
Abstract
The increasing average age in developed societies is paralleled by an increase in the prevalence of many age-related diseases such as osteoarthritis (OA), which is characterized by deformation of the joint due to cartilage damage and increased turnover of subchondral bone. Consequently, deficiency in DNA repair, often associated with premature aging, may lead to increased pathology of these two tissues. To examine this possibility, we analyzed the bone and cartilage phenotype of male and female knee joints derived from 52- to 104-week-old WT C57Bl/6 and trichothiodystrophy (TTD) mice, who carry a defect in the nucleotide excision repair pathway and display many features of premature aging. Using micro-CT, we found bone loss in all groups of 104-week-old compared to 52-week-old mice. Cartilage damage was mild to moderate in all mice. Surprisingly, female TTD mice had less cartilage damage, proteoglycan depletion, and osteophytosis compared to WT controls. OA severity in males did not significantly differ between genotypes, although TTD males had less osteophytosis. These results indicate that in premature aging TTD mice age-related changes in cartilage were not more severe compared to WT mice, in striking contrast with bone and many other tissues. This segmental aging character may be explained by a difference in vasculature and thereby oxygen load in cartilage and bone. Alternatively, a difference in impact of an anti-aging response, previously found to be triggered by accumulation of DNA damage, might help explain why female mice were protected from cartilage damage. These findings underline the exceptional segmental nature of progeroid conditions and provide an explanation for pro- and anti-aging features occurring in the same individual.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During life our DNA is continuously damaged both from external (e.g., UV radiation and numerous chemicals) as well as internal (e.g., reactive oxygen species (ROS)) sources, resulting in numerous types of DNA lesions (e.g., strand interruptions). If left unrepaired, some of these lesions can cause mutations upon replication and consequently trigger carcinogenesis. Others can block transcription, thereby influencing cellular functionality and survival, which may affect lifespan of the organism as a whole (Garinis et al. 2009; Hoeijmakers 2001).
To counteract this continuous genetic erosion, an intricate network of DNA repair systems has evolved that resolves both helix-distorting and non-helix-distorting types of DNA damage. A molecular pathway involved in counteracting helix-distorting types of DNA damage is the nucleotide excision repair (NER) pathway. NER consists of a complex multi-step ‘cut and patch’ reaction involving ~30 or more proteins that act genome wide, designated global genome NER, or specifically in actively transcribed genes, named transcription-coupled NER (TC-NER; Hanawalt and Spivak 2008). TC-NER presumably is part of a broader transcription-coupled repair system that also eliminates non-helix-distorting type of transcription-blocking lesions. Since DNA lesions are thought to affect lifespan, a defective TC(-NE)R contributes to the process of aging (Schumacher et al. 2008a; Hoeijmakers 2009). Indeed, human patients with an inborn defect in TC(-NE)R suffer from a so-called ‘premature aging syndrome’ and show numerous symptoms that normally develop much later in life, including sarcopenia, cachexia, progressive neurodegeneration, deafness, early infertility, and osteoporosis. Many patients die at early age.
An example of such a premature aging syndrome is trichothiodystrophy (TTD). Most patients suffering from this rare, autosomal recessive disease have a mutation in the helicase xeroderma pigmentosum D (XPD). This protein is a subunit of the transcription/repair complex TFIIH, which has a multi-functional role in both normal transcription and in NER. Previously, we mimicked the TTD phenotype by generating a knock in mouse carrying an XPD point mutation, found in several TTD patients (de Boer et al. 1998). The resulting mice showed striking phenotypical similarities with human TTD patients, including characteristic brittle hair; scaly skin; early onset of osteoporosis, kyphosis, and cachexia; and a shortened lifespan (de Boer et al. 2002; Faghri et al. 2008; Wijnhoven et al. 2005). As such, they might present an ideal tool to study the process of aging and age-related diseases.
In the experiment described herein, TTD mice were used to study a common age-related disease: osteoarthritis (OA). In OA, the function of a joint (i.e., to provide friction-less movement) becomes radically disturbed due to drastic deformation of the joint components, which leads to severe mobility problems, pain, and reduction in quality of life. Currently, no curative treatment is available other than analgesia and eventually an operation, in which (part of) the joint is replaced by a prosthesis. Several joints can be affected but knee OA is a leading cause of OA-related impairments, affecting more than 4.3 million individuals in the general US population (Dillon et al. 2006).
The knee joint is composed of several components, to ensure friction-less yet stable movement under loading conditions, and each of these components changes structure and/or becomes damaged in the OA disease process. The most well known is damage to the smooth joint surface that is provided by articular cartilage, but increased turnover of the subchondral bone, located directly underneath the articular cartilage, is of equal importance (Karsdal et al. 2008; Lajeunesse 2004). An additional characteristic of OA is the development of bony outgrowths at the rims of the joint, named osteophytes.
Since TTD mice display many characteristics of age-related diseases which are found in the general human population, among which OA, we analyzed subchondral bone changes and cartilage damage in the knee joint of 52- and 104-week-old wild-type (WT) and DNA repair deficient, premature aging TTD mice.
Materials and methods
Generation of TTD knockout mice and selection of study samples
The generation of the mice used in this study has been described before (de Boer et al. 1998; Dollé et al. 2006; Wijnhoven et al. 2005). Briefly, gene-targeted mouse 129Ola ES cells, in which the xpd gene was mutated using a point mutation (R722W) found in a human TTD patient, were injected into C57Bl/6J blastocysts. Chimeric male mice were mated with C57Bl/6J females, and heterozygous offspring was bred into an isogenic C57Bl/6J genetic background for over 10 generations. In the second round of breeding, double heterozygous mice were intercrossed to obtain XPDTTD homozygous mice (from now on termed TTD mice), which were then used in the third breeding round to generate the experimental animals analyzed in this study. The mice were compared to C57Bl/6J wild-type mice (from now on termed WT mice). WT mice are known to develop clear signs of OA from about 16 months (~70 weeks) of age (Yamamoto et al. 2005; Stoop et al. 1999).
