
Abstract
The Vibrionaceae family groups genetically and metabolically diverse bacteria thriving in all marine environments. Despite often representing a minor fraction of bacterial assemblages, members of this family can exploit a wide variety of nutritional sources, which makes them important players in biogeochemical dynamics. Furthermore, several Vibrionaceae species are well-known pathogens, posing a threat to human and animal health. Here, we applied the phylogenetic placement coupled with a consensus-based approach using 16S rRNA gene amplicon sequencing, aiming to reach a reliable and fine-level Vibrionaceae characterization and identify the dynamics of blooming, ecologically important, and potentially pathogenic species in different sites of the northern Adriatic Sea. Water samples were collected monthly at a Long-Term Ecological Research network site from 2018 to 2021, and in spring and summer of 2019 and 2020 at two sites affected by depurated sewage discharge. The 41 identified Vibrionaceae species represented generally below 1% of the sampled communities; blooms (up to ~ 11%) mainly formed by Vibrio chagasii and Vibrio owensii occurred in summer, linked to increasing temperature and particulate matter concentration. Pathogenic species such as Vibrio anguilllarum, Vibrio tapetis, and Photobacterium damselae were found in low abundance. Depuration plant samples were characterized by a lower abundance and diversity of Vibrionaceae species compared to seawater, highlighting that Vibrionaceae dynamics at sea are unlikely to be related to wastewater inputs. Our work represents a further step to improve the molecular approach based on short reads, toward a shared, updated, and curated phylogeny of the Vibrionaceae family.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bacteria belonging to the Vibrionaceae family (Gammaproteobacteria) are present in all marine environments, with few species also found in brackish and freshwater ones (Thompson et al. 2004; Thompson and Polz 2006). They represent one of the best-studied models for the ecology and evolution of bacterial populations, also due to their easy culturability (Gomez-Gil et al. 2014; Takemura et al. 2014), and are an important component of marine ecosystems, contributing to the main biogeochemical cycles and supporting food webs (Chen et al. 2020).
Vibrionaceae are genetically and metabolically diverse, showing a wide range of lifestyles and niche specializations. Representatives of this family range from planktonic free-living organisms, to symbionts, to sessile forms attached to abiotic and biotic marine surfaces (e.g., suspended or sinking particles) including biofilm-forming species (Reen et al. 2006; Gomez-Gil et al. 2014; Palit and Nair 2013).
Vibrionaceae commonly represent a low fraction of bacterial assemblages in seawater. However, in response to different environmental factors (Takemura et al. 2014), they can reach very high densities forming transient (days) or long-lasting (weeks) blooms (Fuhrman et al. 2015; Westrich et al. 2016). Furthermore, several members of this family are recognized as human and animal pathogens (Thompson et al. 2004). Different Vibrionaceae species are associated with human intestinal and extraintestinal infections (Gomez-Gil et al. 2014; Monsreal et al. 2021) linked to the water microbiota and/or seafood; the most common pathogenic species are Vibrio cholerae, Vibrio parahaemolyticus, Vibrio vulnificus, and Vibrio alginolyticus (Baker-Austin et al. 2018). Other species such as Vibrio harveyi, Vibrio anguillarum, and Vibrio splendidus are commonly correlated with disease events in marine organisms (Austin and Zhang 2006; Rubio-Portillo et al. 2014), including commercially (e.g., aquaculture) and environmentally important fish, mollusc, and crustacean species (Frans et al. 2011; Vezzulli et al. 2015). Furthermore, Vibrionaceae represent the most abundant group in marine fish gut (Egerton et al. 2018) not only as pathogens, but also as symbionts in light organs, where they can exhibit bioluminescence (Urbanczyk et al. 2008; Burtseva et al. 2021).
Given the importance of this bacterial family, there is a high interest in studying its environmental distribution, dynamics, and public health implications (Stewart et al. 2008). In this perspective, culture-independent approaches such as amplicon sequencing (i.e., DNA metabarcoding), which allow to describe the biological diversity as a whole (Deiner et al. 2017), can be a valuable resource.
Amplicon sequencing uses high-throughput sequencing (HTS) to target a species-specific barcode region (Taberlet et al. 2012). The most common HTS platforms, such as Illumina and Thermo Fisher, can reach a read length of a few hundred (~ 600) nucleotides, not enough to cover the full length of the standard barcode regions (e.g., 16S rRNA, ITS) and therefore lowering their informative power. In fact, amplicon sequencing-derived data are generally assigned, when compared with taxonomy reference databases, up to the family or the genus level (Earl et al. 2018).
This limitation has brought to the development of tools that can increase the taxonomic resolution of HTS short reads (Matsen et al. 2012; Gwak and Rho 2020). Among these, the phylogenetic placement is one of the most interesting and increasingly used approach (Rajter and Dunthorn 2021). It has been successfully applied to place the short reads on phylogenetic trees built using full-length reference sequences, allowing their assignment at species level (Matsen et al. 2012; Janssen et al. 2018), showing a greater accuracy than de novo (i.e., based only on short reads) tree construction (Balaban et al. 2020).
Different biomonitoring studies in aquatic environments have used this approach for the survey of metazoans (Mitsi et al. 2019), protists (Elferink et al. 2017; Keck et al. 2018; Gottschling et al. 2021), and prokaryotes (López-Cortés et al. 2020; Iniesto et al. 2021). Recently, a comprehensive and curated cyanobacterial reference database that can be readily used for the phylogenetic placement of these organisms was released (Cydrasil; Roush et al. 2021), widening the biomonitoring potential of this tool.
Here, we present the application of the phylogenetic placement coupled with a consensus-based approach intending to reach a reliable and fine (i.e., at species level) taxonomic characterization within the family Vibrionaceae in seawater and depuration plants at different sites of the northern Adriatic Sea using amplicon sequencing of the 16S rRNA gene. The aim of this work is to characterize their composition and relative abundance to identify the dynamics of blooming, ecologically important, and potentially pathogenic species.
Materials and methods
Study area and sampling
In this work, we studied three coastal marine sites in the northern Adriatic Sea (Fig. 1; Schlitzer 2018).

Sampling sites in the North Adriatic. LS, Lignano Sabbiadoro; SG, San Giorgio di Nogaro; DP, depuration plant
The C1 monitoring station, under study within the Long-Term Ecological Research network (LTER), is located in the Gulf of Trieste (C1, 45° 42′ 2″ N, 13° 42′ 36″ E). Samples investigated at this station were collected monthly between October 2018 and July 2021, at ~ 1 m (surface, S) and ~ 15 m (at the bottom of the water column, B) depths, using 5-L Niskin bottles.
The other two sampling sites (Lignano Sabbiadoro, LS, 45° 38′ 35″ N, 13° 10′ 14.5″ E and San Giorgio di Nogaro, SG, 45° 39′ 19″ N, 13° 14′ 39″ E) are directly affected by the discharge of depurated sewage from the wastewater treatment plants (WWTP) located in their respective municipalities. Both WWTP include primary and secondary treatment of urban and industrial wastewater and a final disinfection step (with UV plus peracetic acid in LS WWTP, with only peracetic acid in SG WWTP). Their marine outfall pipes discharge at 7.5 and 10 km from the shoreline, at 13.7 and 14.3 m depth, respectively. Both sites were sampled monthly in spring and summer 2019 and 2020 (i.e., from April to September/October), using 5-L Niskin bottles. In both cases, seawater samples were collected close to the WWTP submarine outfall pipelines, 1 m above the main diffusion point.
In addition to seawater samples, in LS and SG, we analyzed samples of treated sewage collected upstream the injection into the discharging pipeline of depurators.
Seawater and depuration plant (DP) samples were filtered through 0.2-μm PES membrane filters (PALL Laboratory) and stored at − 80 °C until further processing. Filtration was performed until clogging of membrane pores. Filtered volumes were in the range of 1–3 L for seawater (SW) and 70–500 mL for DP.
Ancillary parameters
At C1 station, a set of biogeochemical parameters, including water temperature, salinity, chlorophyll a (Chl a) and particulate organic carbon (POC) concentration, and prokaryotic abundance were determined contextually to DNA sampling (see the “Study area and sampling” section).
Temperature and salinity were measured by means of a multiparametric probe (SBE 19plus SEACAT), calibrated every 6–12 months.
Chl a concentration was determined fluorometrically according to Lorenzen and Jeffrey (1980) as detailed in Manna et al. (2021). POC concentration was measured using an elemental Analyser CHNO-S Costech mod. ECS 4010 applying the methods by Pella and Colombo (1973) and Sharp (1974), as detailed in Celussi et al. (2017).
