
Abstract
Alzheimer’s disease (AD) is traditionally defined by the presence of two types of protein aggregates in the brain: amyloid plaques comprised of the protein amyloid-β (Aβ) and neurofibrillary tangles containing the protein tau. However, a large proportion (up to 57%) of AD patients also have TDP-43 aggregates present as an additional comorbid pathology. The presence of TDP-43 aggregates in AD correlates with hippocampal sclerosis, worse brain atrophy, more severe cognitive impairment, and more rapid cognitive decline. In patients with mixed Aβ, tau, and TDP-43 pathology, TDP-43 may interact with neurodegenerative processes in AD, worsening outcomes. While considerable progress has been made to characterize TDP-43 pathology in AD and late-onset dementia, there remains a critical need for mechanistic studies to understand underlying disease biology and develop therapeutic interventions. This perspectives article reviews the current understanding of these processes from autopsy cohort studies and model organism-based research, and proposes targeting neurotoxic synergies between tau and TDP-43 as a new therapeutic strategy for AD with comorbid TDP-43 pathology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Perspectives
Pathophysiological spectrum in Alzheimer’s disease
Alzheimer’s disease (AD), a devastating, fatal neurodegenerative disease of aging, features the presence of extracellular amyloid-β (Aβ) plaques and intracellular tangles of hyperphosphorylated tau in the brain [1]. For decades, prevention or clearance of Aβ plaques have been the major therapeutic goals in the pursuit of a treatment for AD. This was a rational approach given that mutations in genes involved in amyloid processes cause some inherited (familial) cases of AD [2]. Unfortunately, most clinical trials have failed thus far to demonstrate a significant benefit to patients, raising the need to pursue additional strategies [3]. Familial mutations driving amyloidogenesis may represent a distinct form of AD in which amyloid pathology is both necessary and sufficient to trigger a cascade of events including pathological tau deposition and neurodegeneration. However, in aging brains that lack these genetic triggers for amyloid pathology, the Aβ plaques that accumulate may still be necessary but perhaps not sufficient to drive neurodegeneration. Accumulation of tau tangles strongly correlates with cognitive impairment and neurodegeneration in AD, and this recognition has stimulated a shift toward the development of tau-targeting therapies [4]. Although tau-targeting therapies hold great promise, to date only a few have made it to clinical trials [5].
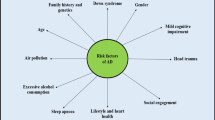
While historically AD has been thought of as a single disease entity driven by dysfunction or accumulation of Aβ and tau, it may more accurately resemble a spectrum of diseases with variable but overlapping clinical and neuropathological features [6]. Amyloid plaques and tau tangles remain the defining pathologic constants of AD, but their respective contributions to neurodegeneration and clinical presentation may differ depending on other genetic, environmental, and pathological factors in play (Fig. 1). Rare mutations in amyloid processing genes cause AD, implicating Aβ in the disease pathophysiology. However, these mutations usually result in much earlier onset of disease distinct from the more common sporadic late-onset AD (LOAD). In LOAD, genome-wide association studies (GWAS) and genome sequencing studies have identified numerous alleles that modify AD risk [7], the strongest modifier being apolipoprotein E [8]. A variety of lifestyle and environmental factors are also believed to influence the risk of LOAD, including level of education, diet, and physical activity [9,10,11,12]. Furthermore, comorbid pathologies, such as vascular brain injury, Lewy body disease, or TDP-43-positive inclusions, are a common observation in the brains of older individuals.

Pathways of neurodegeneration and cognitive impairment in AD. The human brain is subject to a variety of factors, both genetic and environmental, that either incite (orange) or mitigate (blue) the risk of developing the neuropathologic hallmarks of Alzheimer’s disease (amyloid plaques and neurofibrillary tangles). It is currently unknown whether these factors also influence the development of TDP-43 proteinopathy. While initiation of amyloid, tau, or TDP-43 pathology may occur through independent or overlapping events, the presence of multiple pathologies likely drives synergistic neurotoxicity that leads to cognitive decline and neurodegeneration. Observational data from human autopsy studies, as well as work from model systems, suggest that TDP-43 pathology enhances tau deposition and exacerbates the clinical severity of AD
One outstanding question is whether these pathologies have a contributory effect on AD neuropathology or the clinical expression of the disease [13,14,15]. In particular, TDP-43 has become increasingly recognized as a frequent co-pathology in the brains of individuals with the clinical and neuropathologic features of AD.
