
Abstract
Purpose
Over the past 20 years, neuroinflammation (NI) has increasingly been recognised as having an important role in many neurodegenerative diseases, including Alzheimer’s disease. As such, being able to image NI non-invasively in patients is critical to monitor pathological processes and potential therapies targeting neuroinflammation. The translocator protein (TSPO) has proven a reliable NI biomarker for positron emission tomography (PET) imaging. However, if TSPO imaging in acute conditions such as stroke provides strong and reliable signals, TSPO imaging in neurodegenerative diseases has proven more challenging. Here, we report results comparing the recently developed TSPO tracers [18F]GE-180 and [18F]DPA-714 with (R)-[11C]PK11195 in a rodent model of subtle focal inflammation.
Procedures
Adult male Wistar rats were stereotactically injected with 1 μg lipopolysaccharide in the right striatum. Three days later, animals underwent a 60-min PET scan with (R)-[11C]PK11195 and [18F]GE-180 (n = 6) or [18F]DPA-714 (n = 6). Ten animals were scanned with either [18F]GE-180 (n = 5) or [18F]DPA-714 (n = 5) only. Kinetic analysis of PET data was performed using the simplified reference tissue model (SRTM) with a contralateral reference region or a novel data-driven input to estimate binding potential BPND. Autoradiography and immunohistochemistry were performed to confirm in vivo results.
Results
At 40–60 min post-injection, [18F]GE-180 dual-scanned animals showed a significantly increased core/contralateral uptake ratio vs. the same animals scanned with (R)-[11C]PK11195 (3.41 ± 1.09 vs. 2.43 ± 0.39, p = 0.03); [18]DPA-714 did not (2.80 ± 0.69 vs. 2.26 ± 0.41). Kinetic modelling with a contralateral reference region identified significantly higher binding potential (BPND) in the core of the LPS injection site with [18F]GE-180 but not with [18F]DPA-714 vs. (R)-[11C]PK11195. A cerebellar reference region and novel data-driven input to the SRTM were unable to distinguish differences in tracer BPND.
Conclusions
Second-generation TSPO-PET tracers are able to accurately detect mild-level NI. In this model, [18F]GE-180 shows a higher core/contralateral ratio and BPND when compared to (R)-[11C]PK11195, while [18F]DPA-714 did not.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammation is known to play a role in the development and progression of a wide variety of neuropathological conditions, including stroke [1], multiple sclerosis [2] and neurodegenerative diseases [3, 4]. Sensitive and spatially accurate imaging of neuroinflammation (NI) in vivo, however, has long been a challenge. Microglia, which are resident immune cells of the brain, are in a state of ramification and constantly probing against potential insult or injury, even in the healthy brain [5–7]. Once activated, they begin to alter their morphology, becoming increasingly macrophage-like and releasing pro-inflammatory cytokines such as interleukin-1 (IL-1) and tumour necrosis factor-α (TNF-α). Whether this cytokine cascade exacerbates damage or helps to restore equilibrium conditions varies depending on the nature of the insult and with time [8]. The 18 kDa translocator protein (TSPO), a mitochondrial protein which is primarily located in the membranes of mitochondria [9], is expressed at a low level in the healthy mammalian brain. Although the true role of TSPO in NI remains to be fully understood, an increase in expression of the protein is well established as a correlate with the level of microglial activation, proliferation and/or level of infiltrated macrophages, and to some extent astrogliosis [2, 10–12].
Positron emission tomography (PET) studies of neuroinflammation have been conducted for over 30 years. The original and still most widely used radioligand for TSPO in clinical imaging is the C-11 labelled R-enantiomer of 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline carboxamide, (R)-[11C]PK11195. Despite its application to a wide range of diseases in the clinic, the sensitivity and quantification of this tracer have been hampered by its high level of non-specific binding and low free fraction in plasma in humans, which lead to a poor signal to noise ratio [13]. Additionally, labelling of the ligand with C-11 requires an on-site cyclotron and its radiosynthesis is well-known to be troublesome. In recent years, therefore, efforts have been made to develop and characterise so-called ‘second generation’ TSPO ligands with lower non-specific binding, which have exhibited varying levels of success in clinical and preclinical studies. Unfortunately, and unlike (R)-[11C]PK11195, second-generation TSPO ligands are affected by a single-nucleotide polymorphism (rs6971, Ala147Thr) in the TSPO gene which results in variable binding affinity in humans [14] but which has never been reported in animals. One such ligand is N,N-diethyl-2-[4-(2-[18F]fluoroethoxy)phenyl]-5,7-dimethyl-pyrazolo[1,5-a]pyrimidine-3-acetamide ([18F]DPA-714) [15], which was initially evaluated in vivo in a rodent model of acute neuroinflammation using α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) [16] and then in models of stroke and Herpes encephalitis [17, 18]. Compared to (R)-[11C]PK11195, the tracer showed lower non-specific binding and improved bioavailability in brain tissue in the AMPA and stroke models, leading to a better signal to noise ratio than (R)-[11C]PK11195; however, this was not the case in the Herpes encephalitis model. Moreover, a more recent preclinical study using [18F]DPA-714 in a longitudinal model of HIV-1 in transgenic rats showed no significant group differences in tracer uptake between diseased and wild-type animals [19]. Altogether, these reports support the idea that lower and diffuse NI as observed in neurodegenerative diseases is more variable and difficult to detect [16–18]. Nevertheless, the initial evaluation in humans also suggests that this tracer is a promising TSPO ligand [20] and recently a large clinical study in Alzheimer’s disease (AD) patients showed that neuroinflammation was present in both prodromal and confirmed AD patients [21]. Another second generation tracer, S-N,N-diethyl-9-[2-[18F]fluoroethyl]-5-methoxy 2,3,4,9-tetrahydro-1H-carbazole-4-carboxamide ([18F]GE-180), has been used in in vitro, ex vivo and in vivo studies on the detection of microglial activation in acute models of NI [22, 23]. Both studies found an improved signal to noise ratio of [18F]GE-180 over (R)- [11C]PK11195, but both also used models that induced strong and acute NI (i.e., high dose of lipopolysaccharide (LPS) and stroke) and not the more challenging low/mild NI. Another study in a transgenic AD mouse model, however, did identify increased binding in aged wild-type and AD animals, although this result was semi-quantitative and did not directly compare the behaviour of [18F]GE-180 with other tracers [24].
As mentioned above, most of the previous preclinical studies testing second-generation TSPO tracers have primarily used either acute excitotoxic lesion models or models of stroke relevant to the strong levels of NI observed in stroke or brain trauma. In neurodegenerative diseases and models of neurodegeneration, however, the level of TSPO expression is much lower and more widespread than in focal lesions [25–27] and is thus more difficult to detect and quantify. Hence, there is a need to develop an easy-to-perform, robust preclinical model, with low levels of TSPO expression that are clinically relevant to those observed in neurodegeneration. To this end, we performed a comparative evaluation of (R)-[11C]PK11195, [18F]DPA-714 and [18F]GE-180 in rodents injected in the striatum with a low dose (1 μg) of LPS that induces a lower level of neuroinflammation when compared to previously published doses (50 μg [28]), and animals were scanned 3 days post-injection which is after the peak of neuroinflammation induced by LPS between 6 and 12 h post-injection [22, 28].