Total prokaryotes were counted by flow cytometry, using a FACSCanto II (Becton Dickinson) instrument, equipped with an air-cooled laser at 488 nm and standard filter setup. The method by Marie et al (1999) was used, as detailed in Manna et al. (2021).
DNA extraction, 16S amplicon sequencing, and taxonomic assignment
DNA was extracted from membrane filters using the DNeasy PowerWater Kit (Qiagen) with some modifications to increase the DNA yield and quality (detailed in Celussi et al. 2018). Extracted DNA was quantified with a Qubit Fluorimeter (Thermo Fisher Scientific).
For the amplicon sequencing, the V4–V5 region of 16S rRNA gene was amplified using 515-Y (5′-GTGYCAGCMGCCGCGGTAA-3′) and 926R (5′-CCGYCAATTYMTTTRAGTTT-3′) primers (Parada et al. 2016) provided with Illumina adaptors. Libraries were prepared following the 16S Metagenomic Sequencing Library Preparation protocol (Amplicon 2013) and run on an Illumina MiSeq System for a read length of 2 × 250 bp at the genetic and epigenetic ARGO Open Lab Platform, Area Science Park, Trieste, Italy.
Raw sequences were quality filtered and denoised with DADA2 v. 1.20.0 (Callahan et al. 2016) in R (v. 4.4.1; R Core Team 2021) with pseudo-pooling method. After primer removal and visual inspection of the read quality profiles, forward and reverse reads were truncated at positions 220 and 190, respectively. Chimeric sequences were identified and removed in consensus mode. Amplicon-sequence variants (ASVs) with frequency < 2 (singletons) were removed.
Taxonomy was assigned using the Sklearn Naïve Bayes taxonomy classifier (Bokulich et al. 2018) against the SILVA 99% reference database with 7-level taxonomy release 138 (Quast et al. 2012) in QIIME2 2020.6 (Bolyen et al. 2019). We identified 132 oligotypes (ASVs) belonging to Vibrionaceae family: 57 in C1 surface (C1_S), 83 in C1 bottom (C1_B), 66 in Lignano Sabbiadoro (LS), 62 in San Giorgio di Nogaro (SG), 14 in Lignano Sabbiadoro depurator plant (LS_DP), and 9 in San Giorgio di Nogaro depurator plant (SG_DP).
Vibrionaceae oligotypes identified with SILVA were further taxonomically assigned with other reference databases: GTDB ssu_r86.1_20180911 (https://osf.io/25djp/wiki/home/) using the same approach described above in QIIME2, with SILVA release 138 (Quast et al. 2012) with last common ancestor method (LCA), and Ribosomal Database Project (RDP; Cole et al. 2007) using SINA v1.2.11 at default parameters (Pruesse et al. 2012). BLASTN 2.12.0 + (Altschul et al. 1997) was also used, aligning the oligotypes against the nucleotide collection and excluding uncultured and environmental sample sequences: the hits with the lower e-value, 100% query cover, and > 97% of identity were selected.
Consensus-based workflow
To maximize the reliability of the species-level assignment, we applied a consensus-based approach (Fig. S1). Firstly, the Vibrionaceae oligotypes were used for the phylogenetic placement (see the following section). Then, an oligotype selection was performed, retaining only the ones in which the phylogenetic placement at the genus level agreed with at least 3 out of the 5 reference databases, while the others were considered “unassigned” Vibrionaceae. In this way, we exploited the power of the phylogenetic placement to reach the lower taxonomic level, while acknowledging the limits of short read sequencing.
Phylogenetic placement and phylogenetic tree construction
The phylogenetic placement was used to assign the Vibrionaceae oligotype sequences to the near full-length 16S rRNA gene reference phylogeny. The input to the phylogenetic placement algorithm consists of a reference tree, a corresponding reference multiple sequence alignment, and a collection of query sequences. The output is a set of assignments of the query sequences to branches of the tree (Matsen et al. 2012).
For the reference tree, Vibrionaceae sequences were retrieved from Gomez-Gil et al. (2014) and integrated with other lineages and species more recently characterized. The final dataset (Table S1) included 173 sequences coming from published, curated, and verified data, 169 of which were type strains. Reference sequences were aligned with mafft v. 7 and G‐INS‐I strategy (Katoh and Standley 2013). The resulting alignment was used for tree construction. A maximum likelihood (ML; Felsenstein 1981) tree was inferred using RAxML v.8.2.10 (Stamatakis 2014) with the parameters suggested for the evolutionary placement algorithm (EPA-ng; Barbera et al. 2019; Czech et al. 2022). The general time-reversible model and the discrete gamma distribution of nucleotide substitution frequencies (GTR + GAMMA) model of nucleotide substitution were used; supporting values were estimated from 500 bootstraps (Felsenstein 1985). The query sequences were the 132 Vibrionaceae oligotypes. To verify the reliability of the phylogenetic placement and the influence of query length, sequences belonging to three Vibrio species isolated in the Gulf of Trieste (Fabbro et al. 2010, 2012), V. anguillarum (accession number GU120676.1, 843 bp), V. chagasii (GU120679.1, 923 bp), and V. parahaemolyticus (GQ332281.1, 363 bp) were added as controls. The V. anguillarum and V. chagasii sequences were included in the query list both in their original length and in a trimmed version comparable to the sequences coming from NGS (~ 374 bp, named “short”) after aligning them with MUSCLE at default parameters (Edgar 2004) in MEGA X (Kumar et al. 2018).
The query and the control sequences were aligned against the reference alignment using the PArsimony-based Phylogeny-Aware Read Alignment program (PaPaRa) v. 2.5 (Berger and Stamatakis 2012). Then, EPA-ng (Barbera et al. 2019) was used to split query and reference from the PaPaRa alignment. The best scoring RAxML reference tree was selected as input for the phylogenetic placement of the query sequences. For each oligotypes, the placement with the higher likelihood was selected, using a conservative consensus approach following Hoffmann et al. (2020). The “jplace” file (Berger and Stamatakis 2011) was visualized and edited with Interactive Tree Of Life (iTOL) v5 (Letunic and Bork 2021).
To further explore the relationship among Vibrionaceae oligotypes and check their similarity with control sequences, a phylogenetic tree was constructed with mafft v. 7 and G‐INS‐I strategy (Katoh and Standley 2013) and inferring a maximum likelihood (ML) tree using RAxML v.8.2.10 (Stamatakis 2014), with the GTR + GAMMA model of nucleotide substitution.
Statistical analyses
To investigate potential environmental drivers of Vibrionaceae distribution, we analyzed the correlation patterns between the absolute abundance of Vibrionaceae ASVs and temperature, salinity, Chl a, and POC concentration. Absolute abundance was calculated by multiplying the total prokaryotic abundance, measured by flow cytometry (see the “Ancillary parameters” section), by the relative abundance of Vibrionaceae ASVs in the LTER-C1 dataset (Mena et al. 2021).
Since data revealed to be not normally distributed (Shapiro–Wilk test, p < 0.05), we used Spearman’s correlation metrics. A p-value ≤ 0.05 was considered significant, under the null hypothesis that ranks of absolute Vibrionaceae abundance did not covary with the ranks of the environmental parameters considered.
Data handling, visualization, and analysis were conducted in the R environment (v. 4.4.1; R Core Team 2021) using the packages tidyverse (Wickham et al. 2019) and phyloseq (McMurdie and Holmes 2013).
Results
Phylogenetic placement and Vibrionaceae species-level assignment
The phylogenetic tree was composed by 173 sequences with a length of 1770 bp (Fig. 2). Significant bootstrap values (≥ 70; Van de Peer and Salemi 2009) were concentrated in clades belonging to Aliivibrio, Enterovibrio, Grimontia, and Salinivibrio, whereas Vibrio clades were generally less supported. This has already been noted in the 16S rRNA gene-based phylogenies of the Vibrionaceae, which showed low divergence particularly in the genus Vibrio (Gabriel et al. 2014; Ashok Kumar et al. 2020).

Phylogenetic placement of 132 Vibrionaceae oligotypes. The phylogeny was inferred from 173 reference sequences, using maximum likelihood under the GTR + GAMMA model. Support values ≥ 50% (bootstrap n = 500) are showed. Type strains are indicated by the index “T.” The radius of red circles is proportional to the number of oligotypes placed (min = 1, max = 12). Asterisks represent the placement of the known Vibrionaceae species used as controls
The phylogenetic placement of the Vibrionaceae oligotypes was performed using the reference tree. The 132 Vibrionaceae oligotypes were placed in 9 genera and 39 species (Fig. 2); 97 oligotypes were assigned at the species level, 12 were assigned to more than one species of the same genus, while for 23 oligotypes, the placement was possible only up to the genus level (Table S2).
