
Abstract
Partial Least Squares-Discriminant Analysis (PLS-DA) is a PLS regression method with a special binary ‘dummy’ y-variable and it is commonly used for classification purposes and biomarker selection in metabolomics studies. Several statistical approaches are currently in use to validate outcomes of PLS-DA analyses e.g. double cross validation procedures or permutation testing. However, there is a great inconsistency in the optimization and the assessment of performance of PLS-DA models due to many different diagnostic statistics currently employed in metabolomics data analyses. In this paper, properties of four diagnostic statistics of PLS-DA, namely the number of misclassifications (NMC), the Area Under the Receiver Operating Characteristic (AUROC), Q 2 and Discriminant Q 2 (DQ 2) are discussed. All four diagnostic statistics are used in the optimization and the performance assessment of PLS-DA models of three different-size metabolomics data sets obtained with two different types of analytical platforms and with different levels of known differences between two groups: control and case groups. Statistical significance of obtained PLS-DA models was evaluated with permutation testing. PLS-DA models obtained with NMC and AUROC are more powerful in detecting very small differences between groups than models obtained with Q 2 and Discriminant Q 2 (DQ 2). Reproducibility of obtained PLS-DA models outcomes, models complexity and permutation test distributions are also investigated to explain this phenomenon. DQ 2 and Q 2 (in contrary to NMC and AUROC) prefer PLS-DA models with lower complexity and require higher number of permutation tests and submodels to accurately estimate statistical significance of the model performance. NMC and AUROC seem more efficient and more reliable diagnostic statistics and should be recommended in two group discrimination metabolomic studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The goal of systems biology is to explore the interaction between various components in a biological system. Metabolomics measurements provide quantitative information on the metabolic level of the system. This metabolic level has proven an important area of systems biology with the aim to pinpoint putative metabolites related to disease, genetic variation or nutritional interventions (Weckwerth et al. 2004; Yang et al. 2004; Kind et al. 2007; van Velzen et al. 2008; Bernini et al. 2009).
In metabolomics studies different analytical platforms are often used to provide information on large groups of metabolites. Most metabolomics studies result in complex multivariate datasets with varying correlations between the measured metabolite levels so that multivariate data analysis methods are needed to explore these complex datasets.
In the search for metabolic biomarkers, multivariate discrimination models between two classes of subjects/samples are used. One of the most used methods is Partial Least Squares-Discriminant Analysis (PLS-DA) (Barker and Rayens 2003; van Velzen et al. 2008). If a statistically significant discrimination between two classes e.g. the cases and controls classes can be found, then the model parameters can be interpreted for their discriminating power and metabolic biomarkers can be found. In PLS-DA models, a relationship between the metabolomics data and the categorical variable y is developed in such a way that categorical variable values can be predicted for samples of unknown origin given the metabolomics data. Here, the categorical variable y is a vector which values indicate class membership of each sample included in the study e.g. a vector with values of −1 and 1 where −1 represents each sample belonging to the class of controls and 1 represents each sample belonging to the class of cases. However, due to the properties of regression models, the prediction ŷ i of the i-th element of y can take any value, not necessarily exactly −1 or 1. Translation of these values of ŷ to class membership (classification procedure) is a critical point of PLS-DA analysis and can be done, e.g. by applying a threshold above which the sample will be assigned to the cases class and below to the control class.
Another challenge of PLS-DA analysis is the accurate estimation of the quality of the obtained models and thereby differences between two classes. Many diagnostic statistics have been introduced over the time to convert values of ŷ obtained for all the study samples into a single number representing the overall quality of the discrimination model. In this paper we investigate the performance of the four different diagnostic statistics which are usually used for this purpose in metabolomics when PLS-DA is applied. They are: the number of misclassifications (NMC), the Area Under the Receiver Operating Characteristics (AUROC), Q 2 and Discriminant Q 2 (DQ 2). The natures of these diagnostic statistics are very different. Whereas the Q 2/DQ 2 are derived directly from the (ratio-scaled) model predictions ŷ of y, the NMC/AUROC are derived from the (nominal-scaled) class memberships translated from ŷ. It is debatable which measurement scale should be used for diagnostic statistic of PLS-DA (Stevens 1946).
The power of each of diagnostic statistics is investigated in terms of its ability to provide a statistically significant measure of the discrimination between two classes of subjects (e.g. the cases and the controls) when known multivariate effects of different magnitudes are present in the data. This is accomplished by superimposing known multivariate effects of increasing magnitude on the metabolic profiles of subjects from the cases class and calculating the PLS-DA models: one PLS-DA model per each data set with different magnitude of superimposed effect and diagnostic statistics used. In order to obtain unbiased estimates of model performance, PLS-DA is applied in a double cross validation scheme. This means that the four diagnostic statistics are used not only to assess the final quality of the PLS-DA models but also for the optimization of the model, e.g. to select the optimal complexity of model (optimal number of latent variables, #LV). Statistical significance of each PLS-DA model is estimated by comparing the value of the diagnostic statistics (Q 2, DQ 2, NMC or AUROC) to values of its null reference distribution H0 obtained by permutation tests.