Kinetic analysis and modelling of preclinical PET data is difficult; as well as being technically challenging, arterial sampling of rodents is terminal, while reference tissue models using the contralateral hemisphere [16, 29] as input are hampered by the fact that the region may not be devoid of specific binding in certain disease models. Moreover, Folkersma et al. [30] suggest that PET data from any disease model where blood brain barrier (BBB) damage is present cannot be quantified using a reference tissue approach with anatomical reference input. Many studies have resorted to semi-quantitation, including use of standardised uptake values (SUVs) [31] and lesion-to-background ratios [32]. There is no consensus over tracer or model-specific quantification approaches, but previous studies involving a unilateral effect have used the cerebellum [33] or a contralateral reference region [22, 23] together with the simplified reference tissue model (SRTM) or MRTM (multilinear reference tissue model) [34]. However, there is, to date, no such evaluation in a model of low-level inflammation where the target vs. the contralateral reference region is lower and identification and quantification of the lesion is more challenging.
Indeed, due to the presence of potentially widespread brain inflammation, these anatomically defined reference regions may also contain TSPO in disease. Therefore, data-driven methods have previously been employed in clinical (R)-[11C]PK11195 brain PET studies to extract reference tissue kinetics in place of anatomically defined regions (see “Methods” section for more details and [35] for a review). Here, we sought to adapt one such approach [36] for the analysis of rodent PET brain scans in our model of modified LPS injection with the three different TSPO tracers.
Thus, the overall aims of this study were to (i) assess the performance of second-generation TSPO tracers [18F]GE-180 and [18F]DPA-714, directly compared to (R)-[11C]PK11195 in a modified LPS model of mild-level neuroinflammation and (ii) test various quantification approaches in this model of NI.
Materials and Methods
Tracer Synthesis
(R)-[11C]PK11195, [18F]DPA-714 and [18F]GE-180 (Fig. 1) were synthesised as described elsewhere [15, 23, 37–39]. Briefly, (R)-[11C]PK11195 was formed via N-[11C]methylation of an (R)-demethyl precursor [37, 40]; [18F]GE-180 was synthesised on a FASTlab™ platform by nucleophilic fluorination of an S-enantiomer mesylate precursor [39, 41] and [18F]DPA-714 by nucleophilic aliphatic substitution of a tosylate precursor [15].

Chemical structures of TSPO PET radiotracers
Animals
Thirty-seven adult male Wistar rats (396 ± 52 g, Charles River, Margate, Kent, UK) were used in this study: 22 received an LPS injection, 2 received an AMPA injection, 5 naïve (no stereotactic injection) rats were scanned at baseline as controls with [18F]GE-180 (data re-analysed from that previously published [23]), 4 with [18F]DPA-714 and 4 with (R)-[11C]PK11195. All procedures were carried out in accordance with the Animals (Scientific Procedures) Act 1986, and the project was approved specifically by the UK Home Office. Animals were kept under a 12-h light-dark cycle with free access to food and water.
LPS and AMPA Administration
For all procedures, animals were anaesthetised by isoflurane inhalation (induction 5 % and thereafter 2–2.5 %) in O2/NO2 (30 %/70 %). Animals underwent a stereotactic injection of 1 μg lipopolysaccharide (lipopolysaccharide from Escherichia coli 055:B5, Sigma, ref. L2880, Lot 102M4017V) or 7.5 nmol (0.5 μl of a 15 mM solution) of AMPA (n = 2) in the right striatum via a craniotomy (bregma +0.7 mm, lateral −3.0 mm, depth 5.5 mm from the surface of the brain) using a 2 μl Neuros™ microsyringe (Hamilton, USA) and micropump (injection rate 0.5 μl/min, UltraMicroPump II® and Micro4® Controller, WPI Inc., USA). Animals were maintained normothermic (body temperature 36.5 ± 0.5 °C, mean ± SD) during the surgery through the use of a heating blanket (Homeothermic Blanket Control Unit, Harvard Apparatus Limited®, Edenbridge, Kent, UK).
Scanning Protocol
Supplementary Fig. 1 presents a summary of study design; full details of the tracer injected doses, specific activities and purities can be found in Supplementary Table 1. Animals were split into four groups: those scanned sequentially with (R)-[11C]PK11195 then [18F]GE-180 (group 1, n = 6) or (R)-[11C]PK11195 then [18F]DPA-714 (group 2, n = 6) and those scanned with only [18F]GE-180 (group 3, n = 5) or [18F]DPA-714 (group 4, n = 5). In groups 1 and 2, animals were scanned on the same day with (R)-[11C]PK11195 in the morning and an F-18 labelled tracer in the afternoon. Supplementary Fig. 2 shows a correlation plot between specific activity and percentage injected dose of tracer in the inflammatory core at 40–60 min. Although the range of specific activities was quite broad, especially for (R)-[11C]PK11195, this did not significantly affect brain uptake (Figs. 2, 3, 4 and Supplementary Fig. 2).

Average core and contralateral tissue time activity curves (TACs) of LPS-injected animals that underwent dual scans with each tracer are shown. TACs are for a (R)-[11C]PK11195[ 18 F]GE-180, b [18F]GE-180, c (R)-[11C]PK11195[ 18 F]DPA-714 and d [18F]DPA-714 over 60 min duration of scans. Core/contralateral ratios indicated on the figures are calculated over the 40–60 min time frame. Dual scan [18F]GE-180 core/contralateral ratios were significantly higher than e (R)-[11C]PK11195 (p = 0.03), while f [18F]DPA-714 values were not (Wilcoxon signed rank, p < 0.05). Individual animals are represented by different symbols, which correspond between (R)-[11C]PK11195 and the respective F-18 tracer. Data are presented as mean ± SD

Average TACs for cerebellum (black) and contralateral (grey) reference regions for (R)-[11C]PK11195, [18F]GE-180 and [18F]DPA-714 in LPS animals. At 40–60 min, the cerebellar uptake (%ID/cm3) was significantly higher for all three tracers (p = 0.001) than the contralateral uptake

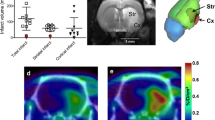
Co-registered (sum 40–60 min) PET/CT images of representative animals dual-scanned with a and c (R)-[11C]PK11195 followed by either b [18F]GE-180 (top) or d [18F]DPA-714 (bottom). Inset top: example of regions defined by automatic segmentation. Inset bottom: [18F]DPA-714 autoradiographic image showing increased specific uptake in the right striatum
All animals were anaesthetised by isoflurane inhalation (induction 5 % and thereafter 2–2.5 %) in O2/NO2 (30 %/70 %) at 3 days post-LPS injection. All tracers were injected intravenously as a bolus through a tail vein. They were then scanned on a Siemens Inveon® small animal PET-CT as described previously [23]. The following data acquisition protocol was used: a CT scan was performed immediately prior to the PET scan for each animal to acquire attenuation correction factors. The time coincidence window was set to 3.432 ns and levels of energy discrimination to 350 and 650 keV. List mode data from emission scans were histogrammed into 16 dynamic frames (5 × 1 min, 5 × 2 min, 3 × 5 min and 3 × 10 min) and emission sinograms were normalised, corrected for attenuation, scattering and radioactivity decay and reconstructed using OSEM3D (16 subsets, 4 iterations) into 128 × 128 × 159 images with 0.776 × 0.776 × 0.796 mm3 voxel size. Respiration and temperature were monitored throughout the scans using a pressure-sensitive pad and rectal probe (BioVet, M2M Imaging Corp, USA). Body temperature was maintained through the use of a heating and fan module controlled by the rectal probe via the interface of the BioVet system.