The placement of control sequences of known attribution allowed us to obtain a measure of the goodness of the workflow (Fig. 2). The V. parahaemolyticus sequence was correctly attributed to V. parahaemolyticus clade. Both the Vibrio anguillarum original and trimmed (“short”) sequences were attributed to V. anguillarum/V. ordalii clade. Vibrio chagasii original sequence was attributed correctly up to the species level, while the trimmed one (i.e., simulating the corresponding short reads in our dataset) was placed also in the upper node. This helped us to place 8 query sequences correctly in V. chagasii species.
Different levels of agreement were detected from the comparison of the taxonomic assignment at genus species between the phylogenetic placement and the 5 databases (Table S3).
Of the 132 oligotypes, 71 (54%) presented a total consensus. The phylogenetic placement assignments at the genus level agreed with BLAST assignment for 98 (74%) oligotypes, with SILVA LCA for 93 (70%), with SILVA for 89 (67%), with GTDB for 81 (61%), and with RDP for 78 (59%).
After this comparison, 103 (78%) oligotypes were retained as they presented a consensus between the phylogenetic placement in at least 3 out of 5 databases.
The oligotypes that were removed at this step belonged to 10 species (Table S4). The fraction of removed reads ranged from low (0–5%) for Vibrio ippocampi, Photobacterium sanguinicancri, and V. anguillarum, to medium (5–30%) for P. aphoticum, Thaumasiovibrio subtropicus, and P. damselae, and to high (30–90%) for Enterovibrio pacificus, Salinivibrio proteolyticus, and Paraphotobacterium marinum (this latter as the only species that was removed). Overall, they represented ~ 3% of the total Vibrionaceae reads.
Finally, we filtered out the oligotypes (18) not assigned at the species level; we reached a final dataset composed by 85 oligotypes (65% of the initial dataset, representing the ~ 75% of the total Vibrionaceae reads; Table S3).
The oligotypes’ phylogenetic tree (Fig. S2) was used to check whether the ASVs assigned to the same clades of the control sequences were correctly retained in the final dataset. No oligotype clustered with the V. parahaemolyticus control sequence in the phylogenetic tree and none was assigned to this species by the phylogenetic placement. For V. anguillarum, 10 out of 10 oligotypes retained (ASV_33,44,56,71,75,77,81,99,100,125) clustered together with this sequence in the phylogenetic tree. ASV_90 that was assigned to this species with the phylogenetic placement and discarded within the consensus-based approach was placed outside the clade. For V. chagasii, 7 out of 7 oligotypes retained (ASV_01,23,29,40,70,94,131) clustered together with this sequence in the phylogenetic tree. ASV_130, which was assigned to this species with the phylogenetic placement and discarded with our consensus-based approach, was placed outside the clade.
Presence and distribution of Vibrionaceae species
The final dataset was composed by 85 oligotypes (out of the initial 132) corresponding to 47 species (including 6 clades): one species of Aliivibrio, four of Enterovibrio, one of Grimontia, one of Salinivibrio, one of Thaumasiovibrio, 14 of Photobacterium, and 25 of Vibrio. Overall, in C1_S and C1_B, 28 and 31 species were detected, respectively. In LS and SG, 36 and 33 species were detected. In LS_DP and SG_DP, 11 and 6 species were detected.
The taxonomic composition and relative abundance in each dataset are reported in Figs. 3, 4, and 5 and Table S5.

Relative abundance of Vibrionaceae species in seawater samples of C1 surface (a and b) and bottom (c and d). The panels b and d zoom on the abundances up to 2%, dashed in panels a and b, respectively

Relative abundance of Vibrionaceae species in seawater samples of Lignano Sabbiadoro (a and b) and San Giorgio di Nogaro (c and d). The panels b and d zoom on the abundances up to 1%, dashed in panels a and b, respectively. Gray areas indicate months that were not sampled

Relative abundance of Vibrionaceae species in depuration plant samples of Lignano Sabbiadoro (a) and San Giorgio di Nogaro (b). Gray areas indicate months that were not sampled
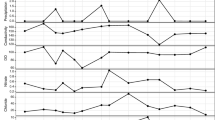
Regarding seawater samples, in C1 surface water (S), the average abundance of Vibrionaceae was 0.4 ± 0.9% with a peak of ~ 3% in June, July, August, and October 2019 (Fig. 3). In bottom water (B), the average abundance was 0.7 ± 1.9% with a peak of ~ 10% in June 2019 (Fig. 3).
In LS, the average abundance was 0.5 ± 0.6%, while in SG was 0.5 ± 0.6%. In seawater of both sites, a peak (~ 8%) was recoded in April 2020 (Fig. 4).
In all sites, the Vibrionaceae blooms were mainly composed by Vibrio chagasii, and, in minor proportion in C1 site, by V. owensii (Fig. 3 and 4).
The relative abundances of Vibrionaceae in the depurator samples were lower than in seawater samples by a few orders of magnitude, with no relevant peaks over time (Fig. 5). The average relative abundances were 9.7 ± 0.5 × 10−4% and 5.0 ± 0.3 × 10−5%, for LS_DP and SG_DP, respectively.
Relationships with environmental variables
A significant positive correlation was found between absolute abundance of Vibrionaceae and water temperature in samples collected at LTER-C1 station (Spearman’s ρ = 0.62; p < 0.001). While we did not find any significant association between Vibrionaceae oligotypes and salinity or Chl a, a positive correlation emerged with POC concentration (Spearman’s ρ = 0.34; p < 0.05).
Discussion
Phylogenetic assignment and consensus-based approach
This study used the phylogenetic placement combined within a consensus-based approach to achieve a fine, species-level characterization of bacteria belonging to the Vibrionaceae family in different sites of the Northern Adriatic Sea.
Although we acknowledge that the term “species” is often inappropriate in the context of prokaryotes, particularly when referring to uncultured organisms, the use of the term “species” for bacterial groups of clinical relevance (including Vibrionaceae) is common practice (Oliver et al. 2012). Furthermore, in this work, we refer to the “species” level to highlight the potential ecological role or pathogenicity of the detected oligotypes.
Our analysis contributed to the identification of the components of bacterial blooms, and to detect potential human and animal pathogens.
Given the importance of the Vibrionaceae family in terms of human health and ecosystem functioning, a better understanding of its dynamics is fundamental. This is even more important in coastal areas, typically characterized by the presence of urbanization, aquaculture, industries, touristic pressure, sewage- and wastewater-related pollution.
Our workflow included the use of (i) control sequences, (ii) different reference databases, and (iii) the comparison between the phylogenetic placement and de novo oligotype phylogenies. This pipeline increased the reliability and the accuracy of the taxonomic assignment at the lower levels (i.e., “species”), when compared to the one obtained using only a reference database (e.g., SILVA) or using the phylogenetic placement alone. The phylogenetic placement algorithms require a reference tree, a corresponding reference multiple sequence alignment, and a collection of query sequences: the output is a set of assignments of the query sequences to branches of the tree (Matsen et al 2012). This tool has been recently used to finely characterize microbial communities in coastal marine (Wilson et al. 2021) and freshwater (Iniesto et al. 2021) environments. Moreover, it has been applied to profile the gut microbiota in toxic and non-toxic puffer fish species (Li et al. 2020) and to assign taxonomy to the white shrimp Litopenaeus vannamei gut oligotypes allowing to study the link between the presence and abundance of pathogenic opportunistic Vibrio species and atypical mass mortality (López-Cortés et al. 2020). Likewise, in our study, this tool allowed us to assign the vast majority of Vibrionaceae oligotypes at the species level (Fig. 2). The use of “long” and “short” control sequences previously obtained from the same sites permitted to estimate the effect of the length of the query sequences, a key factor in amplicon sequence data, and, at the same time, to help with their correct assignment.
The workflow we used included a step in which the taxonomy assigned by the phylogenetic placement was compared with the one obtained through different reference databases at the genus level (i.e., the lowest level reached by most amplicon sequence data), and some discrepancies emerged (Table S3). Disagreements in taxonomic assignments have been mostly attributed to differences in database size and release version, structure, annotation approach (Balvočiūtė and Huson 2017; Edgar 2018) (e.g., SILVA uses a phylogeny-based combination of automated and manual curation, RDP uses a Naïve Bayesian Classifier, BLAST uses local alignments), and updates in bacterial systematics, which affect also the Vibrionaceae family (Ast et al. 2009; Labella et al. 2017). In these cases, we tested and intersected the different approaches to achieve a consensus-based assignment. The removal of ambiguous oligotypes resulted in the removal of ~ 20% of the oligotypes in the final dataset. This step was crucial for reaching the lower taxonomic levels in the most accurate way; even if in our dataset, the ambiguous oligotypes represented a small portion in terms of relative abundance (~ 3%), in other datasets, the proportion could be higher and therefore we recommend including this comparison and selection in the analysis workflow.