TDP-43 clinicopathological correlates in Alzheimer’s disease
TDP-43, an essential protein, has a variety of critical cellular functions that include regulating pre-mRNA splicing, mRNA transport, and forming and stabilizing stress granules [16]. TDP-43 was first linked to neurodegenerative disease when it was identified as the major pathologic protein comprising ubiquitin-positive inclusions in the majority of amyotrophic lateral sclerosis (ALS) and approximately half of frontotemporal lobar degeneration (FTLD) cases [17, 18]. Mutations in the gene coding for TDP-43, TARDBP, cause ~ 4% of familial inherited ALS, directly linking TDP-43 dysfunction with disease [19, 20]. Pathological TDP-43 has since been described in association with numerous other neurodegenerative diseases, most notably Alzheimer’s disease, where it is present in up to 57% of cases in some autopsy cohorts [21,22,23]. In these cohorts, TDP-43 pathology is found in the limbic structures of older individuals with AD neuropathologic change (ADNC) [24,25,26,27]. TDP-43 often associates with hippocampal sclerosis [28, 29], and while it can occur in the absence of AD pathology, particularly in the oldest old (90 +), this is much less common and of uncertain clinical significance [30, 31]. Individuals with both ADNC and TDP-43 pathology typically have more severe cognitive impairment, faster rates of decline, and more rapid hippocampal atrophy over time [24, 32]. At the opposite end of the spectrum, a rarer number of so-called “resilient” individuals develop some degree of amyloid and tau pathology but are able to maintain their cognitive function [33,34,35]. Recent work suggests that one such factor determining resilience versus susceptibility to AD may be the presence of pathological TDP-43 [24, 33, 36, 37].
In a small but well-characterized group of resilient individuals with ADNC from the Adult Changes in Thought (ACT) autopsy cohort, the absence of TDP-43 distinguished those without cognitive impairment (resilient) from those with cognitive decline (AD dementia). When compared to individuals with dementia and matched levels of ADNC, resilient individuals did not have any differences in Lewy bodies or vascular brain injury (including arteriolosclerosis, atherosclerosis, macro- and micro-infarcts, and cerebral amyloid angiopathy). The key difference between the resilient and AD dementia cohorts was the presence of TDP-43 pathology, indicating aberrant or aggregated TDP-43 may be a significant contributing factor to developing dementia [33]. Other community-based cohorts have demonstrated similar associations between the presence of TDP-43 and the likelihood of dementia. In fact, staging criteria for TDP-43 pathology in AD has been developed, with higher stages correlating with worse cognitive impairment [24, 38, 39]. These associations are intriguing but raise new questions. It is currently unknown why some individuals with AD develop TDP-43 pathology while others do not, or why clinical manifestations of AD are more severe in individuals with TDP-43 pathology. Autopsy studies suggest the presence of TDP-43 pathology may lower the threshold for developing the clinical symptoms of AD [26]. However, additional research is needed to understand the mechanisms underlying TDP-43 deposition and AD phenotypes.
Evidence for synergism among TDP-43, Aβ, and tau
In addition to studying the correlations between TDP-43 and cognitive decline, cohort studies have also evaluated the relationship between TDP-43 and the neuropathologic hallmarks of AD, amyloid and tau pathology. Data from the Religious Orders Study and the Memory and Aging Project (ROS/MAP) inform models that implicate amyloid pathology, particularly neuritic plaques, in TDP-43 pathology [40]. The relationship between Aβ and TDP-43 is not well understood. Given the observation that TDP-43 co-pathology is much more common in the presence of ADNC than in pure tauopathies such as FTLD-tau, progressive supranuclear palsy, or corticobasal syndrome, there may be an interaction between amyloid and TDP-43. There is some limited evidence supporting this hypothesis. In vitro, TDP-43 does not regulate expression or processing of amyloid precursor protein but accelerates Aβ aggregation, and in mouse models, it modulates Aβ fibrillization and worsens cognitive outcomes [41,42,43]. However, in an autopsy cohort of AD patients who underwent antemortem amyloid sensitive PET imaging, there were no correlates between TDP-43 pathology and global amyloid PET signal [44]. It is possible there is a reciprocal effect where Aβ also influences the initiation or development of TDP-43 pathology, although this remains to be determined.