Datasets obtained by two different analytical platforms commonly used in the metabolomics studies: UPLC-MS and NMR were used to evaluate properties of the four diagnostic statistics. The multivariate effects superimposed into data sets were intended to represent two situations that can occur in real life metabolomics data analysis: investigating a nutritional effect (in the case of the UPLC-MS data set) and investigating an effect of exposure to a chemical pollutant (in the case of the NMR data set). Moreover, datasets of different size were used to draw general conclusions independent of data set size.
2 Theory
2.1 PLS-DA modeling with a double-cross validation scheme
2.1.1 PLS-DA
Partial least squares discriminant analysis (PLS-DA) and its extensions like multilevel PLS-DA (MPLS-DA, (van Velzen et al. 2008)) and orthogonal PLS-DA (OPLS-DA, (Trygg and Wold 2002)) are the most used classification methods in metabolomics. PLS-DA consists of a classical PLS regression where the dependent variable y is categorical and represents samples class membership e.g. y can be a vector with values of −1 and 1 where −1 represents each sample belonging to the class of controls and 1 represents each sample belonging to the class of cases (Barker and Rayens 2003). By making use of class information, PLS-DA tends to improve the separation between the (two) groups of samples.
2.1.2 PLS-DA with double cross validation schema
Two steps are critical when building a PLS-DA model: the selection of the optimal model complexity e.g. optimal number of latent variables (#LV) and the assessment of the overall quality of the model. In the PLS-DA context, the #LV needs to be optimized in such a way that a suitable number of latent variables is used to build the model. Suitable means that it provides the best description of data thus the best discrimination between samples from two different classes.
Model optimization (i.e. selection of the optimal #LV) and model quality assessment should be always carried out in a double cross validation schema because then assessment of model quality and the model optimization are independent. Samples which are used in final model assessment are not used in the model optimization (calibration): moreover the calibration of the model is carried on in a similar unbiased way (Smit et al. 2007; Westerhuis et al. 2008).
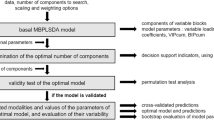
A double cross validation scheme consists of two nested loops CV1 and CV2, (see Smit et al. 2007). The aim of CV1 is to optimize complexity of the PLS-DA model and the aim of CV2 is to assess final model performance. In the outer loop (CV2) the complete dataset is split into a test set and a rest set: the test set is set aside and the rest set is used in a single cross validation (inner loop, CV1). In the CV1 the rest set is again split into a validation (sometimes called optimization) set and a training set. Then, training set is used to develop a series of PLS-DA models with 1 to n latent variables (#LVs) and these PLS-DA models are used to calculate a series of ŷ in for validation set samples which is further used in the selection of an optimal #LV (Fig. 1a). The selection depends on the values of the diagnostics statistics used: #LV with the highest values of AUROC, Q 2 and DQ 2 and the lowest values of NMC are selected. The CV1 procedure is repeated until all samples from rest set have been in the validation set once and only once. For each rest set a separate PLS-DA model with optimal #LV is obtained and this model is further used in CV2 loop to predict ŷ i for each test set sample. The CV2 procedure is repeated until each sample has been in test set once and only once. On the basis of the ŷ i obtained for all the samples vector ŷ is obtained and used in assessment of the overall PLS-DA model quality (see Fig. 1b).

Graphical illustration of use of diagnostic statistics: NMC, AUROC, Q 2 and DQ 2 in double cross validation procedure of PLS-DA. a Use of diagnostics statistics in selection of optimal number of latent variables in CV1, b use of diagnostics statistics in assessment of overall PLS-DA model quality after double cross validation procedure (CV2)
Training, validation and test sets (in both CV1 and CV2 loops) are defined by partitioning the samples in k disjoint subsets. In this study, k = 8 was chosen for the outer loop (CV2) and k = 7 for the inner loop (CV1). This is the most commonly used partition in double cross validation procedure applied to metabolomic data sets. Samples of both classes were always represented in a 1:1 ratio in test, validation and training sets.
As many different disjoint partitions of a data set are possible, the overall procedure was repeated M times (30 in the case of the UPLC-MS dataset and 20 in the case of the NMR data set) resulting in M submodels. That gives M repetitions of the ŷ vector: ŷ 1 ,…, ŷ M . (Fig. 1b). This procedure enables to track the reproducibility of the PLS-DA output (see Sect. 4.2.3). The final measures of quality are given as average values over the M values of chosen diagnostics statistics. The choice of an 8:7 data split and M = 30 (20) is a tradeoff between accuracy and computational time.
2.2 Diagnostic statistics
2.2.1 The Q2 statistic
The Q 2 is de facto the default diagnostic statistic to validate PLS-DA models in metabolomics included in commercial or academic statistical packages like SIMCA (Umetrics Inc, Kinnelon NJ), the PLS-toolbox for Matlab (Eigenvector Research Inc, Wenatchee WA), SAS (SAS Institute Inc, Cary NC) or Metaboanalyst (Xia et al. 2009).
The Q 2 is based on the evaluation of the error between the predicted categorical variable ŷ and the known y. The prediction error is summed over all the samples (PRESS) and referred to the total sum of squares (TSS) (Cruciani et al. 1992):
Q 2 is then defined as:
In a PLS regression the values of ŷ i are not bounded in the range [−1, +1] but, in principle, can assume any value in the range [−∞, +∞]. Any deviation of ŷ i from y i contributes to the PRESS: for instance, a prediction of ŷ i = −2 for a sample with y i = −1 will result in a contribution of 12 to the PRESS even if this corresponds to a correct classification when the discrimination border is set at ŷ i = 0. The same happens if a prediction of 0 (ŷ i = 0) is given to this sample, then the contribution to the PRESS is still 1 = (−1)2. This drawback is (partially) overcome by the so called Discriminant Q 2 (DQ 2).