Interestingly, the taxonomic assignment disagreement mostly affected only few species of Vibrionaceae: Enterovibrio pacificus, Salinivibrio proteolyticus, and Paraphotobacterium marinum (Table S4). Further investigations, considering broader datasets and/or mock communities, should be performed to test whether these taxa are consistently more prone than others to disagreements, for instance due to the lack of reference sequences, and/or incorrect, old, or spurious nomenclature (as suggested by Park and Won 2018). We could exclude that this was due to an under-representation of these taxa in the reference databases, as all were present except for P. marinum in GTDB ssu_r86.1_20180911 (https://osf.io/25djp/wiki/home/) and RDP (Cole et al. 2007).
Our workflow was designed to extract maximum information from datasets generated by 16S rRNA gene amplicon sequencing, the gold standard in the study of prokaryotic environmental communities (Yarza et al. 2014; Goodwin et al. 2017). However, additional markers can be used to increase the taxonomic resolution of targeted taxonomic groups. In Vibrionaceae, heat shock protein 60 (hsp60; Jesser and Noble 2018) has been successfully used and, in combination with the 16S rRNA gene, has provided useful information about the potential emergence of pathogens.
Vibrionaceae communities in North Adriatic coastal sites
At the sampling sites investigated in the present study, Vibrionaceae family members generally represented less than 1% of the total microbial community (Figs. 3 and 4). It is commonly recognized that this family is generally low abundant in seawater microbial communities (Takemura et al. 2014; Fuhrman et al. 2015). However, despite their low relative abundance, members of this family have repeatedly demonstrated to be leading actors in organic matter dynamics, especially in marginal seas and in freshwater-influenced areas (Takemura et al. 2014; Vezzulli et al. 2016; Zhang et al. 2018). Phenotypic characterizations of culturable Vibrio representatives indicate that most members of this genus can degrade and utilize over 40 organic compounds (Farmer et al. 2005), including sugars (mono-, di-, and polysaccharides), alkanes, and lipids (Johnson 2013; Zhang et al. 2018).
Due to this capability of exploiting a wide array of substrates, Vibrionaceae representatives are able to form transient blooms in response to favorable environmental conditions, like micronutrients (e.g., following Saharan dust deposition events, Westrich et al. 2016) or organic matter pulses (e.g., phytoplankton or jellyfish blooms, Miller et al. 2005; Gilbert et al. 2012; Tinta et al., 2020). In our time-series, Vibrionaceae blooms were observed between June and October in both surface and bottom samples at the C1 station (Fig. 3) as already detected in coastal areas, including the northern Adriatic Sea (Gilbert et al. 2012; Tinta et al. 2015). Vibrionaceae diversity and abundance in water are known to be influenced by different biotic and abiotic factors (Kingsley 2014; Palit and Nair 2013). We found a significant positive correlation between Vibrionaceae abundance and temperature (Fig. 6), in line with growing observations that temperature is among the main drivers of Vibrionaceae dynamics in marine environments (Baker-Austin et al. 2018; Zhang et al. 2018; Wong et al. 2019).

Scatterplots of the correlation (Spearman) between Vibrionaceae absolute abundance and (a) temperature, (b) salinity, (c) chlorophyll a, and (d) particulate organic carbon (POC) concentration. Spearman’s ρ and p values are shown only for significant correlations
While salinity is often regarded as an important factor in explaining the abundance patterns of Vibrionaceae family members (see Zhang et al. 2018 for a review), we did not identify a significant relationship with salinity in our dataset. A likely explanation for this lack of relationship would rely on the narrow salinity gradient at our sampling station (31.6–38.6 and 36.7–38.5 in surface and bottom samples, respectively). In fact, studies reporting salinity as an important factor in explaining Vibrionaceae dynamics are often conducted across transitional environments or in estuarine habitats, encompassing a much larger variability range (e.g., 0–27, Takemura et al. 2014).
Besides temperature, POC concentration represented a significant positive driver of Vibrionaceae abundance in samples in the C1 station. Members of this family are often found to be tightly associated with a variety of particulate substrates, from plastic fragments to phytoplankton cells (Hsieh et al. 2007; Kesy et al. 2021; Sampaio et al. 2022). Experimental and genomic investigations of their metabolic capabilities have highlighted the presence of an advanced exoenzymatic machinery for polysaccharide degradation (Zhang et al. 2018). Therefore, given the significative presence of carbohydrates in organic particles (Kharbush et al. 2020), it is likely that they represent an important nutritional source for members of Vibrionaceae.
Time-series analyses (also in the Gulf of Trieste; Celussi and Cataletto 2007; Tinta et al. 2015) showed that species of this family can occasionally bloom, even becoming the prevalent members of the bacterial community for a short period of time or several weeks, before declining (Vergin et al. 2013; Fuhrman et al. 2015).
At the sampled marine sites, Vibrionaceae blooms were mainly formed by two species, identified by the phylogenetic placement as V. chagasii and V. owensii (the latter found only in C1 samples, Fig. 3 and 4), in agreement with previous observations that identified few Vibrionaceae oligotypes “blooming” in response to changes in environmental conditions (Gilbert et al. 2012).
V. chagasii is found to be ubiquitous in aquatic environments (Thomson et al. 2004) as well as among the components of molluscan microbiomes (Romalde et al. 2014). Although its pathogenicity has not been clarified yet, several studies point out its capability to infect marine invertebrates (Teng et al. 2012; Liang et al. 2019) and fishes (Fabbro et al. 2012). The ecological role of this bacterium in the pelagic environment is still poorly understood, but further information is expected to be acquired due to the recent sequencing of its full genome (Sanches-Fernandes et al. 2021). V. owensii (synonym V. communis) belongs to the Harveyi clade (Cano-Gomez et al. 2011). It represents one of the four species of the group (together with V. harveyi, V. campbellii, and V. rotiferianus) acknowledged as pathogens in farmed fish, shellfish, and crustaceans (Goulden et al. 2012; Liu et al. 2018; Pretto 2020), as well as a putative coral pathogen (Amin et al. 2016).
In our dataset, among pathogenic species (that, apart from V. owensii, were present in very low abundances, Fig. 3), we detected V. anguillarum, known to cause vibriosis in fish and shellfish and frequently retrieved in marine and estuarine environments (Kim et al. 2012; Ma et al. 2017; Gao et al. 2018) and Vibrio fluvialis, commonly found in coastal environments and considered an emerging human foodborne pathogen (Ramamurthy et al. 2014), in all sites. Other identified species were Photobacterium damselae, a free-living animal pathogen that can be found in coastal environments (Novianty and Budiarti 2014; Terceti et al. 2018), and Vibrio tapetis, causing brown ring disease in various bivalve species and other vibriosis in fishes (Gomez-Gil et al. 2014). V. parahaemolyticus, identified in the northern Adriatic coastal waters with biochemical and molecular-based methodologies (Fabbro et al. 2010) if present, was probably in abundances too low to be detected with amplicon sequencing.
Together with the high biodiversity of the coastal zone of the Gulf of Trieste (Nasi et al. 2017), we speculate that the presence of animal pathogens could also be due to the abundant mussel farms in the area (Franzo et al. 2014), which could represent a potential reservoir and source of molluscs-associated Vibrionaceae.
Depuration plants’ Vibrionaceae
Our investigation of the Vibrionaceae family revealed a wide taxonomical and phylogenetic diversity in the three marine sampling sites, in contrast with low numbers of oligotypes retrieved in the treated wastewater samples (Fig. 5). In the same way, the Vibrionaceae relative abundances in the treated sewage were much lower than in seawater samples, especially in the case of San Giorgio DP. Although the treated sewage from the two DP shared a few Vibrionaceae species (Vibrio chagasii, V. orientalis, Photobacterium swingsii), they differed in terms of community structure (Fig. S1). Notably, only a few species retrieved in DP samples were also found at sea, in the proximity of the respective pipeline outfalls. This, together with the low number of retrieved reads, suggests that DP may represent (if any) only a minor source of Vibrionaceae for the surrounding marine coastal environment. The two DP investigated in this study provided for final disinfection prior to unloading the treated sewage in the outlet pipeline. Similarly, in a recent investigation in the central Adriatic Sea, we found that DP contributed only partially to the pool of Vibrionaceae retrieved at sea (next to their outfalls) independently on the presence/absence of processes aimed at decreasing the microbiological load, such as disinfection or activated sludge treatments (Fonti et al. 2021).