An aging cohort was designed containing WT and TTD, both males and females (Wijnhoven et al. 2005). All mice were kept in animal facilities of the RIVM (Bilthoven, The Netherlands) under specific pathogen-free conditions in groups of four or less per cage after weaning. The room temperature was 20°C and the light/dark cycle was 12 h/12 h. Standard lab chow (Hope Farms, The Netherlands) and water were supplied ad libitum. Female TTD animals had a significantly reduced lifespan, with a median survival age of 103 weeks for WT and 93 weeks for TTD females (Wijnhoven et al. 2005). Mice were sacrificed at scheduled ages, including 52-week-old and 104-week-old mice. The physical condition of all animals used in the current study was assessed by a pathologist at sacrifice and only those animals that had no abnormal characteristics (e.g., tumor growth, infection) were used in this study. All procedures were performed in accordance with applicable federal guidelines and institutional policies.
Micro-computed tomographic scanning and quantification of bone morphometric parameters
Tibiae from male and female mice, both WT and TTD were selected for analysis, generating a total of eight groups of mice (n = 6–7 per group). Methods used to scan and analyze the proximal portion of the tibiae (see Fig. 1) are described elsewhere (Botter et al. 2006). Briefly, tibiae were fixed in 4% formalin and scanned in the Skyscan 1072 micro-CT scanner (Skyscan, Kontich, Belgium) with a voxel size of 10 μm. To distinguish calcified tissue from non-calcified tissue and noise, the reconstructed greyscale images were segmented by an automated algorithm using local thresholds (Waarsing et al. 2004), resulting in a 3D dataset consisting of stacked black/white cross-sections. In order to study subchondral bone, the epiphysis of the tibia was chosen as the region of interest. The outline of the epiphysis was manually selected using 3D data analysis software (CTAnalyser, Skyscan) excluding any outgrowing mineralized osteophytes. Next, using in-house developed software (available upon request), the subchondral bone plate was separated from the subchondral trabeculae for each cross-section in the 3D dataset. For the epiphysis, the following 3D morphometric parameters were calculated using the freely available software package 3D-Calculator (http://www.erasmusmc.nl/orthopaedie/research/labor/downloads): Trabecular bone volume fraction (Trab BV/TV), which is the ratio of trabecular bone volume (Trab BV, in cubic millimeters) over endocortical tissue volume (TV, in cubic millimeters), trabecular thickness (Tb.Th., in micrometers), trabecular spacing (in micrometers), and connectivity density (dimensionless), indicating the number of trabecular connections per unit volume (Odgaard and Gundersen 1993). Subchondral bone plate thickness was measured in the weight-bearing region of the medial and lateral tibial plateau. At each location, a subset (0.5 mm mediolateral width, 1.0 mm ventrodorsal length) was taken as region of interest. We chose to present the thickness of the subchondral bone plate as an average of medial and lateral values, since changes in thickness were very similar for both anatomical locations, and for reasons of clarity. Thus, values presented here (Fig. 3c) represent average subchondral bone plate thickness (Sb.Pl.Th., in micrometers).

Analysis of bone phenotype of WT and TTD mice. Anatomical context with the analyzed region (proximal tibia) is indicated in the smaller panel. The larger panel shows a transparent 3D model of the proximal tibia. The subchondral (epiphyseal) trabecular bone structure is indicated in orange (left: frontal view, right: top view). The model was build from a transaxial stack of cross-sections of 10 μm thickness. Med medial, lat lateral, pos posterior, ant anterior
As a reference to other published work (especially with regard to the bone phenotype of WT mice), we also analyzed the amount of bone (i.e., Trab BV/TV) of the proximal metaphysis. For this, we selected a volume of interest located directly underneath the growth plate, of 1.0 mm height.
Histological analysis
Following decalcification in 10% EDTA for 14 days and embedding in paraffin, 6-μm thick coronal (frontal) histological sections were taken through the joint at 100 μm intervals. Sections were stained with Safranin O and Fast green and cartilage damage was scored using the semi-quantitative grading and staging system devised by the OARSI Working Group (Pritzker et al. 2006). In this semi-quantitative system, a grade score reflects severity of damage (0—no damage, 6—complete disorganization and deformation of the joint), and a stage score indicates extent of damage (0—cartilage surface intact, 4—>50% of cartilage surface affected). Grade and stage scores are then multiplied, resulting in a maximum obtainable cartilage damage score of 24 for each of the cartilage surfaces analyzed. Four locations were analyzed (medial tibial plateau, medial femoral condyle, lateral tibial plateau, and lateral femoral condyle) in three histological sections per joint. The cartilage damage scores thus obtained were averaged over the three sections and summed over the four locations, yielding a maximum obtainable summed cartilage damage score of 96.
Loss of proteoglycans (stained by Safranin O) was scored using a four-point scoring system from 0 to 3, where 0 represents no loss of proteoglycans and three indicates complete loss of staining for proteoglycans in more than half of the cartilage layer. Scores from the four anatomical locations were summed to reach a maximum obtainable score of 12.
As in mice osteophytes are most readily observed at the medial tibia (Blaney Davidson et al. 2007), this location was taken to score osteophytosis according to the 4-point scoring system from Kamekura et al. (2005), with 0—no osteophytes, 1—formation of cartilage-like tissues, 2—increase of cartilaginous matrix, and 3—endochondral ossification.
To measure cartilage thickness, microphotographs were taken of the medial tibial plateau and cartilage thickness was measured using Bioquant Osteo v7.20 (Bioquant, Nashville, TN). This program measures thickness of a selected area of interest (here the cartilage layer) every 10 μm, producing roughly 80 measurements, which were then averaged. Thickness of the calcified cartilage (CC) was measured separately, after which thickness of the non-calcified cartilage (NCC) could be calculated.