In other AD cohorts, there is evidence for a relationship between TDP-43 and tau pathology, which appear to develop in a similar pattern throughout the brain, beginning in limbic structures and later advancing to neocortex [21]. Josephs et al. at the Mayo Clinic describe higher Braak stage of tau pathology in subjects with comorbid TDP-43 pathology [23], and in the ACT study it was shown that for individuals matched by Braak stage, those with TDP-43 co-pathology had a higher density of tau pathology in frontal cortex [33]. Although different assessments of tau pathology were used in these two studies, this complementary data from two different cohorts suggest that the presence of TDP-43 pathology is associated with increased tau burden. Additional studies have found that a subset of tau and TDP-43 positive inclusions are co-present or co-localized in the same neurons in AD brains [45, 46]. In addition, AD brain–derived phosphorylated tau and TDP-43 can co-immunoprecipitate indicating a strong physical interaction [45, 47, 48]. Given the previously described associations between tau pathology and cognitive dysfunction, a pathological relationship between tau and TDP-43 may underlie the increase in severity of cognitive symptoms when comorbid TDP-43 is identified in AD. Despite these associations, however, human cohort studies cannot be used to determine what the mechanisms of this relationship are, including whether TDP-43 enhances tau pathology, or vice versa. In addition, it remains to be determined whether development of TDP-43 pathology represents an early event in AD pathogenesis or occurs later as disease progresses. For this, it is necessary to develop and study models that can recapitulate both tau and TDP-43 proteinopathies simultaneously.
The models used to study Alzheimer’s disease focus on amyloid and tau pathology, either alone or in combination [49]. TDP-43 proteinopathy has predominantly been studied as a mono-proteinopathy modeling ALS and FTLD [50]. Only recently has TDP-43 proteinopathy been considered an important contributing variable in AD, and to date there are limited studies exploring the relationship between tau and TDP-43 in vitro or in vivo. Data generated by our lab using a C. elegans co-expression model of tau and TDP-43 found that tau and TDP-43 synergize leading to increased pathological protein accumulation, neuronal dysfunction, and neurodegeneration [33]. In cell and mouse models, TDP-43 regulates mRNA splicing of tau exon 10, shifting the ratio of tau microtubule-binding repeats from the normal balanced ratio of 3R/4R-tau to a higher proportion of 4R-tau [51]. Recent work found that tau oligomers promote accumulation of cytoplasmic TDP-43 in HEK293 cells, and brain-derived TDP-43 oligomers can cross-seed tau aggregates in vitro [52]. Finally, in transgenic rats expressing human familial ALS-mutant TDP-43(M337V), hippocampal injections of AAV9 virus carrying phosphorylation-mimic human tau(T175D) led to significantly increased burden of tau pathology than control animals with either TDP-43(M337V) or tau(T175D) alone [53]. Although additional studies are necessary, the current data supports a biological relationship between tau and TDP-43 that drives neurodegeneration in sporadic LOAD. Indeed, if a biomarker of TDP-43 pathology could be developed, this may prove to be a crucial addition to the A,T(N) research framework that currently exists for characterizing risk of dementia [54].
Potential mechanisms of tau and TDP-43 synergy
While the observations from both human cohorts and limited studies using model systems suggest an interaction between tau and TDP-43, the mechanisms that underlie that interaction are unknown. However, there is a wealth of information known about the physiological roles of both tau and TDP-43, as well as their respective roles in other neurodegenerative disease, that serve as a guide and potential place to start the investigation. As a component of tubulin assembly, tau binds and stabilizes microtubules. Tau localizes throughout the cytoplasm, and is enriched in axons, where it assists with cargo trafficking along the microtubule network [55]. Phosphorylation of tau decreases its microtubule-binding affinity, resulting in destabilization of microtubules. Although predominantly found in the cytoplasm, tau is also present in the nucleus where it can stabilize heterochromatin [56]. Recently, tau aggregates have been found to include components of nuclear speckles, which are protein and RNA-containing structures within the nucleus [57]. TDP-43 has demonstrated roles in DNA binding, transcription regulation, pre-mRNA splicing of the majority of transcripts, alternative splice site selection, mRNA stability and transport, and microRNA biogenesis [58]. TDP-43 is typically nuclear, but it can be transported to the cytoplasm. There are numerous possibilities for biological or disease-state interactions between tau and TDP-43 throughout the cell (Fig. 2). In the nucleus, this could extend to DNA or RNA associating functions of tau and TDP-43 including mRNA splicing regulation, nuclear speckle activities, or nuclear-cytoplasmic transport [57, 59,60,61,62,63,64]. In the cytoplasm, TDP-43 may impact tau-associated microtubule stability and transport by binding dynactin [65]. Furthermore, cellular reactions to pathological tau or TDP-43 alone, including formation of stress granules, post-translational modification based signaling such as kinase activation, or changes in proteostasis pathway dynamics, may promote dysfunction and synergistic neurotoxicity from the other protein.