Lignano Sabbiadoro DP showed the presence, in very low abundance, of Vibrio cholerae and of V. navarrensis, which we never detected in seawater. Vibrio cholerae, besides being autochthonous in aquatic environment, finds favorable survival and growth conditions in oligohaline, organic matter-rich, neutral to alkaline sewage (Takemura et al. 2014; Baron et al. 2017). Among the over 200 O serogroups of this species, only two strains (O1 and O139) are linked to severe disease and cholera (Momba and Azab El-Liethy 2017). Even if our analysis could not lead to the identification of the serogroup, we can exclude the presence of the cholera disease-associated strains, as the vast majority of the forms of V. cholerae detected in the environment are harmless estuarine bacteria (Rivera et al. 2001). Vibrio navarrensis has been found to be associated with low salinity condition in Thyrrenian coastal environment (Matteucci et al. 2015). Firstly isolated in sewage (Urdaci et al. 1991), and later also in human clinical specimens (Gladney et al. 2014; Schwartz et al. 2017), it is considered a rare human pathogen, characterized by distinctive molecular makers and virulence-associated genes (Lee and Raghunath 2018).
Interestingly, the most abundant species found in treated wastewater samples (V. chagasii, P. swingsii, V. anguillarum) are of marine origins, being carried into the plants by seawater intrusion (N. De Bortoli, pers. comm.).
Taken together, our results highlight that, differently from other potential pathogens (Fonti et al. 2021), members of the Vibrionaceae family retrieved at sea are unlikely to be related to wastewater inputs. Therefore, we suggest to carefully consider the inclusion of the genus Vibrio in the “blacklist” of potentially pathogenic genera in surveys of microbiological pollution in coastal waters (e.g., Paliaga et al. 2017; Fonti et al. 2021; Orel et al. 2022), also in light of the fact that the distribution of pathogenic traits within a single species (e.g., V. parahaemolyticus, V. vulnificus) carried by specific genes (e.g., tdh, trh, vcgC) is highly variable (Fabbro et al. 2010; Nigro et al. 2022).
Conclusions
Our workflow proved to be useful for a fine-level biodiversity analysis of specific target organisms in environmental studies based on amplicon sequencing. This approach represents a further step toward building a shared, updated, and curated phylogeny of the Vibrionaceae family that could be applied to new and old data for a continuous improvement of molecular tools (Kublanovskaya et al. 2019; Roush et al. 2021). It allows the capitalization of data generated at LTER sites or within genomic observatories that use 16S rRNA to study the prokaryotic communities. The construction of smaller, ad hoc reference databases has been proved useful in enhancing the 16S rRNA gene amplicon sequencing taxonomic resolution, reducing the massive size of the search space, and lowering competition among similar DNA sequences (Ritari et al. 2015).
In the near future, the third generation of sequencing platforms such as Pacific Biosciences (Rhoads and Au 2015) and Oxford Nanopore Technologies (Jain et al. 2018) that allow for longer (up to thousands kb) read length will become more and more common. Even if their error rate is still high (Fu et al. 2019), their contribution will be useful in the perspective of constructing consensus sequences covering the full length of barcode regions, combining short and long reads.
Even if our study was focused on seawater, our approach is also applicable to other target matrices such as sediment and marine metazoan microbiomes (e.g., fish gut), where Vibrionaceae play a significant role.
Access to reliable and fine-level taxonomic data is essential not only for the assessment of the diversity and the composition of the microbial communities, but also for ecological research. The possibility to link specific taxonomic groups with biological, chemical, and physical parameters is fundamental to understand the behavior and the response of microbial assemblages to environmental changes.
Data availability
The sequences generated for this study can be found in the Sequence Reads Archive (SRA) at NCBI under the accession numbers PRJNA767222, PRJNA818117, PRJNA818144, PRJNA818839, and PRJNA818858.
References
Altschul SF, Madden TL, Schäffer A et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. https://doi.org/10.1093/nar/25.17.3389
Amin AKMR, Feng G, Al-Saari N et al (2016) The first temporal and spatial assessment of Vibrio diversity of the surrounding seawater of coral reefs in Ishigaki. Japan Front Microbiol 7:1185. https://doi.org/10.3389/fmicb.2016.01185
Amplicon PCR, Clean-Up PCR, Index PCR (2013) 16S metagenomic sequencing library preparation. https://www.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf
Ashok Kumar J, Vinaya Kumar K, Avunje S et al (2020) Phylogenetic relationship among brackishwater Vibrio species. Evol Bioinform Online 16:1176934320903288. https://doi.org/10.1177/1176934320903288
Ast JC, Urbanczyk H, Dunlap PV (2009) Multi-gene analysis reveals previously unrecognized phylogenetic diversity in Aliivibrio. Syst Appl Microbiol 32:379–386. https://doi.org/10.1016/j.syapm.2009.04.005
Austin B, Zhang XH (2006) Vibrio harveyi: a significant pathogen of marine vertebrates and invertebrates. Lett Appl Microbiol 43:119–124. https://doi.org/10.1111/j.1472-765X.2006.01989.x
Baker-Austin C, Oliver JD, Alam M et al (2018) Vibrio spp. infections. Nat Rev Dis Primers 4:1–19. https://doi.org/10.1038/s41572-018-0005-8
Balaban M, Sarmashghi S, Mirarab S (2020) APPLES: scalable distance-based phylogenetic placement with or without alignments. Syst Biol 69:566–578. https://doi.org/10.1093/sysbio/syz063
Balvočiūtė M, Huson DH (2017) SILVA, RDP, Greengenes, NCBI and OTT—how do these taxonomies compare? BMC Genomics 182:1–8. https://doi.org/10.1186/s12864-017-3501-4
Barbera P, Kozlov AM, Czech L et al (2019) EPA-ng: massively parallel evolutionary placement of genetic sequences. Syst Biol 682:365–369. https://doi.org/10.1093/sysbio/syy054
Baron S, Larvor E, Chevalier S et al (2017) Antimicrobial susceptibility among urban wastewater and wild shellfish isolates of non-O1/Non-O139 Vibrio cholerae from La Rance Estuary (Brittany. France Front Microbiol 8:1637. https://doi.org/10.3389/fmicb.2017.01637
Berger SA, Stamatakis A (2012) PaPaRa 2.0: a vectorized algorithm for probabilistic phylogeny-aware alignment extension. Heidelberg Institute for Theoretical Studies 12.