At the end of the PET scan, rats were rapidly decapitated and brains removed and frozen immediately in isopentane in dry ice. Brains were stored at −80 °C before being cut with a cryomicrotome into adjacent 20 μm coronal slices. Sections were then stored again at −80 °C until immunohistochemistry and autoradiography were performed.
Image Analysis
Images were segmented automatically using local means analysis (LMA) in the BrainVisa/Anatomist framework (http://brainvisa.info, [42–44]). Automatic segmentation was preferred to manually drawn regions of interest (ROIs), which can introduce a user-dependent bias. In short, the LMA algorithm first extracts the whole body of the rodent from the background before identifying voxels in the core of organs based on the notion that their variation in PET signal should be lower than those voxels at the organ borders, which are more subject to variations driven by physiological movement and spillover from neighbouring organs/vessels. Neighbouring voxels are then identified based on the similarity of their kinetics to these organ ‘cores.’ Ultimately, the implementation in BrainVisa segments the whole body image into 200 regions based on the similarity of kinetics of the voxels contained within them. Within the brain (delineated using the CT images), segmented ROIs were then manually selected and labelled under the following scheme: (1) core of the LPS lesion (covering the right striatum and/or the region with the highest uptake), (2) edge 1 (ROI around the core with the second highest uptake of tracer), (3) edge 2 (ROI with the third highest uptake), (4) a contralateral region (ROI with the lowest uptake, used as a reference region in kinetic analysis), (5) cerebellum and (6) skull edges (voxels on the edges of the skull taken to be noise and/or spillover from regions outside the brain, not included in the data analysis) (see top-right corner inset Fig. 4). Where applicable, a region of fourth highest uptake (edge 3) was also included in ROI segmentation. In the control animals, regions of high and low brain uptakes, as well as cerebellum and skull edges, were defined. ROIs’ volumes were similar for all tracers (core 86 ± 20 mm3, edge 1 202 ± 3 mm3, edge 2 498 ± 114 mm3, edge 3 268 ± 31 mm3 and contralateral 217 ± 16 mm3, with a maximum variation in ROIs’ volumes across tracers of 23 %).
The previous work published by our group has presented results with partial volume correction (PVC). Although in our previous study with high amplitude response in TSPO expression, group differences were not affected by PVC being applied or not [23], for low level NI models PVC might actually reduce the signal to background ratio and therefore damp the PET signal. This is due to PVC inherently amplifying noisy, high-frequency signals to reduce spill-over effects. Therefore, in our model of low/mild NI, applying PVC might be somewhat counter-productive. Having analysed results with and without PVC, here we selected to present raw (uncorrected) data, which showed consistent group differences between tracers.
Data-Driven Method for the Extraction of Reference Tissue Kinetics
The method by Yaqub et al. [36] is a successor to that by Turkheimer et al. [45], using a pre-defined brain mask to segment all masked voxel PET time activity curves (TACs) into four classes (grey matter with specific binding, grey matter without specific binding, white matter and blood). Subsequently, in dynamic PET data, the reference tissue is selected as the tissue class with the kinetic most similar to that of grey matter without specific binding. This method requires segmented MR data for the generation of a (camera-specific) population database. Since the spatial resolution of the Inveon® small animal PET-CT camera is between 1 and 2 mm, it is not possible to resolve distinct grey and white matter kinetics in the rat brain. Therefore, we replaced the MR-guided tissue segmentation [36, 45] with an automated kinetic data segmentation approach developed for preclinical PET data (LMA) [42–44]. The LMA method, implemented in BrainVisa, produced three classes of sufficient size (over 10 % of total brain voxels) in the rat brain and with voxels which were located in spatial proximity to each other. We additionally attempted to define more classes (as used in clinical supervised clustering [36, 45]); however, no more than three kinetically distinct classes were visible in the stroke rodent brain (identifiable by TAC shape), suggesting that this was the optimum number for class definition. This was consistent for (R)-[11C]PK11195, [18F]GE-180 and [18F]DPA-714.
In our implementation, a population of adult male Wistar rats in a model of focal cerebral ischaemia were used for class definition. Six animals underwent transient right middle cerebral artery occlusion (MCAO) for 60 min (data published previously and re-analysed [23]) and were dual scanned with (R)-[11C]PK11195 and [18F]GE-180 5 or 6 days later. In addition, four animals with MCAO were scanned with (R)-[11C]PK11195 and [18F]DPA-714 (data re-analysed from [17]). All data were reconstructed and pre-processed as described above. Brain masks [36] were extracted from the individual CT images for each animal. The classes were defined within the brain as ‘activated tissue’ represented by the core of the infarct; ‘normal tissue’ from the contralateral ROI; and ‘tissue with intermediate binding’, represented by cerebral tissue located between the core and contralateral tissue and in the cerebellum. As described above, these regions were defined using the BrainVISA LMA segmentation, for each animal, and TACs were normalised (by subtracting from each frame its mean and dividing it by its standard deviation, SD, to create a unit input) and averaged to produce a population database. The algorithm was then implemented on the dynamic images using the Matlab code, SuperPK Software (Imperial Innovations, Imperial College London, UK [45, 46], developed for clinical studies and adapted here for preclinical class definitions), from the LPS animals to produce cluster maps for normal, intermediate and activated tissue, as well as a reference kinetic, based on the class of voxel most kinetically similar to normal tissue identified in the training data set, for input to the SRTM.
PET Data Modelling
Kinetic analysis of data was performed using the SRTM [47] in the PMOD software package (version 3.6, PMOD Technologies Ltd.) for ROIs (1) to (5). The model assumes a 1-tissue compartment arrangement in the target region (lesion) and the reference region. Input functions were derived using the contralateral hemisphere ROI (region 4), cerebellum (region 5) and the supervised clustering approach. R 1 (ratio of tracer delivery in the target ROI vs. that in all respective reference regions) and BPND were calculated for all ROIs.