Potential mechanisms for tau and TDP-43 synergy in AD. There are numerous pathways that potentially underlie tau and TDP-43 synergistic neurotoxicity. These include interactions within the nucleus during regulated transport between the nucleus and cytoplasm, or associations with DNA, RNA, or nuclear speckles. In the cytoplasm, alterations in microtubule dynamics, stability, or transport along microtubules, derangement of cellular signaling including post-translational modifications such as phosphorylation, ubiqitination, acetylation, or SUMOylation, or formation of stress granules could lead to phenotypic enhancement of tau and TDP-43. Finally, misfolded tau or TDP-43 protein can impair normal cellular proteostasis mechanisms, which may have a broader impact on other aggregation prone proteins
Conclusions and future studies
A majority of patients with sporadic late-onset AD exhibit comorbid TDP-43 pathology, which strongly correlates with cognitive outcomes. The presence of pathologic TDP-43 may enhance tau pathology and concomitant neurotoxicity, either independent of or in concert with amyloid pathology. If true, this may help explain the limited efficacy of amyloid focused therapeutics and argue for the need to develop a multitargeted approach to preventing tau progression and subsequent neurodegeneration. However, while there are clearly intriguing associations observed between tau and TDP-43 in human autopsy and model organism studies, additional work is needed to broaden understanding of these processes. Some suggested areas for future study are described below.
Human cohort studies
Additional community-based cohort studies that include cognitive assessments in life paired with complete neuropathological examination after death are need to supplement the published literature, which currently relies on relatively few cohorts. There are several detailed examinations of TDP-43 pathology described in the literature [38, 39, 45, 66]. However, additional descriptions of the regional distribution, cell type vulnerability, cell compartment localization, and biochemistry of TDP-43 pathology are necessary to better characterize TDP-43 in the context of ADNC, and if possible, distinguish it from other TDP-43 proteinopathies. There is also a need to develop biomarkers for TDP-43 proteinopathy in order to identify these individuals during life, rather than requiring an autopsy. Furthermore, transcriptomic or proteomic studies from these human cohorts will be imperative to identify underlying mechanisms and pathophysiological pathways. Finally, with the addition of more cohorts with pathologically confirmed AD and TDP-43 pathology, GWAS can be performed to identify underlying genetic risk factors that may predispose an individual to developing combined tau, Aβ, and TDP-43 pathology. As we better characterize this disease in humans clinically, pathologically and genetically, progress can be made to generate model system that more accurately represent the disease and used to probe potential pathways and molecular mechanisms.
Model systems
Further development of model organisms that express multiple pathologic proteins is necessary, including those that combine amyloid, tau, and TDP-43 pathologies. Early work in model systems suggests that the presence of TDP-43 exacerbates tau neurotoxicity, worsening disease phenotypes. Therefore, TDP-43 may be a highly relevant therapeutic target. However, these models are only beginning to emerge. It will be necessary to integrate data from human studies into the development of more refined model systems, allowing dissection of interactions between tau and TDP-43. These models should include simple cell culture or invertebrate systems allowing large-scale screening and rapid progress, as well as vertebrate models to investigate disease relevant phenotypes in a mammalian brain. Studies should incorporate consideration of aging as a factor influencing disease. Additionally, conditional expression models will be necessary to study the temporal relationships between tau and TDP-43 and better understand whether perturbation of one is upstream and capable of inducing pathology of the other. Finally, models co-expressing all three proteins (Aβ, TDP-43, and tau) are also necessary to better understand how TDP-43 fits into the A,T(N) framework. As understanding of the pathological role of TDP-43 in AD progresses, essential future work will be to develop therapeutic strategies that prevent or remove disease-promoting TDP-43 and restore healthy brain function.
Overall, determining the contributory roles TDP-43 plays in the progression of AD will require combining autopsy studies of the human pathology with large-scale human genetics and model organism-based experiments. The rewards are great, as determining the nature of TDP-43 contributions to AD may open up a novel path in the pursuit of AD therapeutics.
References
Montine TJ, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11.
Behl T, et al. Exploring the potential of therapeutic agents targeted towards mitigating the events associated with amyloid-beta cascade in Alzheimer's disease. Int J Mol Sci. 2020;21(20).