Berger SA, Stamatakis A (2011) Aligning short reads to reference alignments and trees. Bioinformatics 27:2068–2075. https://doi.org/10.1093/bioinformatics/btr320
Bokulich NA, Dillon MR, Zhang Y, et al (2018) q2-longitudinal: longitudinal and paired-sample analyses of microbiome data. mSystems 3:e00219–18. https://doi.org/10.1128/mSystems.00219-18
Bolyen E, Rideout JR, Dillon MR et al (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. https://doi.org/10.1038/s41587-019-0209-9
Burtseva O, Kublanovskaya A, Baulina O, Fedorenko T, Lobakova E, Chekanov K (2021) The strains of bioluminescent bacteria isolated from the White Sea finfishes: genera Photobacterium, Aliivibrio, Vibrio, Shewanella, and first luminous Kosakonia. J Photochem Photobiol B 208:111895. https://doi.org/10.1016/j.jphotobiol.2020.111895
Callahan BJ, McMurdie PJ, Rosen MJ et al (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Cano-Gomez A, Høj L, Owens L, Andreakis N (2011) Multilocus sequence analysis provides basis for fast and reliable identification of Vibrio harveyi-related species and reveals previous misidentification of important marine pathogens. Syst Appl Microbiol 34:561–565. https://doi.org/10.1016/j.syapm.2011.09.001
Celussi M, Cataletto B (2007) Annual dynamics of bacterioplankton assemblages in the Gulf of Trieste (Northern Adriatic Sea). Gene 406:113–123. https://doi.org/10.1016/j.gene.2007.07.010
Celussi M, Malfatti F, Ziveri P, Giani M, Del Negro P (2017) Uptake-release dynamics of the inorganic and organic carbon pool mediated by planktonic prokaryotes in the deep Mediterranean Sea. Environ Microbiol 19:1163–1175. https://doi.org/10.1111/1462-2920.13641
Celussi M, Quero GM, Zoccarato L et al (2018) Planktonic prokaryote and protist communities in a submarine canyon system in the Ligurian Sea (NW Mediterranean). Prog Oceanog 168:210–221. https://doi.org/10.1016/j.pocean.2018.10.002
Chen X, Zhao H, Jiang G et al (2020) Responses of free-living vibrio community to seasonal environmental variation in a Subtropical Inland Bay. Front Microbiol 3069. https://doi.org/10.3389/fmicb.2020.610974
Cole JR, Chai B, Farris R et al (2007) The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res 35:169–172. https://doi.org/10.1093/nar/gkl889
Czech L, Stamatakis A, Dunthorn M, Barbera P (2022) Metagenomic analysis using phylogenetic placement - a review of the first decade. Front Bioinform 2022. https://doi.org/10.3389/fbinf.2022.871393
Deiner K, Bik HM, Machler E et al (2017) Environmental DNA metabarcoding: transforming how we survey animal and plant communities. Mol Ecol 26:5872–5895. https://doi.org/10.1111/mec.14350
Earl JP, Adappa ND, Krol J et al (2018) Species-level bacterial community profiling of the healthy sinonasal microbiome using Pacific Biosciences sequencing of full-length 16S rRNA genes. Microbiome 6:1–26. https://doi.org/10.1186/s40168-018-0569-2
Edgar R (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Edgar R (2018) Taxonomy annotation and guide tree errors in 16S rRNA databases. PeerJ 6:e5030. https://doi.org/10.7717/peerj.5030
Egerton S, Culloty S, Whooley J, Stanton C, Ross RP (2018) The gut microbiota of marine fish. Front Microbiol 9:873. https://doi.org/10.3389/fmicb.2018.00873
Elferink S, Neuhaus S, Wohlrab S et al (2017) Molecular diversity patterns among various phytoplankton size-fractions in West Greenland in late summer. Deep Sea Res Part I 121:54–69. https://doi.org/10.1016/j.dsr.2016.11.002
Fabbro C, Cataletto B, Del Negro P (2010) Detection of pathogenic Vibrio parahaemolyticus through biochemical and molecular-based methodologies in coastal waters of the Gulf of Trieste (North Adriatic Sea). FEMS Microbiol Lett 3072:158–164. https://doi.org/10.1111/j.1574-6968.2010.01969.x
Fabbro C, Celussi M, Russell H, Del Negro P (2012) Phenotypic and genetic diversity of coexisting Listonella anguillarum, Vibrio harveyi and Vibrio chagassi recovered from skin haemorrhages of diseased sand smelt, Atherina boyeri, in the Gulf of Trieste (NE Adriatic Sea). Lett Appl Microbiol 542:153–159. https://doi.org/10.1111/j.1472-765X.2011.03188.x
Farmer JJ, Janda JM, Brenner FW et al (2005) Genus I. Vibrio Pacini 1854. Bergey’s Manual of Systematic Bacteriology. Springer, New York, pp 494–546
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376. https://doi.org/10.1007/BF01734359
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. https://doi.org/10.1111/j.1558-5646.1985.tb00420.x
Fonti V, Di Cesare A, Šangulin J, Del Negro P, Celussi M (2021) Antibiotic resistance genes and potentially pathogenic bacteria in the central Adriatic Sea: are they connected to urban wastewater inputs? Water 1323:3335. https://doi.org/10.3390/w13233335
Frans I, Michiels CW, Bossier P, Willems KA, Lievens B, Rediers H (2011) Vibrio anguillarum as a fish pathogen: virulence factors, diagnosis and prevention. J Fish Dis 34:643–661. https://doi.org/10.1111/j.1365-2761.2011.01279.x
Franzo A, Cibic T, Del Negro P, Solidoro C (2014) Microphytobenthic response to mussel farm biodeposition in coastal sediments of the northern Adriatic Sea. Mar Pollut Bull 79:379–388. https://doi.org/10.1016/j.marpolbul.2013.11.002
Fu S, Wang A, Au KF (2019) A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol 20:1–17. https://doi.org/10.1186/s13059-018-1605-z
Fuhrman JA, Cram JA, Needham DM (2015) Marine microbial community dynamics and their ecological interpretation. Nat Rev Microbiol 13:133–146. https://doi.org/10.1038/nrmicro3417
Gabriel MW, Matsui GY, Friedman R, Lovell CR (2014) Optimization of multilocus sequence analysis for identification of species in the genus Vibrio. Appl Environ Microbiol 17:5359. https://doi.org/10.1128/AEM.01206-14
Gao X, Pi D, Chen N et al (2018) Survival, virulent characteristics, and transcriptomic analyses of the pathogenic Vibrio anguillarum under starvation stress. Front cell infect microbiol 389. https://doi.org/10.3389/fcimb.2018.00389
Gilbert JA, Steele JA, Caporaso JG et al (2012) Defining seasonal marine microbial community dynamics. ISME J 6:298–308. https://doi.org/10.1038/ismej.2011.107
Gladney LM, Katz LS, Knipe KM et al (2014) Genome sequences of Vibrio navarrensis, a potential human pathogen. Genome Announc 2:e01188-e1214. https://doi.org/10.1128/genomeA.01188-14
Gomez-Gil B, Thompson CC, Matsumura Y et al (2014) The Family Vibrionaceae. Prokaryotes 9:659–747. https://doi.org/10.1007/978-3-642-38922-1_225
Goodwin KD, Thompson LR, Duarte B et al (2017) DNA sequencing as a tool to monitor marine ecological status. Front Mar Sci 4:107. https://doi.org/10.3389/fmars.2017.00107
Gottschling M, Czech L, Mahé F, Adl S, Dunthorn M (2021) The windblown: possible explanations for dinophyte DNA in forest soils. J Eukaryot Microbiol 68:e12833. https://doi.org/10.1111/jeu.12833
Goulden EF, Hall MR, Bourne DG, Pereg LL, Høj L (2012) Pathogenicity and infection cycle of Vibrio owensii in larviculture of the ornate spiny lobster (Panulirus ornatus). Appl Environ Microbiol 78:2841–2849. https://doi.org/10.1128/AEM.07274-11
Gwak HJ, Rho M (2020) Data-driven modeling for species-level taxonomic assignment from 16S rRNA: application to human microbiomes. Front microbiol 2866. https://doi.org/10.3389/fmicb.2020.570825
Hoffmann K, Bienhold C, Buttigieg PL et al (2020) Diversity and metabolism of Woeseiales bacteria, global members of marine sediment communities. ISME J 14:1042–1056. https://doi.org/10.1038/s41396-020-0588-4
Hsieh JL, Fries JS, Noble RT (2007) Vibrio and phytoplankton dynamics during the summer of 2004 in a eutrophying estuary. Ecol Appl 17:102–109
Iniesto M, Moreira D, Reboul G et al (2021) Core microbial communities of lacustrine microbialites sampled along an alkalinity gradient. Environ Microbiol 23:51–68. https://doi.org/10.1111/1462-2920.15252
Jain M, Koren S, Miga KH et al (2018) Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36:338–345. https://doi.org/10.1038/nbt.4060
Janssen S, McDonald D, Gonzalez A et al (2018) Phylogenetic placement of exact amplicon sequences improves associations with clinical information. Msystems 3:e00021-18. https://doi.org/10.1128/mSystems.00021-18
Jesser KJ, Noble RT (2018) Vibrio ecology in the Neuse river estuary, North Carolina, characterized by next-generation amplicon sequencing of the gene encoding Heat Shock Protein 60 (hsp60). Appl Environ Microbiol 84:e00333–18. https://doi.org/10.1128/AEM.00333-18