Immunohistochemistry
Brain sections, including the striatum, from five LPS animals which were also scanned in vivo were stained for CD11b and glial fibrillary acidic protein (GFAP) to confirm the presence of microglia and astrocytes respectively. Staining for neuronal nuclei was also completed using an anti-NeuN antibody. Sections from four animals were also stained to investigate BBB integrity: visualising tight junctions with claudin-5 and large protein diffusion with IgG.
For all procedures described below, phosphate buffered saline (PBS) at 100 mM was used. Rat brain sections were snap frozen in isopentane and stored at −80 °C. Sections were then post-fixed in paraformaldehyde (4 % in PBS) for 10 min and washed (3 × 5 min) in PBS. Sections were incubated in 1 mg/ml sodium borohydride in PBS (3 × 5 min) to reduce auto-fluorescence and washed again in PBS (3 × 5 min). They were then permeabilised with 2 h of incubation in 0.1 % Triton X-100 containing 3 % normal donkey serum in PBS to block non-specific binding. Without further washing, sections were incubated overnight at 4 °C with primary antibodies in 3 % normal donkey serum/0.1 % Triton X-100 in PBS. Double immunohistochemical staining was performed against GFAP with rabbit anti-cow GFAP (Dako, 1:400) and CD11b (OX42) with mouse anti-rat CD11b (Serotec, 1:1000). Adjacent sections from the same animals were stained with mouse anti-mouse NeuN (Chemicon, 1:1000). Sections were then washed (3 × 10 min) in PBS/Triton and incubated for 2 h at room temperature with secondary antibodies (AlexaFluor® 488 nm donkey anti-rabbit for GFAP, AlexaFluor® 594 nm donkey anti-mouse for CD11b, or AlexaFluor® 594 nm donkey anti-mouse for NeuN (Molecular Probes, Invitrogen)), all 1:500 in 3 % normal donkey serum/0.1 % Triton X-100 in PBS and then washed again (3 × 10 min) in PBS.
Following the same protocol, brain sections from two animals injected with LPS from the in vivo study and two animals injected with AMPA (positive control) were incubated with monoclonal rabbit anti-claudin-5 (Sigma-Aldrich, 1:100) primary antibody and AlexaFluor® 488 nm donkey anti-rabbit. To stain for endogenous rat IgG infiltration in the brain parenchyma, adjacent sections were incubated with AlexaFluor® 594 nm donkey anti-rat. AMPA-injected animals were of the same strain and comparative age/weight to LPS animals. Supplementary Table 2 provides a summary of primary and secondary antibodies used during immunohistochemistry.
All sections were mounted with a Prolong® Gold Antifade kit (Molecular Probes, Invitrogen). Images were collected on an Olympus BX51 upright microscope using 4×/0.13, 10×/0.30 or 40×/0.50 UPlanFLN objectives and captured using a Q-Capture Retiga 6000 camera through Q-Capture Pro Software (Molecular Devices). Specific band pass filter sets were used to prevent bleed through from one channel to the next. Images were processed and analysed using ImageJ (http://rsb.info.nih.gov/ij).
Autoradiography
In order to confirm the presence of specific TSPO binding, [18F]DPA-714 autoradiography was performed with 20-μm brain sections from five LPS animals (adjacent to those used for immunohistochemistry). Sections were first incubated for 5 min in cold Tris buffer (Trizma crystals (Sigma, UK), 50 mM adjusted to pH 7.4 at 4 °C with 120 mM NaCl) and then incubated for 1 h at room temperature with either [18F]DPA-714 (total binding) (98.7 GBq/μmol; 5 nM) or [18F]DPA-714 with excess PK11195 (5 μM) to assess non-specific binding. Sections were rinsed twice for 2 min with cold buffer and then once quickly with cold distilled water, before being dried and exposed overnight to a Phosphor-Imager screen (Fujifilm BAS-1800II, Raytek Scientific Ltd., Sheffield, UK). Autoradiographs were visualised and analysed using AIDA software (Raytest GmbH, Germany). ROIs were drawn manually on the lesion and the contralateral area, and binding is expressed as the intensity of photostimulated luminescence (PSL) per pixel.
Statistical Analysis
All data are expressed as mean ± SD. Paired Wilcoxon signed rank tests were used to compare (R)-[11C]PK11195 and the corresponding F-18 tracer uptake or BPND values obtained in the same animal (dual tracer scans). Non-paired (R)-[11C]PK11195, [18F]GE-180 and [18F]DPA-714 results were compared using the Mann-Whitney U test. Non-paired comparisons of tracers were performed as (R)-[11C]PK11195[ 18 F]GE-180 (group 1) vs. [18F]DPA-714only (group 4) and (R)-[11C]PK11195[ 18 F]DPA-714 (group 2) vs. [18F]GE-180only (group 3) (see Supplementary Table 1). Comparisons between all animals scanned with [18F]GE-180 and [18F]DPA-714 (n = 11 vs. n = 11) were also performed. Statistics were performed in GraphPad Prism for Windows (v6.04, GraphPad Software, Inc., San Diego, CA, USA).
Results
PET Imaging
All PET images in LPS animals (groups 1–4) showed localised increase in tracer uptake (%ID/cm3) in the core ROI (Figs. 2 and 4, Table 1). Furthermore, the uptake in edges 1, 2 and 3 was lower compared to the core and not statistically different than the contralateral side, indicating a gradual decrease in neuroinflammation with distance from the injection site.
Non-Paired Scans
For non-paired scans, there was no significant difference in 40–60 min core or contralateral uptakes between tracers. There were no significant differences between the core/contralateral uptake ratios at 40–60 min between (R)-[11C]PK11195 and either F-18 tracer when considering non-paired groups of animals: (R)-[11C]PK11195[ 18 F]DPA-714 (2.26 ± 0.41, n = 6) vs. [18F]GE-180only (2.76 ± 0.48, n = 5) or (R)-[11C]PK11195[ 18 F]GE-180 (2.43 ± 0.39, n = 6) vs. [18F]DPA-714only (2.92 ± 0.43, n = 5). The unpaired comparison of animals scanned with [18F]GE-180 (n = 11) and animals scanned with [18F]DPA-714 (n = 11) showed no significant difference in core/contralateral uptake at 40–60 min (p = 0.47) or the core/contralateral ratio (Table 1). Additionally, there were no significant differences between animals scanned with [18F]GE-180only (n = 5) and [18F]GE-180 (n = 6), nor with [18F]DPA-714only (n = 5) and [18F]DPA-714 (n = 6) (Table 1). There was also no significant difference between the contralateral ROI and the edges 1, 2, 3 and cerebellum ROIs (Table 1).