Statistical analysis
Statistics were analyzed with GraphPad Prism v3.05 (San Diego, CA). When comparing group averages, non-parametric Mann–Whitney was applied. For correlations, non-parametric Spearman rank correlation was used. In all cases p < 0.05 was considered significantly different. In all graphs, data are presented as mean ± standard error of the mean (SEM).
Results
Body weights
As noted before by Wijnhoven et al. (2005), female and male WT mice reached a higher body weight than TTD mice, both at 52 and 104 weeks of age (Fig. 2a). No significant changes in weight between these time points were noted within the female and male genotypes. The weight differences were mainly due to the absence of adipose tissue, as became clear at autopsy during sacrifice. The reduction in fat also became clear from histological analysis of the bone marrow: in bone marrow of WT mice many fat vacuoles were present, in contrast to bone marrow of TTD mice, which contained almost no fat vacuoles (Fig. 2b). No differences were found in the length of either the tibia or the femur between WT and TTDs at either age (data not shown), indicating that the TTD animals were skeletally mature and excluding a developmental delay in growth of the animals.

a Body weights of female and male WT and TTD, aged 52 and 104 weeks. b Gross histological overview of the proximal tibia of WT and TTD mice, aged 104 weeks. An enlargement (magnification ×200) of the metaphyseal bone marrow is shown below each of the overview pictures (magnification ×20). Please note the absence of fat vacuoles in the bone marrow of TTD mice. This difference was already visible at 52 weeks, albeit less clearly (images not shown)
Bone changes in knee joints of WT and TTD mice
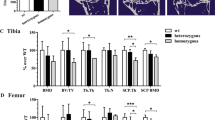
In all groups of mice a significant amount of trabecular bone was lost at 104 weeks of age, as shown from the significant reductions in trabecular bone volume fraction (Trab BV/TV) in the epiphysis (Fig. 3a) and in the more distally located metaphysis (Fig. 3b). These findings are consistent with an aging-related bone decline in both WT and TTD mice (Halloran et al. 2002; Ferguson et al. 2003; Diderich 2010). When comparing genotypes within each of the sexes we noticed that, especially in female mice, the amount of trabecular bone at 52 weeks of age was already lower in female TTDs compared to the age-matched WT mice, although this did not reach statistical significance (females, 52-week-old WT vs. TTD, p = 0.06 for epiphyseal trabecular bone, p = 0.07 for metaphyseal trabecular bone). It is important to note that at young age, the phenotype of TTD animals is not different from WT mice, but starts to deviate from about 13 weeks of age (Wijnhoven et al. 2005; Diderich 2010). The differences observed at 52 weeks are therefore in line with previous findings on accelerated bone loss in female TTD mice.

Bone changes in female and male WT and TTD mice, aged 52 and 104 weeks. a Trabecular bone volume fraction (Trab BV/TV) in tibial epiphysis. b Trab BV/TV in tibial metaphysic. c Subchondral bone plate thickness in the tibial epiphysis. Please note that values refer to the average thickness of the medial and lateral subchondral bone plate. d Thickness of non-calcified (NCC) and calcified (CC) portion of the cartilage layer. Please note the decreased values and increased standard error in both sexes of the 104-week-old WT mice. This was caused due to the severe cartilage damage in two cases (one mouse in each group), for which it was not possible to obtain a thickness value. In these cases ‘0’ was used as value. The histological section (magnification ×200) is provided for anatomical context, the dashed line indicates the border between NCC and CC. Shown are averages ±SEM, significance was tested within genders, with p < 0.05 considered significant. Ages are depicted inside the bars. NCC non-calcified cartilage, CC calcified cartilage, SBP subchondral bone plate, BM bone marrow
Interestingly, at the age of 104 weeks TTD males displayed a further reduction to the same minimum as observed in TTD and WT females. This might indicate that the amount of trabecular bone reached a bottom value (around 15% in epiphysis, 3% in metaphysis) below which no further bone loss occurred. In WT males trabecular bone values were highest at 52 weeks and showed a significant reduction at the age of 104 weeks, but did not reach the low values observed in WT females and both TTD genders. Again, these findings are in agreement with accelerated bone loss in TTD mice.
Consistent with this interpretation, we found that the thickness of the subchondral bone plate (Sb.Pl.Th.) in both sexes of TTD mice decreased significantly between 52 and 104 weeks, whereas this decline was not observed in either female or male WT mice (Fig. 3c). This decrease was caused by thinning of bone tissue, since the thickness of the calcified portion of the cartilage layer (which is also detected by the X-rays used in micro-CT; SMB, personal observations) did not change in either gender or genotype (Fig. 3d). Also, no significant changes were found in the total thickness of the cartilage layer (Fig. 3d, NCC + CC).
Only small, but systematic reductions in average epiphyseal trabecular thickness (Tb.Th.) were measured in both sexes and in both genotypes (female WT: 52 weeks, 80.6 ± 1.7 μm; 104 weeks, 76.1 ± 1.3 μm; female TTD: 52 weeks, 78.6 ± 1.1 μm; 104 weeks, 75.0 ± 1.9 μm; male WT: 52 weeks, 77.9 ± 1.1 μm; 104 weeks, 75.1 ± 1.2 μm; male TTD: 52 weeks, 74.2 ± 1.5 μm; 104 weeks, 69.1 ± 0.5 μm) and this reduction was statistically significant for the 104-week-old TTD males (p < 0.01 compared to 52-week-old TTD male, p < 0.01 compared to 104-week-old WT male). The frequency distribution of the measured trabecular thicknesses (Fig. 4, upper panel) shows an approximate normal distribution. The area under the curve of each distribution is equal to the total amount of trabecular bone (Trab BV) inside the epiphysis. These areas are smaller in all groups of 104-week-old mice, indicating loss of trabecular bone. Thus, thinning of trabeculae does not fully explain the decreased amount of subchondral trabecular bone seen in all groups of mice. Equally important, trabeculae were lost, especially in the thickness range of 90–150 μm. This was further substantiated by the trabecular connectivity density, which dropped significantly in female WT mice (52 weeks, 58.8 ± 17.7; 104 weeks, 0.4 ± 0.4, p < 0.05) and male TTD mice (52 weeks, 78.4 ± 14.4; 104 weeks, 26.7 ± 10.5, p < 0.05), and also had a downward trend in female TTD mice (52 weeks, 39.2 ± 8.3; 104 weeks, 22.6 ± 9.1, p = 0.24) and male WT mice (52 weeks, 78.8 ± 7.6; 104 weeks, 49.3 ± 9.4, p = 0.05). In line with these findings, trabecular spacing was significantly increased in all 104-week-old groups of mice, except for the 104-week-old WT males (data not shown), who accordingly had the smallest decrease in subchondral trabecular bone (shown in Fig. 3a), consistent with the idea that bone aging in this group was lowest. The changes in trabecular bone of the tibial epiphysis are visualized by 3D reconstructions in Fig. 4 (bottom panel).