Johnson CN (2013) Fitness factors in Vibrios: a mini-review. Microb Ecol 65:826–851. https://doi.org/10.1007/s00248-012-0168-x
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Keck F, Vasselon V, Rimet F, Bouchez A, Kahlert M (2018) Boosting DNA metabarcoding for biomonitoring with phylogenetic estimation of operational taxonomic units’ ecological profiles. Mol Ecol Resour 18:1299–1309. https://doi.org/10.1111/1755-0998.12919
Kesy K, Labrenz M, Scales BS, Kreikemeyer B, Oberbeckmann S (2021) Vibrio colonization is highly dynamic in early microplastic-associated biofilms as well as on field–collected microplastics. Microorganisms 9:76. https://doi.org/10.3390/microorganisms9010076
Kharbush JJ, Close HG, Van Mooy BAS et al (2020) Particulate organic carbon deconstructed: molecular and chemical composition of particulate organic carbon in the ocean. Front Mar Sci 7:518. https://doi.org/10.3389/fmars.2020.00518
Kim EY, Kim YR, Kim DG, Kong IS (2012) A susceptible protein by proteomic analysis from Vibrio anguillarum under various environmental conditions. Bioprocess Biosyst Eng 35:273–282. https://doi.org/10.1007/s00449-011-0636-6
Kingsley DH (2014) Shellfish contamination and spoilage. Encyclopedia of Food Microbiology. Elsevier, Amsterdam, pp 389–396
Kublanovskaya A, Chekanov K, Solovchenko A, Lobakova E (2019) Cyanobacterial diversity in the algal–bacterial consortia from Subarctic regions: new insights from the rock baths at White Sea Coast. Hydrobiologia 830:17–31. https://doi.org/10.1007/s10750-018-3844-0
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547. https://doi.org/10.1093/molbev/msy096
Labella AM, Arahal DR, Castro D, Lemos ML, Borrego JJ (2017) Revisiting the genus Photobacterium: taxonomy, ecology and pathogenesis. Int Microbiol 20:1–10. https://doi.org/10.2436/20.1501.01.280
Lee L, Raghunath P (2018) Vibrionaceae diversity, multidrug resistance and management. Front Microbiol 9:563. https://doi.org/10.3389/fmicb.2018.00563
Letunic I, Bork P (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49:293–296. https://doi.org/10.1093/nar/gkab301
Li Z, Tian J, Lai Y, Lee CH, Cai Z, Yu CF (2020) Puffer fish gut microbiota studies revealed unique bacterial co-occurrence patterns and new insights on tetrodotoxin producers. Mar Drugs 18:278. https://doi.org/10.3390/md18050278
Liang X, Wang JS, Liu YZ et al (2019) Complete genome of a marine bacterium Vibrio chagasii ECSMB14107 with the ability to infect mussels. Mar Genomics 48:100683. https://doi.org/10.1016/j.margen.2019.04.008
Liu L, Xiao L, Zhang M et al (2018) A Vibrio owensii strain as the causative agent of AHPND in cultured shrimp, Litopenaeus vannamei. J Invert Pathol 153:156–164. https://doi.org/10.1016/j.jip.2018.02.005
López-Cortés A, Latisnere-Barragán H, García-Maldonado JQ, Martínez MA, Munguía-Vega A (2020) Intestinal microbiota analyses of Litopenaeus vannamei during a case of atypical massive mortality in Northwestern Mexico. Curr Microbiol 77:2312–2321. https://doi.org/10.1007/s00284-020-02079-z
Lorenzen CJ, Jeffrey SW (1980) Determination of chlorophyll in seawater. UNESCO Tech Pap Mar Sci 35:1–20
Ma Y, Wang Q, Xu W, Liu X, Gao X, Zhang Y (2017) Stationary phase-dependent accumulation of ectoine is an efficient adaptation strategy in Vibrio anguillarum against cold stress. Microbiol Res 205:8–18. https://doi.org/10.1016/j.micres.2017.08.005
Manna V, De Vittor C, Giani M, Del Negro P, Celussi M (2021) Long-term patterns and drivers of microbial organic matter utilization in the northernmost basin of the Mediterranean Sea. Mar Environ Res 164:105245. https://doi.org/10.1016/j.marenvres.2020.105245
Marie D, Brussaard CP, Thyrhaug R, Bratbak G, Vaulot D (1999) Enumeration of marine viruses in culture and natural samples by flow cytometry. Appl Environ Microbiol 65:45–52. https://doi.org/10.1128/AEM.65.1.45-52.1999
Matsen FA, Hoffman NG, Gallagher A, Stamatakis A (2012) A format for phylogenetic placements. PLoS ONE 72:e31009. https://doi.org/10.1371/journal.pone.0031009
Matteucci G, Schippa S, Di Lallo G, Migliore L, Thaller MC (2015) Species diversity, spatial distribution, and virulence associated genes of culturable vibrios in a brackish coastal Mediterranean environment. Ann Microbiol 65:2311–2321. https://doi.org/10.1007/s13213-015-1073-6
McMurdie PJ, Holmes S (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. https://doi.org/10.1371/journal.pone.0061217
Mena C, Balbín R, Reglero P, Martín M, Santiago R, Sintes E (2021) Dynamic prokaryotic communities in the dark western Mediterranean Sea. Sci Rep 11:17859. https://doi.org/10.1038/s41598-021-96992-3
Miller SD, Haddock SHD, Elvidge CD, Lee TF (2005) Detection of a bioluminescent milky sea from space. Proc Natl Acad Sci USA 102:14181–14184. https://doi.org/10.1073/pnas.0507253102
Mitsi K, Arroyo AS, Ruiz-Trillo I (2019) A global metabarcoding analysis expands molecular diversity of Platyhelminthes and reveals novel early-branching clades. Biol Lett 15:20190182. https://doi.org/10.1098/rsbl.2019.0182
Momba M, Azab El-Liethy M (2017) Vibrio cholerae and Cholera biotypes. Global water pathogen project. https://www.waterpathogens.org/sites/default/files/Vibrio%20cholerae%20and%20Cholera%20biotypes_0.pdf. Accessed 10 Apr 2022
Monsreal JF, Serralta-Peraza LEDS, Abuxapqui JJF (2021) Species of the genus Vibrio of clinical-epidemiological importance. MOJ Biol Med 6:116–125. https://doi.org/10.15406/mojbm.2021.06.00142
Nasi F, Auriemma R, Bonsdorff E (2017) Biodiversity, feeding habits and reproductive strategies of benthic macrofauna in a protected area of the northern Adriatic Sea: a three-year study. Mediterr Mar Sci 18:292–309. https://doi.org/10.12681/mms.1897
Nigro OD, James-Davis LTI, De Carlo EH, Li YH, Steward GF (2022) Variable freshwater influences on the abundance of Vibrio vulnificus in a tropical estuary. Appl Environ Microbiol 88:e01884-e1921. https://doi.org/10.1128/aem.01884-21
Novianty RI, Budiarti S (2014) Lytic bacteriophage for Photobacterium damselae isolated from water environment. Int J Innov Res Sci Engineer 2:549–553
Oliver JD, Pruzzo C, Vezzulli L, Kaper JB (2012) Vibrio species. Food microbiol 401-439. https://doi.org/10.1128/9781555818463.ch16
Orel N, Fadeev E, Klun K, Ličer M, Tinta T, Turk V (2022) Bacterial indicators are ubiquitous members of pelagic microbiome in anthropogenically impacted coastal ecosystem. Front Microbiol 12:765091. https://doi.org/10.3389/fmicb.2021.765091
Paliaga P, Korlević M, Ivančić I, Najdek M (2017) Limited influence of primary treated sewage waters on bacterial abundance, production and community composition in coastal seawaters. Mar Environ Res 131:215–226. https://doi.org/10.1016/j.marenvres.2017.09.012
Palit A, Nair NB (2013) Bacteria: Other Vibrios. In: Motarjemi Y, Moy G, Todd E (eds) Encyclopedia of Food Safety, vol 1. History. Science and Methods. Elsevier, London, pp 570–573
Parada AE, Needham DM, Fuhrman JA (2016) Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol 18:1403–1414. https://doi.org/10.1111/1462-2920.13023
Park SC, Won S (2018) Evaluation of 16S rRNA databases for taxonomic assignments using a mock community. Genomics Inform 16:e24. https://doi.org/10.5808/GI.2018.16.4.e24
Pella E, Colombo B (1973) Study of carbon, hydrogen and nitrogen determination by combustion-gas chromatography. Mikrochim Acta 61:697–719. https://doi.org/10.1007/BF01218130
Pretto T (2020) Vibrio harveyi group. In: Zrnic S (ed) Diagnostic Manual for the main pathogens in European seabass and Gilthead seabream aquaculture. CIHEAM, Zaragoza, pp 75–82
Pruesse E, Peplies J, Glöckner FO (2012) SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinf 28:1823–1829. https://doi.org/10.1093/bioinformatics/bts252
Quast C, Pruesse E, Yilmaz P et al (2012) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. nucleic Acid Res 41:590–596. https://doi.org/10.1093/nar/gks1219
R Core Team (2021) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Rajter L, Dunthorn M (2021) Ciliate SSU-rDNA reference alignments and trees for phylogenetic placements of metabarcoding data. Metabarcoding Metagenom 5:e69602. https://doi.org/10.3897/mbmg.5.69602
Ramamurthy T, Chowdhury G, PazhaniGP SS (2014) Vibrio fluvialis: an emerging human pathogen. Front Microbiol 5:91. https://doi.org/10.3389/fmicb.2014.00091