2.2.2 Discriminant Q2 statistics, DQ2
Discriminant Q 2, DQ 2 (Westerhuis et al. 2008), is based on the fact that the prediction error is disregarded when the prediction is beyond the class label (i.e. >1 or <−1). PRESS is then redefined as PRESSD:
and the definition of DQ 2 straightforwardly follow from (3):
This correction is effective only when the prediction is in the direction of the true class label, for instance when a sample with y i = −1 is predicted to be ŷ i = −1.5. If this sample is predicted with ŷ i = 0 or +1, the prediction error contributes to the PRESSD. It is then clear that the larger the prediction error, the larger the PRESSD which in turn implies a smaller value of DQ 2.
2.2.3 Number of misclassifications (NMC)
In the PLS-DA predicted values of ŷ i can be transformed into a class membership (i.e. cases/controls) by relating them to a set discrimination threshold (classification boundary). This threshold is usually set at 0 when two classes have similar size and variance and when y is a vector of −1 (for samples from class of controls) and 1 (for samples from class of cases). If these conditions are not met the discriminative threshold can be adjusted to other values (Lloyd et al. 2009). The predicted values ŷ i for the i-th sample is related to the 0 threshold: the sample is assigned to class of cases if ŷ i ≥ 0 or to class of controls if ŷ i < 0. The assigned class is then compared with the true class membership and classified either as a True Positive (TP), a True Negative (TN), a False Positive (FP) or a False Negative (FN). When all samples have been predicted and assigned to a class, the total number of True Negatives, False Positives, False Negatives, and True Positives can be computed to create a Confusion Matrix (Broadhurst and Kell 2006) (see also Supplementary Fig. 1) which summarizes the prediction ability of the model.
The number of misclassification (NMC) is calculated as the sum of False Positive and False Negative:
The NMC is the most intuitive of all diagnostic statistics as it simply indicates the number of samples which are wrongly classified by the model.
2.2.4 Area under the receiver operator characteristic
Apart from the NMC, several criteria can be derived from the confusion matrix (Lloyd et al. 2009) and the specificity (Sp) and the sensitivity (Se) (Altman and Bland 1994) are two of the mostly used, especially in assessing the performance of diagnostic tests.
The specificity and the sensitivity are defined as
The sensitivity is a measure of how well the model is able to correctly classify samples of the class of cases, while the specificity measures how well the model can predict samples from the class of controls. The Receiver Operator Characteristic (ROC) (Fawcett 2004; Davis and Goadrich 2006) combines these two parameters. By plotting the sensitivity against 1-specificity for different values of the discrimination threshold a ROC curve can be defined. The ROC curve provides a spectrum of performance assessments and the area under the ROC (AUROC) is commonly used as diagnostic statistics of PLS-DA models. The AUROC values range from 1 (perfect discrimination between classes) and 0 (0.5 and lower usually means no discrimination at all).
2.2.5 Differences between NMC/AUROC and Q2/DQ2
Class membership can be coded as 1 and −1 in categorical variable y: a sample belongs either to class 1 (e.g. cases) or −1 (e.g. controls). These classes could also have been indicated by class A and B showing that the numerical values 1 and −1 are irrelevant (they are only used as dummy variables). Predicted class memberships (ŷ) are also categorical variables and the NMC/AUROC statistics are directly derived from these memberships and are so-called permissible statistics (Stevens 1946). For instance, the interpretation of means and variances are problematic for categorical variables while they are well-defined for ratio-scaled variables.
The Q 2 and Discriminant Q 2 are derived from predictions (ŷ i ) and are allowable statistics if we assume that the ŷ i values are ratio-scaled variables. It is interesting to note that the definition of Q 2 and DQ 2 relies on the calculation of the mean of the categorical vector y (Eq. 2), a statistic which is not permissible for categorical variables (Stevens 1946). This is a fundamental problem of using these statistics in the PLS-DA.
Errors in the class membership predictions (i.e. deviations from the values −1/+1) have a different impact on the behavior of the four diagnostic statistics Q 2, DQ 2, NMC and AUROC. This can be shown by means of a simple simulation. We simulated the prediction
of a vector
containing the class memberships of six samples. A plot of the four diagnostic statistics as a function of the error ε on the prediction of the first sample is shown in Fig. 2. The error ε ranges from −10 to 10 with ε = 0 corresponding to a perfect prediction, and an increment of 0.25. A value of 0 was used as the discrimination threshold in the simulation. This means that if the prediction (1 + ε) of a sample of class 1 is ≤ 0, the sample is wrongly classified.

Behavior of the diagnostic statistics NMC, AUROC, Q 2 and DQ 2 as a function of the error ε on the simulated prediction ŷ = [−1 −1 −1 1 1 1 + ε] of a vector y = [−1 −1 −1 1 1 1] containing the class membership of six samples
It appears that NMC and AUROC are not sensitive to the magnitude of the error ε while Q 2 and DQ 2 strongly depend on the magnitude ε. For example an error ε = 6 gives a lower Q 2 than an error ε = 2 where NMC is equal to 1 for both errors. Values of Q 2 (and DQ 2) are sensitive to outliers with high errors ε.