For comparison with the control (naïve) animals, all the LPS-injected animals were pooled for each tracer. Average contralateral uptakes in all LPS animals at 40–60 min (0.09 ± 0.02 %, 0.07 ± 0.03 % and 0.07 ± 0.02 % for (R)-[11C]PK11195 ((R)-[11C]PK11195[ 18 F]GE-180 + (R)-[11C]PK11195[ 18 F]DPA-714, n = 12), [18F]GE-180 (+[18F]GE-180only , n = 11) and [18F]DPA-714 (+[18F]DPA-714only, n = 11) respectively) were not significantly different from the low uptake regions in control animals (0.09 ± 0.01 % (n = 4), 0.08 ± 0.01 % (n = 5) and 0.05 ± 0.01 % (n = 4) for (R)-[11C]PK11195, [18F]GE-180 and [18F]DPA-714 respectively), supporting the use of the contralateral ROI as reference region.
Dual Scans
For dual scans, there was no significant difference between all the ROIs studied for all tracers. However, core/contralateral ratios were significantly higher (+40 ± 36 %) for [18F]GE-180 than (R)-[11C]PK11195 (3.41 ± 1.09 vs. 2.43 ± 0.39, p = 0.03), while [18F]DPA-714 values were not (2.80 ± 0.69 vs. 2.26 ± 0.41, p = 0.09, +25 ± 25 %; Table 1 and Fig. 2).
Kinetic Analysis
Previous work [17, 23] has shown that only paired scans can truly account for inter-individual variability; thus, for the kinetic analysis, only dual scan data were considered.
SRTM with Contralateral Reference Input
Table 2 shows BPND results for dual-scanned animals. Average values calculated with a contralateral reference input for animals scanned with (R)-[11C]PK11195 then [18F]GE-180 were 1.25 ± 0.29 and 1.94 ± 0.75 respectively (+57 ± 57 %, p = 0.03), while (R)-[11C]PK11195 vs. [18F]DPA-714 dual scan BPND values were 1.11 ± 0.31 and 1.58 ± 0.54, respectively (+43 ± 34 %, p = 0.06).
SRTM with Cerebellar Reference Input
Overall, using a cerebellar reference input returned lower BPND values than using a contralateral reference input (p = 0.001 for all tracers) and BPND results were not significantly different between tracers (0.60 ± 0.26 vs. 0.84 ± 0.46 for (R)-[11C]PK11195 vs. [18F]GE-180; 0.46 ± 0.17 vs. 0.43 ± 0.26 for (R)-[11C]PK11195 vs. [18F]DPA-714, Table 2).
SRTM with Data-Driven Clustering Reference Input
BPND values with a data-driven reference input function were overall lower than those from a contralateral reference region (p = 0.001 for (R)-[11C]PK11195 and [18F]DPA-714, and p = 0.002 for [18F]GE-180), but were highly correlated (Spearman’s correlation coefficient ρ = 0.83 and 0.90 respectively for dual scans). For dual-scanned animals, (R)-[11C]PK11195 and [18F]GE-180 values were 0.83 ± 0.22 and 0.63 ± 0.43 respectively (p = 0.31, n.s.), while (R)-[11C]PK11195 and [18F]DPA-714 BPND values were 0.75 ± 0.20 and 0.49 ± 0.20 respectively (p = 0.03, Table 2).
Autoradiography
Autoradiographs confirmed the presence of specific [18F]DPA-714 binding in the ipsilateral striatum compared to the contralateral striatum. Quantification of the autoradiography revealed a 5.62 ± 0.72-fold increase in specific binding in the ipsilateral striatum when compared to the contralateral striatum (Fig. 4 (insert bottom right) and Supplementary Fig. 3).
Immunohistochemistry
Anti-rat IgG immunohistochemistry will label rat IgG in blood vessels; as a large protein, IgG should not diffuse into the brain if the BBB is intact. Our staining indicated that rat IgG were still localised in blood vessels in the LPS model, hence supporting presence of an intact BBB, whereas in the AMPA model the staining of blood vessels was lost and was diffuse in the brain parenchyma, indicating BBB leakage. This was confirmed by immunohistochemistry of the tight-junction protein claudin-5, as LPS animals showed similar intact vessels in the ipsilateral and contralateral area, while in AMPA-challenged animals, the vessels were clearly damaged (Fig. 5a) in the ipsilateral side, indicating a compromised BBB.

a Claudin-5 (×60 magnification) and IgG (×20 magnification) staining in ipsilateral and contralateral striatum for LPS and positive control AMPA-challenged animals. Claudin-5 staining revealed no differences between contralateral and ipsilateral sides in the LPS-challenged animals, while for AMPA-challenged animals, there was visible damage to vessels in the ipsilateral side and some disruption in the contralateral hemisphere. IgG appears normal in ipsilateral and contralateral sides of LPS animals but shows signs of leakage in the ipsilateral side of AMPA animals. b Staining for microglia (CD11b, red) and astrocytes (GFAP, green), merged at ×10 and ×20 (white rectangles) magnification; c NeuN staining at ×20 magnification in core and contralateral striatum
Activated microglial cells (CD11b) were observed in large numbers in the core of the inflamed area compared to the contralateral hemisphere, as were astrocytes (GFAP) (Fig. 5b). NeuN stained nuclei showed no obvious difference between contralateral and ipsilateral striatum, supporting that there was no neuronal loss induced by the LPS injection (Fig. 5c).
Discussion
PET Imaging and Ex Vivo Observations
Although results from rodents are not directly transferable to humans due to inter-species differences, preclinical studies are still valuable for assessment and characterisation of tracer behaviour, especially where models can be developed to mimic clinically relevant levels of inflammation. The primary aim of this study was to quantitatively compare the recently developed second-generation TSPO radioligand [18F]GE-180 with the more well-established [18F]DPA-714 and the original (R)-[11C]PK11195 in a rat model of low/mild focal inflammation. Recently published work has shown that both F-18 labelled tracers have an improved SNR over (R)-[11C]PK11195 in models of acute inflammation [17, 22, 23, 28], but to date, there is neither reported investigation comparing these new tracers nor looking at their performance in a model producing a lower level inflammation (∼2-fold increase in TSPO PET signal), which is more similar to the levels observed in neurodegenerative diseases than stroke [17, 23], excitotoxic lesions [16, 48] or high doses of LPS [28, 34] (∼4–6-fold increase in TSPO PET signal). The second aim of this study was to assess the kinetic quantitation methodology via use of the SRTM with different reference input functions. Non-terminal preclinical kinetic modelling has so far been limited to semi-quantitation (SUV) or use of an anatomical reference region in unilateral models. In order to evaluate methods that could be used in neurodegenerative diseases, such as AD transgenic models, which display low levels of neuroinflammation and where a contralateral reference tissue does not exist, we have compared here the BPND values obtained with the SRTM and three different reference input functions: (1) a contralateral region, (2) a cerebellar region, (3) a reference kinetic derived from a data-driven supervised clustering approach.