Top panel thickness distributions of epiphyseal trabecular bone in female and male WT and TTD mice, aged 52 and 104 weeks. The number of observations of a measured thickness were counted and pooled in intervals of 30 μm of thickness. Please note that area under the curve depicts the total amount of trabecular bone (Trab BV). Trab BV in males is higher compared to females, since males are larger animals, but this is compensated by TV (not shown) and both sexes therefore have similar Trab BV/TV ratios as depicted in Fig. 2a, *p < 0.05, **p < 0.01, 52 vs. 104 weeks. Bottom panel 3D visualizations of the epiphyseal trabecular bone structure, as viewed from the top. The anatomical positioning is the same as shown in Fig. 1 (right)
Next to a decreased Trab BV (Fig. 4), the reduction in the amount of trabecular bone (i.e., Trab BV/TV, Fig. 3a) was also caused by an increase in the total amount of endocortical tissue volume (TV), thereby lowering the Trab BV/TV ratio (data not shown). The increased endocortical TV indicated that with aging, more space became present inside the epiphysis. Since the epiphysis itself was not significantly enlarged during aging in neither females nor males, we hypothesized that the endocortical thinning of the subchondral bone plate would explain the increased TV. We therefore studied the relation between the Sb.Pl.Th. and TV and found an inverse correlation in TTD, but not in WT mice (female TTD, 52 and 104 weeks, r = −0.56, p = 0.05; male TTD, 52 and 104 weeks, r = −0.72, p < 0.01). Thus, in 104-week-old TTD mice, which had the thinnest subchondral bone plate (Fig. 2a), TV was large, contributing to a decrease in Trab BV/TV.
Cartilage changes in knee joints of WT and TTD mice
The most commonly observed type of cartilage damage was superficial fibrillation, occasionally accompanied by small fissures down the non-calcified region, mainly at the lateral side of the tibial plateau (Fig. 5). This type of damage is similar what has been found by others in WT mice (Lapvetelainen et al. 1995; Stoop et al. 1999). Cartilage damage values ranged from ~10–30 points out of a maximum of 96 points in all groups of mice (Fig. 6a). In both sexes of WT mice, cartilage damage increased from mild at 52 weeks of age to moderate at 104 weeks of age, although this difference was only statistically significant in the female mice. In two cases clear disorganization throughout the joint was found: 1 out of n = 7 in the group of 104-week-old WT males (summed cartilage damage score 61.3) and 1 out of n = 6 in the group of 104-week-old WT females (summed cartilage damage score 70.8). Compared to WT females, the female TTD mice had significantly less cartilage damage both at 52 and 104 weeks of age. In males the amount of damage was more similar between genotypes, although the 52-week-old TTD mice had a trend towards more cartilage damage compared to age-matched WT mice (p = 0.05).

Representative examples of cartilage damage at the lateral tibial plateau in female and male WT and TTD mice, aged 52 and 104 weeks. Cartilage damage (arrowheads) in 104-week-old female TTD mice was only sporadically observed, contrary to the other groups of 104-week-old mice. In addition, Safranin O depletion indicating proteoglycan loss was observed in all 104-week-old mice, except for the TTD females. Magnification ×200. C cartilage, SBP subchondral bone plate, M meniscus, BM bone marrow

Quantification of histological OA characteristics in knee joints of female and male WT and TTD mice, aged 52 and 104 weeks. a Cartilage damage, scoring was performed according to the OARSI Working Group scoring system. b proteoglycan loss. The staining score is inversely correlated with of the Safranin O staining intensity, a high score therefore indicates more proteoglycan loss. c Osteophyte presence at the medial tibia, a higher score meaning more mature (mineralized) osteophytes. Shown are averages ±SEM, significance was tested within genders, with p < 0.05 considered significant. Ages are depicted inside the bars. n.p. not present
To find out if a relation existed between the amount of cartilage damage and changes observed in the underlying subchondral bone plate, we plotted the average subchondral bone plate thickness against the amount of cartilage damage, but no relation was found between these parameters in any of the mouse groups. Because within every group of mice, the highest cartilage damage scores were consistently found at the lateral side, where the subchondral bone plate was thinner compared to the medial side (data not shown), we checked for a relation by plotting the separate medial and lateral subchondral bone plate and cartilage damage values for each anatomical site (i.e., medial and lateral tibial plateau), but no relation was found.
At 104 weeks of age, loss of proteoglycans as assessed by Safranin O depletion was observed in all but the female TTD mice, in which the cartilage phenotype remained relatively normal during aging (Figs. 5 and 6b). At 104 weeks of age, TTD females had significantly less proteoglycan depletion compared to age-matched female WT mice. In males a similar amount of proteoglycan depletion was found in both genotypes, which proved to be significantly different for TTD males and also had an upward trend for male WT animals (p = 0.07; Fig. 6b).