2.2.6 Permutation test
Although an NMC = 0 or a Q 2 = 0.99 can be thought to correspond to good models with a high discriminating power, these values of the diagnostic statistics can be attained purely by chance due to a lucky random choice of samples in the test, validation and training sets. This means that it is not known which value of these diagnostic statistics really corresponds to a good discrimination between groups (Westerhuis et al. 2008). To overcome these problems and to give a measure of the statistical significance of the diagnostic statistics (P-value), a permutation test was introduced (Lindgren et al. 1996; Golland et al. 2005; Mielke and Berry 2007; Pesarin and Salmaso 2010). Permutation tests assume that there is no difference among two groups that are randomly formed (Westerhuis et al. 2008). In a permutation test the labels of the samples are randomly permuted and a new classification model is calculated (Lindgren et al. 1996). The performance of the model obtained with is assessed by one of the four diagnostic statistics and the values of diagnostic statistics are expected to be higher than for original (unpermuted) data for NMC and lower for Q 2, DQ 2 and AUROC. By repeating this procedure N times, a null distribution of H0 for each of four diagnostic statistic is obtained. H0 is then a distribution of diagnostic statistics of models that are expected to be insignificant (Fisher 1937).
Statistical significance of the PLS-DA model is then assessed by relating the values of the diagnostic statistics for this model calculated with the original data set to the H0 distribution of the diagnostic statistics values obtained for models calculated with the permuted data sets. In case of NMC the upper threshold P for the P-value is calculated as
where #(NMC p ≤ NMC) is the number of element in the null distribution which are smaller or equal to the NMC for the original data set. It is worthy to note that the estimation of P depends, apart N, on the value of NMC which is actually estimated by averaging the M values obtained by M different submodels (Fig. 1b), that is P is also sensitive to the distribution of the values of NMC. In the case of AUROC, Q 2 and DQ 2, P is calculated with a similar formula but inequality ≤ must be replaced with ≥ and similar considerations do apply.
When using the permutation distribution to infer P-values, the left tail (in the case of NMC) and right tail (in the case of of Q 2, DQ 2 and AUROC) are of interest. This means that the number of permutations needs to be “large enough” to sample the tails of the distribution. The lower limit of the number of permutations is dictated by the required statistical significance: for instance, to attain a P-value <0.01 at least 100 permutations are necessary but cannot be sufficient to a proper sampling of the distributions tails. An optimal number is difficult to be inferred: (Churchill and Doerge 1994) suggested that to estimate a permutation P-value of 0.01 as many as 104 permutations are needed in genetics applications. The true permutation P-value can be calculated by taking into account all the possible permutations (Sun and Wright 2010) which is actually dictated by the number of samples: with N samples, N! are permutations possible. With N = 60 (the size of a typical small metabolomics dataset) there are >1080 possible permuted data sets that obviously cannot all be screened. On the other hand, a limited number of samples can hamper the sampling of the tails because extreme values of the distribution may not be detected. This issue is discussed further in Sect. 4.1.
3 Materials and methods
3.1 Data sets
3.1.1 UPLC-MS data set
The UPLC-MS data set consists of 96 samples × 101 lipids levels measured at the Demonstration and Competence Laboratory, Netherlands Metabolomics Centre at Leiden University, Leiden, The Netherlands. Technical details of the UPLC-MS lipidomics platform are described in (Hu, van Dommelen et al. 2008) and in the Supplementary Material. 96 samples are serum samples collected from healthy subjects before the start of the nutritional intervention study in the frame of BCL study (more information available on request). To 48 randomly selected samples nutritional effects were added as described in Sect. 3.1.3.
3.1.2 NMR data set
Ten different data sets, each consisting of 60 NMR spectra (small NMR data sets), have been constructed by randomly selecting the spectra from a pool of 256 homogenous NMR serum spectra of subjects of the DiOGenes study (Larsen et al. 2009). Technical details of 1H NMR spectra acquisition are presented in the Supplementary Material. Each small NMR dataset was composed of 60 spectra (samples) each with 420 data points. A multivariate effect has been subsequently added to the 30 spectra randomly selected from 60 spectra (the case group). Using the same strategy, ten larger NMR data sets (large NMR data sets), consisting of 200 NMR spectra (100 + 100) have also been generated.
3.1.3 Superimposed multivariate effects
3.1.3.1 Nutritional effects
Original multivariate nutritional effects were changes in levels of 101 lipids calculated for each of 33 healthy subjects participating in the nutritional study (group of 33 subjects with the largest nutritional effect in BCL study). For each of the 33 subjects changes between lipid levels before and after nutritional intervention were calculated. On that basis 33 different original multivariate nutritional effects were derived. Ten different magnitudes of these effects were obtained by multiplication of the original effects by constant numbers: 1 (original effects), 0.75, 0.626, 0.55, 0.5, 0.375, 0.25, 0.15, 0.1, 0.05. To each of 48 samples (randomly selected from UPLC-MS data set) one of these 33 multivariate nutritional effects (randomly selected) or their magnitudes were added. In that way ten different data sets with different magnitudes of superimposed nutritional effects were obtained. Each of them consisted of 48 lipid profiles with superimposed effects (the class of cases) and 48 lipid profiles without superimposed effects (the class of controls).