Reen FJ, Almagro-Moreno S, Ussery D, Boyd EF (2006) The genomic code: inferring Vibrionaceae niche specialization. Nat Rev Microbiol 4:697–704. https://doi.org/10.1038/nrmicro1476
Rhoads A, Au KF (2015) PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13:278–289. https://doi.org/10.1016/j.gpb.2015.08.002
Ritari J, Salojärvi J, Lahti L, de Vos WM (2015) Improved taxonomic assignment of human intestinal 16S rRNA sequences by a dedicated reference database. BMC Genom 16:1–10. https://doi.org/10.1186/s12864-015-2265-y
Rivera IN, Chun J, Huq A, Sack RB, Colwell RR (2001) Genotypes associated with virulence in environmental isolates of Vibrio cholerae. Appl Environ Microbiol 67:2421–2429. https://doi.org/10.1128/AEM.67.6.2421-2429.2001
Romalde JL, Diéguez AL, Lasa A, Balboa S (2014) New Vibrio species associated to molluscan microbiota: a review. Front Microbiol 4:413. https://doi.org/10.3389/fmicb.2013.00413
Roush D, Giraldo-Silva A, Garcia-Pichel F (2021) Cydrasil 3, a curated 16S rRNA gene reference package and web app for cyanobacterial phylogenetic placement. Sci Data 8:1–6. https://doi.org/10.1038/s41597-021-01015-5
Rubio-Portillo E, Yarza P, Penalver C, Ramos-Esplá AA, Anton J (2014) New insights into Oculina patagonica coral diseases and their associated Vibrio spp. communities. ISME J 8:1794–1807. https://doi.org/10.1038/ismej.2014.33
Sampaio A, Silva V, Poeta P, Aonofriesei F (2022) Vibrio spp.: life strategies, ecology, and risks in a changing environment. Drivers 14:97. https://doi.org/10.3390/d14020097
Sanches-Fernandes GM, Califano G, Keller-Costa T et al (2021) Draft genome sequence of Vibrio chagasii 18LP, isolated from gilthead seabream (Sparus aurata) larvae reared in aquaculture. Microbiol Resour Announc 10:e00658-e721. https://doi.org/10.1128/MRA.00658-21
Schlitzer R (2018) Ocean data view. https://odv.awi.de
Schwartz K, Kukuc C, Bier N, Taureck K, Hammerl JA, Strauch E (2017) Diversity of Vibrio navarrensis revealed by genomic comparison: veterinary isolates are related to strains associated with human illness and sewage isolates while seawater strains are more distant. Front Microbiol 8:1717. https://doi.org/10.3389/fmicb.2017.01717
Sharp JH (1974) Improved analysis for “particulate” organic carbon and nitrogen from seawater. Limnol Oceanogr 19:984–989. https://doi.org/10.4319/lo.1974.19.6.0984
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinf 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stewart JR, Gast RJ, Fujioka RS, Solo-Gabriele HM et al (2008) The coastal environment and human health: microbial indicators, pathogens, sentinels and reservoirs. Environ Health 72:1–14. https://doi.org/10.1186/1476-069X-7-S2-S3
Taberlet P, Coissac E, Hajibabaei M, Rieseberg LH (2012) Environmental DNA. Mol Ecol 21:1789–1793. https://doi.org/10.1111/j.1365-294X.2012.05542.x
Takemura AF, Chien DM, Polz MF (2014) Associations and dynamics of Vibrionaceae in the environment, from the genus to the population level. Front Microbiol 5:38. https://doi.org/10.3389/fmicb.2014.00038
Teng W, Li W, Zhang M et al (2012) Isolation, identification and pathogenicity of Vibrio chagasii from Patinopecten yessoensis. J Fish China 36:937–943. https://doi.org/10.3724/SP.J.1231.2012.27674
Terceti MS, Vences A, Matanza XM, Dalsgaard I, Pedersen K, Osorio CR (2018) Molecular epidemiology of Photobacterium damselae subsp. damselae outbreaks in marine rainbow trout farms reveals extensive horizontal gene transfer and high genetic diversity. Front Microbiol 9:2155. https://doi.org/10.3389/fmicb.2018.02155
Thompson JR, Polz MF (2006) Dynamics of Vibrio populations and their role in environmental nutrient cycling. Fabiano L, Thompson BA. Swings J The biology of Vibrios. Wiley, New York, pp 190–203
Thompson FL, Iida T, Swings J (2004) Biodiversity of vibrios. Microbiol Mol Biol Rev 68:403–431. https://doi.org/10.1128/MMBR.68.3.403-431.2004
Tinta T, Vojvoda J, Mozetič P et al (2015) Bacterial community shift is induced by dynamic environmental parameters in a changing coastal ecosystem (northern Adriatic, northeastern Mediterranean Sea) –a 2-year time-series study. Environ Microbiol 17:3581–3596. https://doi.org/10.1111/1462-2920.12519
Tinta T, Zhao Z, Escobar A et al (2020) Microbial processing of jellyfish detritus in the ocean. Front Microbiol 11:590995. https://doi.org/10.3389/fmicb.2020.590995
Urbanczyk H, Ast JC, Kaeding AJ, Oliver JD, Dunlap PV (2008) Phylogenetic analysis of the incidence of lux gene horizontal transfer in Vibrionaceae. J Bacteriol 190:3494–3504. https://doi.org/10.1128/JB.00101-08
Urdaci MC, Marchand M, Ageron E, Arcos JM, Sesma B, Grimont PA (1991) Vibrio navarrensis sp. nov., a species from sewage. Int J Syst Evol 412:290–294. https://doi.org/10.1099/00207713-41-2-290
Van de Peer Y, Salemi M (2009) Phylogenetic inference based on distance methods. In: Lemey P, Salemi M, Vandamme A (eds) The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing. Cambridge University Press, Cambridge, pp 142–180
Vergin KL, Done B, Carlson C, Giovannoni SJ (2013) Spatiotemporal distributions of rare bacterioplankton populations indicate adaptive strategies in the oligotrophic ocean. Aquat Microb Ecol 71:1–13. https://doi.org/10.3354/ame01661
Vezzulli L, Pezzati E, Stauder M, Stagnaro L, Venier P, Pruzzo C (2015) Aquatic ecology of the oyster pathogens Vibrio splendidus and Vibrio aestuarianus. Environ Microbiol 17:1065–1080. https://doi.org/10.1111/1462-2920.12484
Vezzulli L, Grande C, Reid PC, Hélaouët P, Edwards M, Höfle MG (2016) Climate influence on Vibrio and associated human diseases during the past half-century in the coastal North Atlantic. Proc Natl Acad Sci USA 113:5062–5071. https://doi.org/10.1073/pnas.1609157113
Westrich JR, Ebling AM, Landing WM et al (2016) Saharan dust nutrients promote Vibrio bloom formation in marine surface waters. Proc Natl Acad Sci USA 113:5964–5969. https://doi.org/10.1073/pnas.1518080113
Wickham H, Averick M, Bryan J et al (2019) Welcome to the tidyverse. J Open Source Softw 4:1686. https://doi.org/10.21105/joss.01686
Wilson JM, Chamberlain E, Erazo N, Carter ML, Bowman JS (2021) Recurrent microbial community types driven by nearshore and seasonal processes in coastal Southern California. Environ Microbiol 23:3225–3239. https://doi.org/10.1111/1462-2920.15548
Wong YY, Lee C, Bong C et al (2019) Environmental control of Vibrio spp. abundance and community structure in tropical waters. FEMS Microbiol Ecol 95:fiz76. https://doi.org/10.1093/femsec/fiz176
Yarza P, Yilmaz P, Pruesse E et al (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635–645. https://doi.org/10.1038/nrmicro3330
Zhang XH, Lin H, Wang X, Austin B (2018) Significance of Vibrio species in the marine organic carbon cycle-a review. Sci China Earth Sci 61:1357–1368. https://doi.org/10.1007/s11430-017-9229-x
Acknowledgements
The authors would like to thank all the personnel involved in field activities: M. Bazzaro and F. Relitti for Chl a and POC data, respectively, M. Kralj and G. Zazo for CTD data, and C. Balestra for flow cytometry data. We acknowledge the two reviewers, whose constructive comments improved the quality of this manuscript.
Funding
Open access funding provided by Istituto Nazionale di Oceanografia e di Geofisica Sperimentale within the CRUI-CARE Agreement. This study has been developed in the framework of the Interreg MED Strategic Project SHAREMED, co-financed by the European Regional Development Fund under the Funding Programme Interreg MED 2014–2020. Website: https://sharemed.interreg-med.eu/.
The study was partially supported by funds from the Interreg Italy-Croatia project AdSWiM (managed use of treated urban wastewater for the quality of the Adriatic Sea), ID 10,046,144, and benefited from CINECA ISCRA C resources (HP10CS2I1S; PI: EB).
Author information
Authors and Affiliations
Contributions
MC conceptualized the study. EB, VM, and VF performed the analyses. EB, VM, VF, CF, and MC processed the data. EB wrote the manuscript with the help and inputs of all co-authors.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Robert Duran
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Banchi, E., Manna, V., Fonti, V. et al. Improving environmental monitoring of Vibrionaceae in coastal ecosystems through 16S rRNA gene amplicon sequencing. Environ Sci Pollut Res 29, 67466–67482 (2022). https://doi.org/10.1007/s11356-022-22752-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-022-22752-z