Previous preclinical studies using an LPS model have typically used larger doses of the endotoxin, inducing high-level inflammation [22, 28]. Moreover, previous publications did not compare (R)-[11C]PK11195 with second-generation TSPO tracers [28, 34] in the more challenging setting of low/mild neuroinflammation and animals were not dual scanned with the tracers [22]. Finally, the comparison between (R)-[11C]PK11195 and [18F]GE-180 performed by Dickens et al. only used the contralateral reference region as input for modelling.
Our study showed that [18F]GE-180 uptake and BPND were significantly higher than those of (R)-[11C]PK11195, while [18F]DPA-714 were not. Figure 2e–f shows the core/contralateral uptake ratios for dual-scanned animals; it is inferable from Fig. 2e that the significant difference seen between [18F]GE-180 and (R)-[11C]PK11195 may be driven by three particular animals of the six scanned with both (R)-[11C]PK11195 and [18F]GE-180. These animals are also likely to drive the increased variability observed with [18F]GE-180 over the other tracers. We also performed a comparison in ‘non-paired’ animals individually scanned with different tracers ((R)-[11C]PK11195[ 18 F]DPA-714 vs. [18F]GE-180, (R)-[11C]PK11195[ 18 F]GE-180 vs. [18F]DPA-714, Table 1). Interestingly, core/contralateral uptake ratios between 40 and 60 min post-injection for unpaired groups were not significantly higher for either [18F]GE-180 or [18F]DPA-714 when compared to (R)-[11C]PK11195, or compared to one another. Altogether, the lack of differences between [18F]DPA-714 and [18F]GE-180 suggests that both tracers provide similar readouts of neuroinflammation.
These observations also highlight (1) that the differences in sensitivity (if any) between the three tracers used in this study are rather small (2) that, as hypothesised, the inherent variability within an experiment or populations makes quantification more challenging when measuring low/mild level of neuroinflammation and (3) further supports the importance of dual scanning in the same subject to truly compare the performance of tracers both in preclinical and clinical settings.
Ex vivo experiments were performed in order to confirm the in vivo observations. Firstly, immunohistochemistry was performed with CD11b to assess microglial presence in the core of the LPS injection site, GFAP staining for astrocytic activation, NeuN to evaluate neuronal loss and claudin-5 and IgG to assess BBB disruption. Results supported the PET findings; both activated microglia and astrocytes were significantly present in the right striatum compared to the left (contralateral) side. Autoradiography also confirmed the presence of TSPO specific binding in the ipsilateral striatum while the NeuN staining showed no neuronal loss in the right striatum. Finally, claudin-5 staining showed no BBB disruption compared to a positive control AMPA model (Fig. 5a).
Although this study was designed to directly compare the three tracers in vivo, comparison of [18F]GE-180 vs. [18F]DPA-714 in the same animal was not practically feasible due to the rapid progression of TSPO expression over 24 h as induced by LPS injection, requiring the scans to be conducted within hours, and the constraints imposed by the half-life of F-18 and the tracer production. Similarly, considering the difference in tracer pharmacokinetics, the comparison of these two tracers by in vitro autoradiography would not have yielded relevant information regarding their in vivo behaviour as PET tracers, hence our comparison of unpaired scans for these two tracers.
PET Data Modelling
In the case of a pathologically damaged BBB, the tracer gains access to the brain parenchyma; thus the extent of binding to TSPO is affected non-trivially and should be accounted for in kinetic modelling. Here, ex vivo investigation of the BBB disruption in LPS-challenged animals vs. positive control AMPA-challenged animals showed no significant BBB disruption in the inflamed area with this low dose of LPS when compared to the AMPA-treated animals (Fig. 5a). Additionally, R 1 values calculated using the contralateral reference region were close to unity in the core of the inflamed area for all animals, suggesting that tracer diffusion to the contralateral (reference) and ipsilateral regions was similar. This is not the case in strong excitotoxic or stroke models, which can affect quantification [23].
When using the contralateral ROI, only the BPND[ 18 F]GE-180 was significantly higher than BPND-(R)-[ 11 C]PK11195, and as for core/contralateral ratio, BPND[ 18 F]DPA-714 showed a non-significant trend to increase (Table 2). When compared to the contralateral region, both a cerebellar input and a supervised clustering derived input produced significantly lower BPND values for all tracers. Using a cerebellar input showed no group differences between (R)-[11C]PK11195 and second-generation tracers in paired comparisons, while a cluster-derived input identified significantly lower BPND values with [18F]DPA-714 in animals dual-scanned with (R)-[11C]PK11195. The blood perfusion of the cerebellum in rodents is different than that in the brain and leads to a different tracer pharmacokinetic (i.e., a higher peak and uptake than in the normal brain/contralateral ROI, Fig. 3). It is also possible that the cerebellum contains some specific TSPO binding. Together, these two factors would lower the BPND values calculated in the core of the inflamed area by over-estimating the input of the reference tissue.
By the same rationale, a cluster-derived reference region could contain some specific binding, therefore introducing a bias; the MCAO model may not be optimal to define a reference cluster, as the ‘healthy’ tissue could itself contain a compartment of specifically bound tracer, further complicating the derivation of TSPO-free reference voxels in the LPS animal brains. However, this is unlikely since, in a comparison of the region of lowest uptake in control animals to the contralateral reference region in LPS animals, no significant difference in uptake was observed, supporting the use of the contralateral ROI as healthy reference tissue in this model. We applied this approach by reducing and reassigning the number of partitionable classes compared to the clinically established method, producing significantly lower BPND values for all tracers than a contralateral reference input to the SRTM. Clinical studies have used four or six class-supervised clustering; having also attempted to define four classes in the rodent brain (data not shown), we saw no obvious improvement to the BPND results from the LPS animals. This could be due to a number of factors: (i) unlike in the stroke model, the lower level of neuroinflammation induced in this LPS model may inherently not be separable into three or more kinetically distinct classes and/or (ii) the supervised clustering algorithm as it stands is not fit to use with preclinical images which have lower number of voxels to be used to identify classes.
Furthermore, [18F]DPA-714 BPND values were significantly lower than those from (R)-[11C]PK11195, while [18F]GE-180 results were not (Table 2). We speculate that this difference could be the result of voxels exhibiting pathology-related signal in LPS animals being mistakenly classed as normal tissue by the supervised clustering algorithm. In defining classes in the training data set, we were unable to identify a blood pool class such as that used in clinical supervised clustering [45], as the venous sinus in the rodent brain is smaller in diameter than the resolution of the PET camera, and external derivation (from, for example, the left ventricle of the heart) was not possible due to field of view limitations. The intermediate binding class defined instead, while reliably identified by the LMA brain segmentation, was kinetically not orthogonal to the normal tissue class. As these three radiotracers have similar affinities for TSPO, it is more likely that the supervised clustering algorithm mis-identified some ‘intermediate’ binding voxels as normal tissue leading to lower and slightly more variable BPND values for all tracers and hence the absence of significant differences between BPND for [18F]GE-180 and (R)-[11C]PK11195. Finally, if [18F]DPA-714 is more sensitive to these effects (i.e., if the voxels classed as normal tissue are more contaminated by TSPO-related signal, e.g., through inherently increased levels of microglial activation in these animals), it may explain the significant difference seen when estimating BPND with the supervised clustering approach.