3.1.3.2 Exposure to a chemical pollutant
Aldrin, an isomer of hexachlorohexahydrodimethanonaphthalene, C12H8Cl6 (Martin 1958; Younos and Weigmann 1988) is an organochlorine pesticide whose use is severely limited in most countries and banned within the EU (http://www.pesticides.gov.uk/approvals.asp?id=55). Despite the strict regulation, the presence of this compound, as well of other organochlorine pollutants, has been reported in the sera of healthy subjects, suggesting that exposure to some organochlorine compounds is strongly related to environmental contamination (Lino and Silveira 2006; Carreño et al. 2007). (Lino and Silveira 2006) reported levels of Aldrin in the blood of healthy subjects ranging from <5 to 400 μg/l with an average concentration of 13 ± 42 μg/l.
The Aldrin spectrum was simulated for the average concentration of this compound in blood (13 μg/L) and was the linear combination of Lorentzian peaks as previously described (Günther and Gleason 1980; Cloarec et al. 2005). Aldrin resonance positions where retrieved from the SDBS online database (SDBSWeb: http://riodb01.ibase.aist.go.jp/sdbs/ (National Institute of Advanced Industrial Science and Technology, accessed in August 2010). A simulated NMR spectrum of Aldrin contains 53 out of 420 data points which are not equal to zero.
Exposure to this pollutant was introduced by superimposing the simulated NMR spectrum of Aldrin to the NMR spectra of serum samples of healthy subjects from the group of cases (randomly selected subjects: 30 out of 60 for small NMR data sets and 100 out of 200 for large NMR data sets). For the small NMR data sets the magnitudes of pollutant levels were chosen to range from 0 (no exposure to pollutant) to 50 times (0, 10, 15, 20, 25, 30, 35, 40, 45, 50) of the average observed concentration of Aldrin in blood. For the large NMR data sets, the exposure intensity ranged from 0 to 20 times (0, 2, 2.5, 5, 7.5, 10, 12.5, 15, 17.5, 20) of average concentration of Aldrin in blood.
3.2 Data analysis procedure and software
PLS-DA with a double cross validation procedure and four different diagnostic statistics was used. This procedure was applied M-times to each of UPLC-MS and NMR data sets with superimposed nutritional or exposure effects (10 UPLC-MS data sets, 100 small NMR data sets and 100 large NMR data sets). That resulted in M submodels for each data set (M = 30 for each of UPLC-MS data set and M = 20 for each of NMR data set). The performance of the PLS-DA model of each data set was evaluated on the basis of means of diagnostic statistics calculated across M submodels (see Fig. 1b) and related to means of diagnostic statistics of permutation tests using Eq. 7 to obtain P-value (for more information see Supplementary Material). A number of 3000 permutation tests for each of UPLC-MS data sets and 2000 permutation tests for each of NMR data sets were calculated using the same procedure as described above but with permuted y. All analyses were done in Matlab 2010a (The Mathworks Inc., Natick, Massachusetts, USA), using in-house routines, partly based on the PLS Toolbox (Eigenvector Research Inc, Wenatchee WA). Permutation tests have been performed on the LISA-SARA Dutch super-computer (www.sara.nl).
4 Results and discussion
4.1 Statistical significance of PLS-DA models vs. magnitudes of superimposed effects and used diagnostic statistics
Performance of PLS-DA model depends not only on the data set used, thus differences between two classes (e.g. magnitude of nutritional effects present in lipid profiles of subjects from the class of cases). It can also depend on values of diagnostic statistics used in optimization and performance assessment of the PLS-DA model (see Fig. 1). When differences between two classes are becoming very small, the power of each of the diagnostic statistics can be easily investigated in terms of their ability to provide a statistically significant measure of the discrimination between the two classes. This is accomplished by superimposing known multivariate effects of decreasing magnitude onto data of subjects from the class of cases and calculating a series of PLS-DA models. In this series, a single PLS-DA model is obtained for each of many data sets with different magnitudes of superimposed effects and one of four diagnostic statistics used in optimization and performance assessment. The most powerful diagnostic statistic is the one which provides a statistically significant PLS-DA model calculated for data with the smallest superimposed effect.
4.1.1 UPLC-MS data sets with superimposed nutritional effects
10 PLS-DA models were calculated for UPLC-MS data sets with different magnitudes of superimposed effects and each of four diagnostic statistics (Q 2, DQ 2, NMC or AUROC) as described in Sect. 3.2. Each of 10 PLS-DA models contained 30 submodels. The ranges of the 30 values of each of the diagnostic statistics (obtained by 30 PLS-DA submodels for each PLS-DA model, see Fig. 1b) are presented in Table 1. It should be notified that (i) the quality of the PLS-DA models decreases when the magnitude of the effects decrease: the values of Q 2, DQ 2 and AUROC decrease and value of NMC increases and (ii) the range of the values of the diagnostic statistics increases when the magnitude of the effect decreases.