Finally, osteophytes were observed in 52-week-old WT mice of both sexes (females, 4 out of n = 6; males, 2 out of n = 5), and prevalence rose to 100% in the 104-week-old animals (females, 6 out of n = 6; males, 7 out of n = 7). TTD mice had less osteophytosis at 52 weeks of age (females, 0 out of n = 6; males, 1 out of n = 6), and significantly less at 104-week-old of age (females, 1 out of n = 7; males, 4 out of n = 6) compared to age-matched WT animals (Fig. 6c).
Discussion
This is the first study that investigated the link between DNA repair deficiency as seen in a mouse model for the DNA repair compromised, premature aging disorder trichothiodystrophy, and OA-related changes in cartilage and subchondral bone with aging. Given the fact that both human TTD patients and TTD mice present a predominantly progeroid clinical picture, including prominent osteoporosis, it was expected that these mice would also display an increased cartilage damage, a hallmark characteristic of osteoarthritis (OA). Remarkably, however, not only did our findings indicate that this was not the case, we also found that female TTD mice were protected from cartilage damage. In sharp contrast, at the level of (subchondral) bone, we did confirm the accelerated bone aging phenotype, especially in TTD females.
The question then is, what explains this apparent tissue difference between bone and cartilage in aging TTD mice, and why are female mice affected more than males? Previous results already showed that other tissues in TTD mice also exhibit a paradoxically slower rate of aging-related pathology compared to WT mice. For instance, incidence of age-related cataract of the eye lens was decreased in TTD mice as compared to WT littermates (Wijnhoven et al. 2005). An interesting common denominator of both cartilage and lens tissue is the absence of any vascular, neuronal, or lymphatic tissue and the post-mitotic state of the cells, whereas bone (as most other tissues) is highly vascularized and has a higher turnover. Epithelial cells of the lens are nourished via the aqueous humor and anaerobic metabolism is the primary source of energy (Harding 1997). Articular cartilage receives its nutrients via the surrounding synovial fluid which is encapsulated by the knee capsule, and via the underlying subchondral bone (Benedek 2006; Huber et al. 2000; Imhof et al. 1999). Embedded inside their extracellular matrix, the chondrocytes survive under relative anoxic conditions, with oxygen tensions varying from around 6% at the joint surface to 1% in the deep layers of healthy articular cartilage (Pfander and Gelse 2007). In contrast, blood vessels are commonly observed in subchondral bone (Imhof et al. 2000), and oxygen is important for the survival of osteoblasts (Sun and Wei 2009).
Vascularization of a tissue is important for nutrient and oxygen supply to tissues and cells and for determining the metabolic capacity of the cells. However, the downside of oxygen is the generation of DNA-damaging ROS, which are produced when oxygen is consumed in the aerobic respiratory chain of mitochondria. ROS are important for some cellular processes in both bone and cartilage (Kim et al. 2010; Rached et al. 2010; Ambrogini et al. 2010; Morita et al. 2007; Gibson et al. 2008), but high levels of ROS (i.e., oxidative stress) can lead to increased levels of oxidative DNA damage which may not be repaired sufficiently, thereby disturbing normal cellular metabolism and increasing the risk for pathology (Wauquier et al. 2009; Garinis et al. 2009; Hoeijmakers 2001; Giorgio et al. 2007). The concept of ROS production in relation to age-related bone loss and development of osteoporosis has recently been carefully reviewed by Manolagas (Manolagas 2010) and the significance of FoxO-mediated defense against oxidative stress for osteoblast function and skeletal homeostasis has recently been demonstrated (Rached et al. 2010; Ambrogini et al. 2010). A role for ROS in OA has also been discussed in the context of DNA damage, together with the effect of oxygen tension on ex vivo chondrocyte function and antioxidant intervention studies (Henrotin et al. 2005). However, it was concluded that the role of ROS in cartilage degradation and development of OA remained an open question.
The concept of ROS-induced oxidative stress leading to pathology is especially relevant for a situation in which the DNA repair system is compromised, as is the case in TTD. However, this concept also implies that less metabolic active and low oxygen consuming tissues (such as cartilage and the eye lens) have reduced sensitivity to age-related accumulation of DNA damage. Following this argument, oxidative stress in the nonvascularized cartilage and lens is expected to be limited compared to bone and other vascularized tissues, and the presence of only a partially functional DNA repair system would therefore suffice. Our current study therefore strongly indicates that oxidative damage does not play a major role in the age-related etiology of cartilage damage and OA. In this respect, the role of antioxidant treatment (either taken as supplements or via enriched dietary intake) may not be effective, and although in the last decades a number of randomized clinical trials have been performed which tested the use of supplements as treatment for OA, antioxidant supplementation so far has not shown clear efficacy (Wluka et al. 2002; Canter et al. 2007; Brien et al. 2008). Subsequent direct proof that oxidative damage is not involved in OA should be provided by genetic studies and age-related cartilage-specific interference in metabolism, ROS production and cartilage degradation.
A second explanation for the observed age-related differences in cartilage and bone between WT and TTD mice has emerged from microarray expression profile analysis of other DNA repair-deficient progeroid mouse mutants, which revealed a ‘survival response’ involving suppression of the somatotrophic (IGF-1/growth hormone) axis (Garinis et al. 2009; Niedernhofer et al. 2006; Schumacher et al. 2008b; Van de Ven et al. 2007). This survival response is a systemic adaptive response aimed at minimizing further damage to the DNA, by shifting the energy equilibrium of the organism from growth and proliferation to preservation of somatic maintenance. In addition to lowering IGF-1 production, the survival response stimulates cellular antioxidant systems and suppresses the production of certain hormones, such as estrogens, which may also explain why TTD mice have a lower bone mass compared to WT mice. This latter finding is of particularly interest, as the association of low amounts of cartilage damage associated with (subchondral) osteopenia point to the ‘old’ theory of an inverse relation between OA and osteoporosis (Dequeker et al. 2003). Here, IGF-1 may be a mechanistic link, since its levels were shown to be elevated in OA cartilage (Schneiderman et al. 1995) but its production is generally found to decline with aging leading to osteoporosis (Perrini et al. 2010). However, studies looking at the relation between IGF-1 levels and OA remain inconclusive (Denko and Malemud 2005). Interestingly, Biermasz and co-workers found that acromegaly patient having abnormally high circulating levels of IGF-1 showed an almost fourfold increased risk for radiological osteoarthritis of the hip (Biermasz et al. 2009). It would therefore be interesting to determine both circulating, but probably more important, local IGF-1 levels and IGF-I signaling inside the articular cartilage of old WT and TTD mice and assess a relation with cartilage damage.