Overall, these results suggest that (1) further work on the development and implementation of this method for preclinical imaging should be performed, and (2) the automatic segmentation of ROIs as implemented in the BrainVisa platform or PMOD® is currently the best approach, although unfortunately best suited for models with clearly identifiable rather than diffuse areas of neuroinflammation.
A recent study by Ory and colleagues using [18F]DPA-714 [34] validated the use of a reference tissue model with the contralateral striatum as input, producing BPND values which correlated well with a two-tissue compartment modelling approach. Interestingly, using a cerebellar input function to the SRTM in their high-dose LPS model produced BPND results which were also highly correlated with blood modelling, albeit with higher bias and producing BPND values lower than any other reference region, similar to what we observed in the present study. Discrepancies in these results, as well as those using the data-driven supervised clustering approach, which was unable to identify a suitable class of reference voxels, are most likely related to differences in the inflammatory responses induced by high (50 μg) and low dose (1 μg) of LPS. However, high doses of LPS inducing a neuroinflammatory response similar to what is observed after acute neurodegeneration are not representative of pathological conditions with lower neuroinflammatory levels, such as neurodegenerative diseases. Interestingly, the study also used a 90-min imaging window with this high-dose LPS injection; in the current study, we were able to show differences between tracers by the 60 min time point, suggesting that it is not necessary to scan for longer time periods with a low-dose LPS model or in a stroke model [17, 23]. Furthermore, it is well described in the TSPO imaging literature [49–51] that models or clinical cases in which a strong neuroinflammation (such as in stroke) are much easier to model consistently, whereas in the case of low or subtle neuroinflammation levels as observed in neurodegenerative diseases, the modelling is far more challenging, leading to discrepancies between studies. Considering the failure of the data-driven supervised clustering approach presented here, it is clear that further work is needed to determine a reliable reference tissue allowing the accurate quantification of TSPO tracer in preclinical studies without the need of performing arterial blood sampling when models of neurodegeneration such as transgenic mice or rats are used.
Conclusions
The second-generation TSPO-PET radiotracer [18F]GE-180 showed an increased signal to noise ratio over (R)-[11C]PK11195 when considering dual scans whereas [18F]DPA-714 did not. However, the comparison of unpaired scans did not reveal such differences. The discrepancy between the statistical group results from paired and non-paired comparisons highlights the importance of performing dual scans for direct tracer comparison. Moreover, no significant differences were found between [18F]DPA-714 and [18F]GE-180. Altogether, these results suggest that when measuring low level of neuroinflammation, the differences between second-generation tracers (between them or when compared to (R)-[11C]PK11195) are small in term of sensitivity, while retaining their attractiveness in term of of-site use due to the longer F-18 half-life. Cerebellar and data-driven inputs to the SRTM were unable to produce comparable BPND values to those using a contralateral reference region. The choice of the contralateral ROI as reference tissue was supported by the fact that comparison between tracer uptakes in the contralateral ROI and healthy animals showed no significant difference and that ex vivo staining showed that the contralateral ROI contained very few activated microglia. Our study however also highlighted the remaining issue that when a low level of neuroinflammation present and no anatomically defined reference region can be identified, there is still no well-established method to identify a reference tissue in preclinical studies.
References
Gerhard A, Schwarz J, Myers R, et al. (2005) Evolution of microglial activation in patients after ischemic stroke: a [11C](R)-PK11195 PET study. NeuroImage 24:591–595
Banati RB, Newcombe J, Gunn RN, et al. (2000) The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain 123(Pt 11):2321–2337
Gerhard A, Banati RB, Cagnin A, Brooks DJ (2001) In vivo imaging of activated microglia with [C-11]PK11195 positron emission tomography (PET) in idiopathic and atypical Parkinson’s disease. Neurology 56:A270–A270
Politis M, Su P, Piccini P (2012) Imaging of microglia in patients with neurodegenerative disorders. Front Pharmacol 3:96
Davalos D, Grutzendler J, Yang G, et al. (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752–758
Raivich G (2005) Like cops on the beat: the active role of resting microglia. Trends Neurosci 28:571–573
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318
Czeh M, Gressens P, Kaindl AM (2011) The yin and yang of microglia. Dev Neurosci 33:199–209
Papadopoulos V, Baraldi M, Guilarte TR, et al. (2006) Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci 27:402–409
Lavisse S, Guillermier M, Herard AS, et al. (2012) Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci 32:10809–10818
Chen MK, Guilarte TR (2008) Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther 118:1–17
Banati RB (2002) Visualising microglial activation in vivo. Glia 40:206–217
Ching AS, Kuhnast B, Damont A, et al. (2012) Current paradigm of the 18-kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation and neurodegenerative diseases. Insights Imaging 3:111–119
Owen DR, Yeo AJ, Gunn RN, et al. (2012) An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab 32:1–5
James ML, Fulton RR, Vercoullie J, et al. (2008) DPA-714, a new translocator protein-specific ligand: synthesis, radiofluorination, and pharmacologic characterization. J Nucl Med 49:814–822
Chauveau F, Van Camp N, Dolle F, et al. (2009) Comparative evaluation of the translocator protein radioligands C-11-DPA-713, F-18-DPA-714, and C-11-PK11195 in a rat model of acute neuroinflammation. J Nucl Med 50:468–476
Boutin H, Prenant C, Maroy R, et al. (2013) [(18) F]DPA-714: direct comparison with [C-11]PK11195 in a model of cerebral ischemia in rats. PLoS One 8
Doorduin J, Klein HC, Dierckx RA, et al. (2009) [11C]-DPA-713 and [18F]-DPA-714 as new PET tracers for TSPO: a comparison with [11C]-(R)-PK11195 in a rat model of herpes encephalitis. Mol Imaging Biol 11:386–398
Lee DE, Yue X, Ibrahim WG, et al. (2015) Lack of neuroinflammation in the HIV-1 transgenic rat: an [(18)F]-DPA714 PET imaging study. J Neuroinflammation 12:171