As already mentioned in the introduction section, the values of diagnostic statistics do not alone indicate if quality of model is good or bad and if differences between two classes are statistically relevant or not. Statistical significance of diagnostic statistics values of any (sub)model can be assessed by comparing them or their means (see Fig. 1b) to values of their null reference distributions H0 obtained by permutation tests (see Sect. 2.2.6). A plot of the P-values for each of the four diagnostic statistics as a function of the effect magnitude (statistical significance profile of each diagnostic statistics) is presented in Fig. 3a. The significance threshold α is usually set to 0.05 in the majority of metabolomics applications. That means that P-value smaller that 0.05 indicates that the null hypothesis H0 (no difference between the two classes) can be rejected and observed difference between groups is assumed to be statistically significant at α = 0.05. By inspection of Fig. 3a it appears that, given an effect magnitude, different diagnostic statistics give different P-values. To infer a statistically significant discrimination between the two classes (P-value ≤ 0.05), the effect magnitude need to be about 1, 0.85, 0.55 and 0.55 when Q 2, DQ 2, AUROC or NMC are used, respectively. Models using NMC and AUROC clearly outperform those based on Q 2 and DQ 2. NMC/AUROC based models give significant discrimination for an effect magnitude (Δ effect ≥0.55 × original effects) which is half of that required for models based on Q 2 and DQ 2 (Δ effect ≥1 × original effects).

Statistical significance profiles for PLS-DA models of UPLC-MS data set (a) and NMR data set (b) when NMC, AUROC, Q 2 and DQ 2 are used. Profiles show ability of each diagnostic statistics to provide a statistically significant measure of the discrimination between two classes (P-value) as a function of the magnitude of the multivariate effects added on the data (∆ effect)
Interestingly, the DQ 2 and Q 2 P-values for very small effects are not equal to 0.5. This fact may be related either to inadequate number of PLS-DA submodels or to an undersampling of the DQ 2 and Q 2 H0 distributions due to a limited number of permutations. The number of PLS-DA submodels (30 in our case) can be insufficient to obtain a representative mean value of the DQ 2 and Q 2 statistics. That is highly probable when distribution of 30 values is not symmetric. On the other hand, distributions of diagnostic statistics in permutation tests can also be essential in estimating P-value. Distributions of permutation tests of ŷ i and diagnostic statistics for models of UPLC-MS data set with 0.75 × effect were plotted in Supplementary Fig. 2. Shapes of distributions of permutation tests of NMC and AUROC are symmetric in contrary to DQ 2 and Q 2 distributions which are left-side skewed. Distributions of permutation tests of DQ 2 and Q 2 should be chi-square distributions because they are distributions of sum of squares (Eqs. 3 and 5) but there are not many values of permutation tests in the right tail of those H0 distributions when 3000 permutation tests are used. That makes an accurate estimation of the P-value of diagnostic statistics such as DQ 2 and Q 2 of the original models difficult and raises a question about the number of submodels and permutation tests required to properly estimate P-values. Another solution can be to apply resampling methods such as bootstrap in combination with permutation testing.
4.1.2 NMR data sets with superimposed exposure effect
The multivariate effects added to the NMR data sets were intended to mimic the exposure to a chemical pollutant. The overall strategy of superimposing known multivariate effects was an analogue to strategy applied to the UPLC-MS data sets (see Sect. 4.1.1) but for each magnitude of superimposed effects, 10 different data sets have been randomly generated for a grand total of 100 data sets. Therefore, the results presented for each magnitude of superimposed effects refer to the average values over the 10 data sets. This extended strategy was chosen to take into account the intrinsic variability when a data set is build by sampling subjects from a larger population. The ranges of the four diagnostic statistics for the different PLS-DA models of small NMR data sets are given in Table 2. Presented ranges show a similar behavior to that observed for the UPLC-MS data sets (Table 1). Figure 3b presents the P-values (averaged over the 10 data sets) as a function of the effect magnitude (statistical significance profile of each diagnostic statistics) for small NMR data sets. Here also, NMC and AUROC outperform Q 2 and DQ 2 in term of providing a statistically significant discrimination between classes. With the α = 0.05, significant statistical discrimination is obtained for effect magnitude ≥20 for NMC/AUROC optimized models. Magnitude ≥35 and 40 is required for PLS-DA models optimized with DQ 2 and Q 2, respectively. Again an effect magnitude ratio 1:2 is observed as in the case of PLS-DA models of UPLC-MS data sets. Similar results and conclusions also apply to the large NMR data sets (see Supplementary Fig. 3).
Interestingly, the averaging over ten different data sets leads to P-values ≈0.5 for the Q 2 and DQ 2 when no effect is present, as it should be when no differences between classes is expected. That was not the case for the UPLC-MS data sets where only one data set is used for each effect magnitude. There number of PLS-DA models and permutation tests was not enough to properly estimate P-values.
4.2 Properties of PLS-DA models and diagnostic statistics
In order to explain observed differences in the performances of the models optimized and assessed by different diagnostic statistics, other properties of obtained models were evaluated further. Models complexity, distributions and reproducibility of models predictions were studied.