Furthermore, it is possible that nonvascularized and non-proliferative tissues, such as the cells in the eye lens and cartilage, profit more from this systemic survival response, than they suffer from the consequences of the DNA repair defect in part derived from endogenous oxidative stress. As a net result these organs and tissues undergo less pathological changes with aging compared to vascularized tissues. However, it is currently unknown whether a (protective) survival response is also present in cartilage of (female) TTD mice, and cartilage-specific microarray studies will be instrumental to answer this question.
A final explanation for the observed differences in cartilage aging between WT and TTD mice, although speculative, is the absence of adipose tissue in the latter. In humans, obesity is one of the major risk factors for developing OA, and recent studies indicate that, next to increased body weight and thus increased mechanical joint load, the adipose tissue itself may also induce or exacerbate OA, by excreting pro-inflammatory cytokines such as leptin (Otero et al. 2005; Gabay et al. 2008; Sandell 2009; Yusuf et al. 2010). However, if this will hold for TTD mice as well will necessitate extensive further analyses.
It is difficult to relate our results directly to observations in the human TTD patients since to our knowledge, no study has ever reported or looked at OA development in TTD patients, probably because of the extreme rarity of the disease and the fact that most patients die at an age too early for OA development. With regard to bone changes, some reports stated bone abnormalities including osteopenia in the long bones and osteosclerosis in the axial skeleton (Faghri et al. 2008), but clear sex differences have not been described.
An intriguing and yet unexplained observation in our study was the difference between female and male TTD mice. Female TTD mice had less OA development than TTD males at both ages, whereas in WT mice the OA severity between both sexes was similar. Studies comparing spontaneous OA in both male and female WT mice are scarce but those that are published (usually serving as controls for genetically modified mice) do not report large differences between genders (Bohm et al. 2005; Lories et al. 2008). However, in some OA susceptible mouse strains such as the STR/ORT mice, known to develop progressive cartilage damage at early age (10–20 weeks of age), males are affected more than females (Munasinghe et al. 1996). This finding also holds for instability-induced OA (van Osch et al. 1993; Ma et al. 2007). These studies indicate that male mice, when challenged (by joint instability or genetic modification) are more prone to develop OA than female mice. This difference between male and female mice might be further enhanced by changes in the levels of sex hormones (Schwartz et al. 1994; Sowers et al. 1996; Sniekers et al. 2008).
To summarize, we found that TTD mice, despite their overall premature aging phenotype, did not have more, or even had less, OA characteristics compared to WT mice. This may be explained by a difference in response of a tissue to the ‘survival response’ triggered by the accumulation of DNA damage, or by a difference in sensitivity to the DNA damage itself (or a combination). The outcome of this process is that some, preferentially nonvascularized, tissues will remain relatively unaffected, giving this disorder a segmental nature. This study therefore provides important clues as to why progeroid syndromes such as TTD show a segmental pattern of aging.
References
Ambrogini E, Almeida M, Martin-Millan M, Paik JH, Depinho RA, Han L, Goellner J, Weinstein RS, Jilka RL, O’Brien CA, Manolagas SC (2010) FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab 11:136–146
Benedek TG (2006) A history of the understanding of cartilage. Osteoarthritis Cartilage 14:203–209
Biermasz NR, Wassenaar MJ, van der Klaauw AA, Pereira AM, Smit JW, Roelfsema F, Wolterbeek R, Kroon HM, Kloppenburg M, Romijn JA (2009) Pretreatment insulin-like growth factor-I concentrations predict radiographic osteoarthritis in acromegalic patients with long-term cured disease. J Clin Endocrinol Metab 94:2374–2379
Blaney Davidson EN, Vitters EL, van Beuningen HM, van de Loo FA, Van den Berg WB, Van der Kraan PM (2007) Resemblance of osteophytes in experimental osteoarthritis to transforming growth factor beta-induced osteophytes: limited role of bone morphogenetic protein in early osteoarthritic osteophyte formation. Arthritis Rheum 56:4065–4073
Bohm BB, Aigner T, Roy B, Brodie TA, Blobel CP, Burkhardt H (2005) Homeostatic effects of the metalloproteinase disintegrin ADAM15 in degenerative cartilage remodeling. Arthritis Rheum 52:1100–1109
Botter SM, van Osch GJ, Waarsing JH, Day JS, Verhaar JA, Pols HA, van Leeuwen JP, Weinans H (2006) Quantification of subchondral bone changes in a murine osteoarthritis model using micro-CT. Biorheology 43:379–388
Brien S, Prescott P, Bashir N, Lewith H, Lewith G (2008) Systematic review of the nutritional supplements dimethyl sulfoxide (DMSO) and methylsulfonylmethane (MSM) in the treatment of osteoarthritis. Osteoarthritis Cartilage 16:1277–1288
Canter PH, Wider B, Ernst E (2007) The antioxidant vitamins A, C, E and selenium in the treatment of arthritis: a systematic review of randomized clinical trials. Rheumatology (Oxford) 46:1223–1233