Arlicot N, Vercouillie J, Ribeiro MJ, et al. (2012) Initial evaluation in healthy humans of [18F]DPA-714, a potential PET biomarker for neuroinflammation. Nucl Med Biol 39:570–578
Hamelin L, Lagarde J, Dorothee G, et al. (2016) Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain 139:1252–1264
Dickens AM, Vainio S, Marjamaki P, et al. (2014) Detection of microglial activation in an acute model of neuroinflammation using PET and radiotracers C-11-(R)-PK11195 and F-18-GE-180. J Nucl Med 55:466–472
Boutin H, Murray K, Pradillo J, et al (2015a) 18F-GE-180: a novel TSPO radiotracer compared to 11C-R-PK11195 in a preclinical model of stroke Eur J Nucl Med Mol Imaging 42:503–511
Liu B, Le KX, Park MA, et al. (2015) In vivo detection of age- and disease-related increases in neuroinflammation by 18F-GE180 TSPO microPET imaging in wild-type and Alzheimer’s transgenic mice. J Neurosci 35:15716–15730
Gerhard A, Banati RB, Goerres GB, et al. (2003) [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology 61:686–689
Cagnin A, Brooks DJ, Kennedy AM, et al. (2001b) In-vivo measurement of activated microglia in dementia. Lancet 358:461–467
Serriere S, Tauber C, Vercouillie J, et al. (2015) Amyloid load and translocator protein 18 kDa in APPswePS1-dE9 mice: a longitudinal study. Neurobiol Aging 36:1639–1652
Ory D, Planas A, Dresselaers T, et al. (2015) PET imaging of TSPO in a rat model of local neuroinflammation induced by intracerebral injection of lipopolysaccharide. Nucl Med Biol 42:753–761
Lartey FM, Ahn GO, Shen B, et al. (2014) PET imaging of stroke-induced neuroinflammation in mice using [18F]PBR06. Mol Imaging Biol 16:109–117
Folkersma H, Boellaard R, Vandertop WP, et al. (2009) Reference tissue models and blood-brain barrier disruption: lessons from (R)-[11C]PK11195 in traumatic brain injury. J Nucl Med 50:1975–1979
Shao X, Wang X, English SJ, et al. (2013) Imaging of carrageenan-induced local inflammation and adjuvant-induced systemic arthritis with [(11)C]PBR28 PET. Nucl Med Biol 40:906–911
Airas L, Dickens AM, Elo P, et al. (2015) In vivo PET imaging demonstrates diminished microglial activation after fingolimod treatment in an animal model of multiple sclerosis. J Nucl Med 56:305–310
Winkeler A, Boisgard R, Awde AR, et al. (2012) The translocator protein ligand [(1)(8)F]DPA-714 images glioma and activated microglia in vivo. Eur J Nucl Med Mol Imaging 39:811–823
Ory D, Postnov A, Koole M, et al (2015) Quantification of TSPO overexpression in a rat model of local neuroinflammation induced by intracerebral injection of LPS by the use of [F]DPA-714 PET. Eur J Nucl Med Mol Imaging 43(1):163-172
Hinz R, Boellaard R (2015) Challenges of quantification of TSPO in the human brain. Clin Transl Imaging 3:403–416
Yaqub M, van Berckel BN, Schuitemaker A, et al. (2012) Optimization of supervised cluster analysis for extracting reference tissue input curves in (R)-[(11)C]PK11195 brain PET studies. J Cereb Blood Flow Metab 32:1600–1608
Camsonne R, Crouzel C, Comar D, et al. (1984) Synthesis of N-(C-11) methyl, N-(methyl-1 propyl), (chloro-2 phenyl)-1 isoquinoleine carboxamide-3 (Pk-11195)–a new ligand for peripheral benzodiazepine receptors. J Label Compd Radiopharm 21:985–991
Cremer JE, Hume SP, Cullen BM, et al. (1992) The distribution of radioactivity in brains of rats given [N-methyl-11C]PK 11195 in vivo after induction of a cortical ischaemic lesion. Int J Rad Appl Instrum B 19:159–166
Wadsworth H, Jones PA, Chau WF, et al. (2012) [(1)(8)F]GE-180: a novel fluorine-18 labelled PET tracer for imaging translocator protein 18 kDa (TSPO). Bioorg Med Chem Lett 22:1308–1313
Landini A (1976) Traumatic lesions of the urinary tract. Radiographic aspects. Radiol Med 62:855–872
Wickstrom T, Clarke A, Gausemel I, et al. (2014) The development of an automated and GMP compliant FASTlab synthesis of [(18) F]GE-180; a radiotracer for imaging translocator protein (TSPO). J Label Compd Radiopharm 57:42–48
Maroy R, Boisgard R, Comtat C, et al. (2008) Segmentation of rodent whole-body dynamic PET images: an unsupervised method based on voxel dynamics. IEEE Trans Med Imaging 27:342–354
Maroy R, Boisgard R, Comtat C, et al. (2010) Quantitative organ time activity curve extraction from rodent PET images without anatomical prior. Med Phys 37:1507–1517
Maroy R, Boisgard R, Comtat C, et al (2007) Local means analysis: an unsupervised method for the segmentation of rodent whole-body dynamic PET images. 2007 4th IEEE International Symposium on Biomedical Imaging: Macro to Nano 1–3:324–327
Turkheimer FE, Edison P, Pavese N, et al. (2007) Reference and target region modeling of [C-11]-(R)-PK11195 brain studies. J Nucl Med 48:158–167
Boellaard R, Turkheimer F, Hinz R, et al (2008) Performace of a modified supervised cluster algorithm for extracting reference region input functions from (R)-[11C]PK11195 brain PET studies [abstract]. IEEE Nuclear Science Symposium 5400-5402P
Lammertsma AA, Hume SP (1996) Simplified reference tissue model for PET receptor studies. NeuroImage 4:153–158
Chauveau F, Boutin H, Van Camp N, et al. (2011) In vivo imaging of neuroinflammation in the rodent brain with [11C]SSR180575, a novel indoleacetamide radioligand of the translocator protein (18 kDa). Eur J Nucl Med Mol Imaging 38:509–514
Chauveau F, Boutin H, Van Camp N, et al. (2008) Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imaging 35:2304–2319
Boutin H, Pinborg LH (2015b) TSPO imaging in stroke: from animal models to human subjects. Clin Transl Imaging 3:423–435
Varrone A, Nordberg A (2015) Molecular imaging of neuroinflammation in Alzheimer’s disease. Clin Transl Imaging 3:437–447
Acknowledgments
This work was supported by the Engineering and Physical Sciences Research Council, UK (EPSRC), the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. HEALTH-F2-2011-278850 (INMiND) and GE Healthcare Ltd. The authors wish to thank all the personnel of the WMIC, especially Ms. Alison Smigova, Ms. Lidan Christie, Mr. Michael Green, Ms. Carol Brough and Ms. Gemma Chapman for facilitating the study. GE Healthcare provided the FASTLab cassettes for [18F]GE-180 production.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
SS is the recipient of an EPSRC CASE PhD studentship with GE Healthcare. GE Healthcare was not involved in the design of the study. All other authors declare no conflicts of interest.
Electronic supplementary material
ESM 1
(PDF 487 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sridharan, S., Lepelletier, FX., Trigg, W. et al. Comparative Evaluation of Three TSPO PET Radiotracers in a LPS-Induced Model of Mild Neuroinflammation in Rats. Mol Imaging Biol 19, 77–89 (2017). https://doi.org/10.1007/s11307-016-0984-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-016-0984-3