4.2.1 Complexity of PLS-DA models in CV1
As illustrated in Fig. 1a, the Q 2, DQ 2, NMC, and AUROC were used in the CV1 loop of the double cross-validation procedure to optimize the number of latent variables of each PLS-DA model generated in the CV1 loop. The use of a particular diagnostic statistic in this place is a critical point for differences in PLS-DA models performances presented in Fig. 3. The type of diagnostic statistics can strongly influence the model complexity, i.e. the number of latent variables used in the model. That becomes even more evident when complexity of models (ranging from 1 to 6 latent variables) is evaluated (Fig. 4a). It can be observed that NMC/AUROC optimized models have usually more latent variables (#LVs: 4–6) than Q 2/DQ 2 optimized models (#LV: 1–3). Moreover, this tendency is even more prominent when differences between classes are getting smaller and performance of models is getting worse (Supplementary Fig. 4). This means that models selected by Q 2 and DQ 2 are usually more simple and conservative than those selected by NMC/AUROC. Taking into account that NMC/AUROC selected models are more powerful in detecting small differences between groups it can be concluded that NMC/AUROC select models which use information about these differences more extensively and for that a higher number of latent variables is required. However, the higher number of latent variables makes interpretation of results more difficult and the number of latent variables should generally be limited to a few latent variables. A maximum number of 6 latent variables used in this study was chosen on the basis of previous analysis of UPLC-MS and NMR data sets where 4–5 latent variables were usually enough.

Complexity of the PLS-DA models. a dependency of the number of latent variables (#LV) upon the diagnostic statistics (NMC, AUROC, Q 2 and DQ 2) used in the model optimization in CV1 of double cross validation procedure for UPLC-MS data set with magnitude of superimposed effects equal 0.75, b Ranks of 53 biomarkers (variables associated with superimposed multivariate effect) obtained by PLS-DA models with different complexity (1–6 latent variables). PLS-DA models were obtained for small NMR data set with magnitude of superimposed effects equal 45. Ranks of biomarkers were obtained over all 420 variables in the data set according to the corresponding absolute values of their PLS regression coefficients. Ranks of biomarkers statistically significant at α = 0.05 are shown as dashed line and non-significant as regular line
The complexity of the PLS-DA models has a direct impact on model interpretation. PLS-DA models can be used for biomarker discovery, for instance by looking at the relative importance variables used in the PLS-DA model. This can done by ranking the variables according the value of their PLS regression coefficients: the variable with the largest (in absolute value) coefficient gets rank 1, the second one rank 2 and so on (Breitling, Armengaud et al. 2004)
In case of NMR data sets where simulated Aldrin spectra was added, 53 biomarkers associated with exposure to Aldrin (non-zero data points of Aldrin spectrum) are expected to be found by the PLS-DA models. Figure 4b shows the ranks of those 53 variables (for small NMR dataset with magnitude of added effects equal 45) for six PLS-DA models with different model complexity (from 1 to 6 latent variables). Each horizontal line presents a rank of one of 53 biomarkers. Minimal rank is in this case 1 (the most important variable out of 420 variables used in PLS-DA model) and maximal rank is 420 (the least important variable out of 420 variables used in PLS-DA model). Statistical significance of presented ranks was assessed by 10000 permutation tests and the corresponding P-values were calculated as detailed in Sect. 2.2.6. Ranks of biomarkers which obtained a P-value <0.05 are marked in blue and those with P-value >0.05 are marked in red.
Figure 4b shows that the complexity of the model does influence ranks of variables but most importantly influences statistical significance of variables with low ranks (see variables with rank 1–30). It appears that simple models built with fewer latent variables (LV from 1 to 3, as those usually selected by Q 2/DQ 2 in CV1) fail in providing statistical significance for a great number of these low rank variables, thus those variables will be omitted during biomarker selection. On the contrary, models built with more latent variables (LV from 4 to 6, as those usually selected by NMC/AUROC in CV1) are able to provide statistical significance to those most important variables. In this light it appears that more complex models (selected by NMC/AUROC) provide not only better discrimination of case and control group but also are more informative and accurate in term of biomarker discovery.
4.2.2 Distribution of predicted class membership vs. model complexity in CV1
In CV1 the diagnostic statistics are calculated on the basis of the predictions of categorical variable ŷ for validation set samples (Fig. 1a). Distributions of ŷ i obtained for all samples of validation sets in CV1 for all 30 submodels of the PLS-DA model of UPLC-MS data set with 0.75 × effect were investigated. They are plotted for 1–6 latent variables (#LV) in Fig. 5. In a case of ideal discrimination of two classes, half of ŷ i should be equal to −1 (class of controls) and other half to 1 (class of cases). This is hardly true in metabolomics studies because of the inherent variation between the individuals within the same class. It was also not the case in our data sets, where a majority of ŷ i has values between −1 and 1. The distributions of ŷ i vary between models with different number of latent variables. The range of values of ŷ i increases and shape of distribution is getting more flat when #LV is increasing. Smaller number of samples with ŷ i close to discrimination threshold value (0 in this case) is observed for models with greater complexity. That means smaller number of ambiguous samples in the model and better model performance for more complex models (usually selected by NMC and AUROC).

Distributions of ŷ i (ypred) of validation set samples in CV1 vs. number of latent variables (#LV) in the PLS-DA model
When NMC and AUROC values are calculated it is only important on which side of discrimination threshold value is ŷ i (< or >0) but not its value itself. In this way the values of ŷ i greater than 1 and lower than −1 do not influence NMC and AUROC values more than the values of ŷ i between −1 and 1 do. This is in contrary to Q 2 and DQ 2 which do not base on threshold value but on prediction error between values of ŷ i and y i treating their values as values of quantitative variable. In this case, the values of ŷ i greater than 1 and lower than −1 do increase prediction error and decrease values of Q 2 (DQ 2) prominently. Complex PLS-DA models (#LV > 3) have wider ranges of ŷ i , greater prediction error and lower values of Q 2 and DQ 2. That explains why when DQ 2 and Q 2 are used in the model optimization in CV1 less complex models are selected. When DQ 2 and Q 2 are used, the phrase “better safe than sorry” is followed. Model with the smallest prediction error e.g. the majority of ŷ i in a “safe” range −1 to 1 is selected and a number of samples with correctly predicted class labels is not taken into account.
