynamin-like GTPase Vps1 drive endosomal recycling. Elife. 2021;10:e69883.
Yong X, Zhao L, Hu W, Sun Q, Ham H, Liu Z, et al. SNX27-FERM-SNX1 complex structure rationalizes divergent trafficking pathways by SNX17 and SNX27. Proc Natl Acad Sci U S A. 2021;118(36):e2105510118.
Yong X, Zhao L, Deng W, Sun H, Zhou X, Mao L, et al. Mechanism of cargo recognition by retromer-linked SNX-BAR proteins. PLoS Biol. 2020;18(3):e3000631.
Schöbel S, Neumann S, Hertweck M, Dislich B, Kuhn PH, Kremmer E, et al. A novel sorting nexin modulates endocytic trafficking and alpha-secretase cleavage of the amyloid precursor protein. J Biol Chem. 2008;283(21):14257–68.
Xu S, Nigam SM, Brodin L. Overexpression of SNX3 decreases amyloid-β peptide production by reducing internalization of amyloid precursor protein. Neurodegener Dis. 2018;18(1):26–37.
Vazquez-Sanchez S, Gonzalez-Lozano MA, Walfenzao A, Li KW, van Weering JRT. The endosomal protein sorting nexin 4 is a synaptic protein. Sci Rep. 2020;10(1):18239.
Li Q, Li X, Wang L, Zhang Y, Chen L. miR-98-5p acts as a target for Alzheimer’s disease by regulating Aβ production Through modulating SNX6 expression. J Mol Neurosci. 2016;60(4):413–20.
Zhao Y, Wang Y, Yang J, Wang X, Zhao Y, Zhang X, et al. Sorting nexin 12 interacts with BACE1 and regulates BACE1-mediated APP processing. Mol Neurodegener. 2012;7:30.
Burrinha T, Almeida CG. Aging impact on amyloid precursor protein neuronal trafficking. Curr Opinion Neurobiol. 2022;73:102524.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zadka, Ł., Sochocka, M., Hachiya, N. et al. Endocytosis and Alzheimer’s disease. GeroScience 46, 71–85 (2024). https://doi.org/10.1007/s11357-023-00923-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-023-00923-1