Fish PV, et al. New approaches for the treatment of Alzheimer’s disease. Bioorg Med Chem Lett. 2019;29(2):125–33.
Nelson PT, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–81.
Pluta R and Ulamek-Koziol M. Tau protein-targeted therapies in Alzheimer's disease: current state and future perspectives, in Alzheimer's Disease: Drug Discovery, X. Huang, Editor. 2020. Brisbane (AU).
Hascup ER, Hascup KN. Toward refining Alzheimer’s disease into overlapping subgroups. Alzheimers Dement (NY). 2020;6(1):e12070.
Bertram L, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39(1):17–23.
Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–3.
Chan D, et al. Lifestyle activities in mid-life contribute to cognitive reserve in late-life, independent of education, occupation, and late-life activities. Neurobiol Aging. 2018;70:180–3.
Gow AJ, Pattie A, Deary IJ. lifecourse activity participation from early, mid, and later adulthood as determinants of cognitive aging: the lothian birth cohort 1921. J Gerontol B Psychol Sci Soc Sci. 2017;72(1):25–37.
Kliegel M, Zimprich D, Rott C. Life-long intellectual activities mediate the predictive effect of early education on cognitive impairment in centenarians: a retrospective study. Aging Ment Health. 2004;8(5):430–7.
Bennett DA, et al. Cognitive and social lifestyle: links with neuropathology and cognition in late life. Acta Neuropathol. 2014;127(1):137–50.
Montine TJ, et al. Adult Changes in Thought study: dementia is an individually varying convergent syndrome with prevalent clinically silent diseases that may be modified by some commonly used therapeutics. Curr Alzheimer Res. 2012;9(6):718–23.
White LR, et al. Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu-Asia aging studies. Neurology. 2016;86(11):1000–8.
Yu L, et al. Residual decline in cognition after adjustment for common neuropathologic conditions. Neuropsychology. 2015;29(3):335–43.
Ratti A, Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem. 2016;138(Suppl 1):95–111.
Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3.
Arai T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–11.
Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8(8):423–34.
Van Deerlin VM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409–16.
Wilson AC, et al. TDP-43 in aging and Alzheimer’s disease - a review. Int J Clin Exp Pathol. 2011;4(2):147–55.
Davidson YS, et al. TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol. 2011;122(6):703–13.
Josephs KA, et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014;127(6):811–24.
Nelson PT, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019.
Nelson PT, et al. "New Old Pathologies": AD, PART, and cerebral age-related TDP-43 With sclerosis (CARTS). J Neuropathol Exp Neurol. 2016;75(6):482–98.
James BD, et al. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain. 2016;139(11):2983–93.
Yang HS, et al. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE epsilon4 haplotype status: a community-based cohort study. Lancet Neurol. 2018;17(9):773–81.
Kawas CH, et al. Multiple pathologies are common and related to dementia in the oldest-old: the 90+ study. Neurology. 2015;85(6):535–42.
Robinson JL, et al. Non-Alzheimer’s contributions to dementia and cognitive resilience in the 90+ study. Acta Neuropathol. 2018;136(3):377–88.
Arnold SJ, Dugger BN, Beach TG. TDP-43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathol. 2013;126(1):51–7.
Nag S, et al. TDP-43 pathology and memory impairment in elders without pathologic diagnoses of AD or FTLD. Neurology. 2017;88(7):653–60.
Josephs KA, et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: a longitudinal retrospective study. Lancet Neurol. 2017;16(11):917–24.
Latimer CS, et al. Resistance and resilience to Alzheimer’s disease pathology are associated with reduced cortical pTau and absence of limbic-predominant age-related TDP-43 encephalopathy in a community-based cohort. Acta Neuropathol Commun. 2019;7(1):91.
Negash S, et al. Resilient brain aging: characterization of discordance between Alzheimer’s disease pathology and cognition. Curr Alzheimer Res. 2013;10(8):844–51.
Montine TJ, et al. Concepts for brain aging: resistance, resilience, reserve, and compensation. Alzheimers Res Ther. 2019;11(1):22.
Josephs KA, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology. 2008.
Wilson RS, et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol. 2013;70(11):1418–24.
Nag S, et al. TDP-43 pathology in anterior temporal pole cortex in aging and Alzheimer’s disease. Acta Neuropathol Commun. 2018;6(1):33.
Josephs KA, et al. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol. 2016;131(4):571–85.
Power MC, et al. Combined neuropathological pathways account for age-related risk of dementia. Ann Neurol. 2018;84(1):10–22.