de Boer J, de Wit J, van Steeg H, Berg RJ, Morreau H, Visser P, Lehmann AR, Duran M, Hoeijmakers JH, Weeda G (1998) A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol Cell 1:981–990
de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, Weeda G, van der Horst GT, van Leeuwen W, Themmen AP, Meradji M, Hoeijmakers JH (2002) Premature aging in mice deficient in DNA repair and transcription. Science 296:1276–1279
Denko CW, Malemud CJ (2005) Role of the growth hormone/insulin-like growth factor-1 paracrine axis in rheumatic diseases. Semin Arthritis Rheum 35:24–34
Dequeker J, Aerssens J, Luyten FP (2003) Osteoarthritis and osteoporosis: clinical and research evidence of inverse relationship. Aging Clin Exp Res 15:426–439
Diderich KE (2010). Bone aging in DNA repair deficient trichothiodystrophy mice. Dissertation, Erasmus University Rotterdam, The Netherlands
Dillon CF, Rasch EK, Gu Q, Hirsch R (2006) Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J Rheumatol 33:2271–2279
Dollé ME, Busuttil RA, Garcia AM, Wijnhoven S, van Drunen E, Niedernhofer LJ, van der Horst G, Hoeijmakers JH, van Steeg H, Vijg J (2006) Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice. Mutat Res 596:22–35
Faghri S, Tamura D, Kraemer KH, Digiovanna JJ (2008) Trichothiodystrophy: a systematic review of 112 published cases characterises a wide spectrum of clinical manifestations. J Med Genet 45:609–621
Ferguson VL, Ayers RA, Bateman TA, Simske SJ (2003) Bone development and age-related bone loss in male C57BL/6J mice. Bone 33:387–398
Gabay O, Hall DJ, Berenbaum F, Henrotin Y, Sanchez C (2008) Osteoarthritis and obesity: experimental models. Joint Bone Spine 75:675–679
Garinis GA, Uittenboogaard LM, Stachelscheid H, Fousteri M, van Ijcken W, Breit TM, van Steeg H, Mullenders LH, van der Horst GT, Bruning JC, Niessen CM, Hoeijmakers JH, Schumacher B (2009) Persistent transcription-blocking DNA lesions trigger somatic growth attenuation associated with longevity. Nat Cell Biol 11:604–615
Gibson JS, Milner PI, White R, Fairfax TP, Wilkins RJ (2008) Oxygen and reactive oxygen species in articular cartilage: modulators of ionic homeostasis. Pflugers Arch 455:563–573
Giorgio M, Trinei M, Migliaccio E, Pelicci PG (2007) Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 8:722–728
Halloran BP, Ferguson VL, Simske SJ, Burghardt A, Venton LL, Majumdar S (2002) Changes in bone structure and mass with advancing age in the male C57BL/6J mouse. J Bone Miner Res 17:1044–1050
Hanawalt PC, Spivak G (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9:958–970
Harding JJ (1997) Biochemistry of the eye. Chapman & Hall Medical, London
Henrotin Y, Kurz B, Aigner T (2005) Oxygen and reactive oxygen species in cartilage degradation: friends or foes? Osteoarthritis Cartilage 13:643–654
Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411:366–374
Hoeijmakers JH (2009) DNA damage, aging, and cancer. N Engl J Med 361:1475–1485
Huber M, Trattnig S, Lintner F (2000) Anatomy, biochemistry, and physiology of articular cartilage. Invest Radiol 35:573–580
Imhof H, Breitenseher M, Kainberger F, Rand T, Trattnig S (1999) Importance of subchondral bone to articular cartilage in health and disease. Top Magn Reson Imaging 10:180–192
Imhof H, Sulzbacher I, Grampp S, Czerny C, Youssefzadeh S, Kainberger F (2000) Subchondral bone and cartilage disease: a rediscovered functional unit. Invest Radiol 35:581–588
Kamekura S, Hoshi K, Shimoaka T, Chung U, Chikuda H, Yamada T, Uchida M, Ogata N, Seichi A, Nakamura K, Kawaguchi H (2005) Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthritis Cartilage 13:632–641
Karsdal MA, Leeming DJ, Dam EB, Henriksen K, Alexandersen P, Pastoureau P, Altman RD, Christiansen C (2008) Should subchondral bone turnover be targeted when treating osteoarthritis? Osteoarthritis Cartilage 16:638–646
Kim MS, Yang YM, Son A, Tian YS, Lee SI, Kang SW, Muallem S, Shin DM (2010) RANKL-mediated ROS pathway that induces long-lasting Ca2+ oscillations essential for osteoclastogenesis. J Biol Chem 285:6913–6921
Lajeunesse D (2004) The role of bone in the treatment of osteoarthritis. Osteoarthritis Cartilage 12(Suppl A):S34–S38
Lapvetelainen T, Nevalainen T, Parkkinen JJ, Arokoski J, Kiraly K, Hyttinen M, Halonen P, Helminen HJ (1995) Lifelong moderate running training increases the incidence and severity of osteoarthritis in the knee joint of C57BL mice. Anat Rec 242:159–165
Lories RJ, Peeters J, Szlufcik K, Hespel P, Luyten FP (2008) Deletion of frizzled-related protein reduces voluntary running exercise performance in mice. Osteoarthritis Cartilage 17:390–396
Ma HL, Blanchet TJ, Peluso D, Hopkins B, Morris EA, Glasson SS (2007) Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage 15:695–700
Manolagas SC (2010) From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev 31:266–300
Morita K, Miyamoto T, Fujita N, Kubota Y, Ito K, Takubo K, Miyamoto K, Ninomiya K, Suzuki T, Iwasaki R, Yagi M, Takaishi H, Toyama Y, Suda T (2007) Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J Exp Med 204:1613–1623
Munasinghe JP, Tyler JA, Hodgson RJ, Barry MA, Gresham GA, Evans R, Hall LD (1996) Magnetic resonance imaging, histology, and x-ray of three stages of damage to the knees of STR/ORT mice. Invest Radiol 31:630–638
Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, Hoeijmakers JH (2006) A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444:1038–1043
Odgaard A, Gundersen HJ (1993) Quantification of connectivity in cancellous bone, with special emphasis on 3-D reconstructions. Bone 14:173–182
Otero M, Lago R, Lago F, Casanueva FF, Dieguez C, Gomez-Reino JJ, Gualillo O (2005) Leptin, from fat to inflammation: old questions and new insights. FEBS Lett 579:295–301
Perrini S, Laviola L, Carreira MC, Cignarelli A, Natalicchio A, Giorgino F (2010) The GH/IGF1 axis and signaling pathways in the muscle and bone: mechanisms underlying age-related skeletal muscle wasting and osteoporosis. J Endocrinol 205:201–210
Pfander D, Gelse K (2007) Hypoxia and osteoarthritis: how chondrocytes survive hypoxic environments. Curr Opin Rheumatol 19:457–462
Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, Salter D, Van den Berg WB (2006) Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage 14:13–29
Rached MT, Kode A, Xu L, Yoshikawa Y, Paik JH, Depinho RA, Kousteni S (2010) FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab 11:147–160
Sandell LJ (2009) Obesity and osteoarthritis: is leptin the link? Arthritis Rheum 60:2858–2860
Schneiderman R, Rosenberg N, Hiss J, Lee P, Liu F, Hintz RL, Maroudas A (1995) Concentration and size distribution of insulin-like growth factor-I in human normal and osteoarthritic synovial fluid and cartilage. Arch Biochem Biophys 324:173–188
Schumacher B, Garinis GA, Hoeijmakers JH (2008a) Age to survive: DNA damage and aging. Trends Genet 24:77–85
Schumacher B, van der Pluijm I, Moorhouse MJ, Kosteas T, Robinson AR, Suh Y, Breit TM, van Steeg H, Niedernhofer LJ, van Ijcken W, Bartke A, Spindler SR, Hoeijmakers JH, van der Horst GT, Garinis GA (2008b) Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet 4:e1000161
Schwartz Z, Nasatzky E, Ornoy A, Brooks BP, Soskolne WA, Boyan BD (1994) Gender-specific, maturation-dependent effects of testosterone on chondrocytes in culture. Endocrinology 134:1640–1647
Sniekers YH, Weinans H, Bierma-Zeinstra SM, van Leeuwen JP, van Osch GJ (2008) Animal models for osteoarthritis: the effect of ovariectomy and estrogen treatment—a systematic approach. Osteoarthritis Cartilage 16:533–541
Sowers MF, Hochberg M, Crabbe JP, Muhich A, Crutchfield M, Updike S (1996) Association of bone mineral density and sex hormone levels with osteoarthritis of the hand and knee in premenopausal women. Am J Epidemiol 143:38–47
Stoop R, Van der Kraan PM, Buma P, Hollander AP, Billinghurst RC, Poole AR, Van den Berg WB (1999) Type II collagen degradation in spontaneous osteoarthritis in C57Bl/6 and BALB/c mice. Arthritis Rheum 42:2381–2389
Sun X, Wei Y (2009) The role of hypoxia-inducible factor in osteogenesis and chondrogenesis. Cytotherapy 11:261–267
Van de Ven M, Andressoo JO, Holcomb VB, Hasty P, Suh Y, van Steeg H, Garinis GA, Hoeijmakers JH, Mitchell JR (2007) Extended longevity mechanisms in short-lived progeroid mice: identification of a preservative stress response associated with successful aging. Mech Ageing Dev 128:58–63
Van Osch GJ, Van der Kraan PM, Vitters EL, Blankevoort L, Van den Berg WB (1993) Induction of osteoarthritis by intra-articular injection of collagenase in mice. Strain and sex related differences. Osteoarthritis Cartilage 1:171–177
Waarsing JH, Day JS, Weinans H (2004) An improved segmentation method for in vivo microCT imaging. J Bone Miner Res 19:1640–1650
Wauquier F, Leotoing L, Coxam V, Guicheux J, Wittrant Y (2009) Oxidative stress in bone remodelling and disease. Trends Mol Med 15:468–477
Wijnhoven SW, Beems RB, Roodbergen M, van den BJ L, PH DK, van der Horst GT, Vijg J, Hoeijmakers JH, van Steeg H (2005) Accelerated aging pathology in ad libitum fed Xpd(TTD) mice is accompanied by features suggestive of caloric restriction. DNA Repair (Amst) 4:1314–1324
Wluka AE, Stuckey S, Brand C, Cicuttini FM (2002) Supplementary vitamin E does not affect the loss of cartilage volume in knee osteoarthritis: a 2 year double blind randomized placebo controlled study. J Rheumatol 29:2585–2591
Yamamoto K, Shishido T, Masaoka T, Imakiire A (2005) Morphological studies on the ageing and osteoarthritis of the articular cartilage in C57 black mice. J Orthop Surg (Hong Kong) 13:8–18
Yusuf E, Nelissen RG, Ioan-Facsinay A, Stojanovic-Susulic V, DeGroot J, van Osch G, Middeldorp S, Huizinga TW, Kloppenburg M (2010) Association between weight or body mass index and hand osteoarthritis: a systematic review. Ann Rheum Dis 69:761–765
Acknowledgments
We are grateful to Piet Kramer and Nicole Kops for their skilful technical assistance. This work was financially supported by National Institute of Health (NIH)/National Institute of Aging (NIA), grant number 1 PO1 AG-17242, NIEHS (1UO1 ES011044), Netherlands Organization for Scientific Research (NWO) through the foundation of the Research Institute Diseases of the Elderly, as well as grants from the Dutch Cancer Society (EUR 99-2004), and EC (QRTL-1999-02002).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Botter, S.M., Zar, M., van Osch, G.J.V.M. et al. Analysis of osteoarthritis in a mouse model of the progeroid human DNA repair syndrome trichothiodystrophy. AGE 33, 247–260 (2011). https://doi.org/10.1007/s11357-010-9175-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-010-9175-3