Laos V, et al. Catalytic Cross talk between key peptide fragments that couple Alzheimer’s disease with amyotrophic lateral sclerosis. J Am Chem Soc. 2021;143(9):3494–502.
Shih YH, et al. TDP-43 interacts with amyloid-beta, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease. Nat Commun. 2020;11(1):5950.
Hicks DA, et al. The cellular expression and proteolytic processing of the amyloid precursor protein is independent of TDP-43. Biosci Rep. 2020;40(4).
Teipel SJ, et al. Association of TDP-43 Pathology with global and regional 18F-florbetapir PET signal in the Alzheimer’s disease Spectrum. J Alzheimers Dis. 2021;79(2):663–70.
Tome SO, et al. TDP-43 interacts with pathological tau protein in Alzheimer’s disease. Acta Neuropathol. 2021;141(5):795–9.
Higashi S, et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–94.
Kadokura A, et al. Regional distribution of TDP-43 inclusions in Alzheimer disease (AD) brains: their relation to AD common pathology. Neuropathology. 2009.
Smith VD, et al. Overlapping but distinct TDP-43 and tau pathologic patterns in aged hippocampi. Brain Pathol. 2018;28(2):264–73.
Drummond E, Wisniewski T. Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 2017;133(2):155–75.
Van Damme P, Robberecht W, Van Den Bosch L. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech. 2017;10(5):537–49.
Gu J, et al. Transactive response DNA-binding protein 43 (TDP-43) regulates alternative splicing of tau exon 10: implications for the pathogenesis of tauopathies. J Biol Chem. 2017;292(25):10600–12.
Montalbano M, et al. TDP-43 and Tau oligomers in Alzheimer's disease, Amyotrophic lateral sclerosis, and frontotemporal dementia. Neurobiol Dis. 2020;146:105130.
Moszczynski AJ, et al. Synergistic toxicity in an in vivo model of neurodegeneration through the co-expression of human TDP-43(M337V) and tau(T175D) protein. Acta Neuropathol Commun. 2019;7(1):170.
Jack CR Jr, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–62.
Barbier P, et al. Role of tau as a microtubule-associated protein: structural and functional aspects. Front Aging Neurosci. 2019;11:204.
BukarMaina M, Al-Hilaly YK, Serpell LC. Nuclear Tau and its potential role in Alzheimer’s disease. Biomolecules. 2016;6(1):9.
Lester E, et al. Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron. 2021;109(10):1675-1691 e9.
Francois-Moutal L, et al. Structural insights Into TDP-43 and effects of post-translational modifications. Front Mol Neurosci. 2019;12:301.
Casafont I, et al. TDP-43 localizes in mRNA transcription and processing sites in mammalian neurons. J Struct Biol. 2009;167(3):235–41.
Apicco DJ, et al. Dysregulation of RNA splicing in tauopathies. Cell Rep. 2019;29(13):4377-4388 e4.
Hsieh YC, et al. Tau-mediated disruption of the spliceosome triggers cryptic RNA splicing and neurodegeneration in Alzheimer’s disease. Cell Rep. 2019;29(2):301-316 e10.
Diez L, Wegmann S. Nuclear Transport deficits in tau-related neurodegenerative diseases. Front Neurol. 2020;11:1056.
Fallini C, et al. Traffic jam at the nuclear pore: all roads lead to nucleocytoplasmic transport defects in ALS/FTD. Neurobiol Dis. 2020;140:104835.
Eck RJ, Kraemer BC, and Liachko NF. Regulation of TDP-43 phosphorylation in aging and disease. Geroscience. 2021.
Deshimaru M, et al. DCTN1 Binds to TDP-43 and regulates TDP-43 Aggregation. Int J Mol Sci. 2021;22(8).
Josephs KA, et al. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 2014;127(3):441–50.
Acknowledgements
We thank the reviewers for helpful comments and suggestions. We thank the researchers who have contributed their efforts to this exciting emerging area, some of whom we were able to highlight here. Figure 2 was created with BioRender.com.
Funding
This work was supported by grants from the Department of Veterans Affairs [Merit Review Grant #I01BX004044 to N.F.L.] and National Institutes of Health [K08 AG065426 to C.S.L.].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Latimer, C.S., Liachko, N.F. Tau and TDP-43 synergy: a novel therapeutic target for sporadic late-onset Alzheimer’s disease. GeroScience 43, 1627–1634 (2021). https://doi.org/10.1007/s11357-021-00407-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-021-00407-0