4.2.3 Reproducibility of predictions of PLS-DA models in CV2
As detailed in Sect. 2.1.2 (see also Fig. 1b), for UPLC-MS data set 30 different prediction vector ŷ of the original class membership vector y are generated by 30 submodels after CV2 procedure. That assures that the finally considered ŷ is independent of random combinations of samples used in double cross-validation procedure. Reproducibility of ŷs across different submodels can be easily employed in describing PLS-DA models stability. For each study sample the variance across 30 prediction values ŷ i of different submodels could be estimated and used in assessment of PLS-DA model stability.
The variance of ŷ i across each of the 96 samples in the UPLC-MS data set was calculated. Obtained variances were averaged to give one mean variance representative for predictions of all samples. This procedure was applied separately to ŷs obtained by the submodels with four different diagnostic statistics and 10 different effects superimposed. The results are graphically shown in Fig. 6. For the largest effect magnitude (∆ effect = 1) UPLC-MS data set all PLS-DA models show a significant (at α = 0.05) discrimination between the two classes. Then the mean variance of ŷ is ≈0.12 and it is independent on diagnostic statistics used in model optimization. When the magnitude of the effect is smaller than 0.55 and the discrimination between the two classes is not significant for all of presented models (Fig. 3a) and mean variance of ŷ is dependent on applied diagnostic statistics. Then, models optimized on the basis of NMC or AUROC statistics are less reproducible than those optimized by Q 2 or DQ 2. The reproducibility of ŷ by the models optimized by DQ 2 and Q 2 decreases when the magnitude of the effect increases. This is an opposite behavior to this of ŷ obtained by models optimized with NMC and AUROC. The mean variance of ŷ for models optimized with Q 2 and DQ 2 is ca. two times smaller than this of models with large significant effects.

Mean variance of the prediction ŷ as a function of the magnitude of superimposed multivariate effect (Δ effect) in the case of UPLC-MS data sets
In conclusion, for small statistically insignificant effects in data sets, Q 2/DQ 2 optimized models tend to give very reproducible predictions what is in contrary to less reproducible predictions NMC and AUROC optimized models. That indicates that Q 2/DQ 2 optimized models are more stable and conservative than NMC and AUROC optimized models. This property can be associated with lower complexity of those models described in Sects. 4.2.1 and 4.2.2.
5 Conclusion remarks
NMC, AUROC and DQ 2, Q 2 belong to two separate groups of diagnostic statistics used in optimization and performance assessment of PLS-DA models. Several theoretical and practical differences between those diagnostic statistics were presented in this paper.
PLS-DA models using NMC or AUROC as diagnostic statistics are more powerful in detecting small differences between two groups than models using DQ 2 or Q 2. This phenomenon is related to two factors: complexity of PLS-DA models optimized during CV1 and distributions of submodels and permutation tests used to calculate P-value. During CV1, due to assumptions of (D)Q 2 diagnostics statistics, models with lowest prediction error of class membership are selected and these are not always the models with best discrimination power. Additionally, number of PLS-DA submodels as well as number of permutation tests sufficient for estimation P-values of NMC and AUROC is usually not enough to properly estimate P-values of DQ 2 or Q 2. Finally, PLS-DA models with NMC or AUROC as diagnostic statistics are more accurate in finding biomarkers responsible for two classes discrimination with PLS-DA method.
Our recommendation for metabolomic studies with two classes discrimination problem is to use NMC or AUROC as diagnostic statistics of PLS-DA models.
References
Altman, D. G., & Bland, J. M. (1994). Diagnostic tests. 1: Sensitivity and specificity. BMJ: British Medical Journal, 308(6943), 1552.
Barker, M., & Rayens, W. (2003). Partial least squares for discrimination. Journal of Chemometrics, 17(3), 166–173.
Bernini, P., Bertini, I., et al. (2009). Individual human phenotypes in metabolic space and time. Journal of Proteome Research, 8(9), 4264–4271.
Breitling, R., Armengaud, P., et al. (2004). Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Letters, 573(1–3), 83–92.
Broadhurst, D. I., & Kell, D. B. (2006). Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics, 2(4), 171–196.
Carreño, J., Rivas, A., et al. (2007). Exposure of young men to organochlorine pesticides in Southern Spain. Environmental Research, 103(1), 55–61.
Churchill, G. A., & Doerge, R. W. (1994). Empirical threshold values for quantitative trait mapping. Genetics, 138(3), 963.
Cloarec, O., Dumas, M. E., et al. (2005). Evaluation of the orthogonal projection on latent structure model limitations caused by chemical shift variability and improved visualization of biomarker changes in 1H NMR spectroscopic metabonomic studies. Analytical Chemistry, 77(2), 517–526.
Cruciani, G., Baroni, M., et al. (1992). Predictive ability of regression models. Part I: Standard deviation of prediction errors (SDEP). Journal of Chemometrics, 6(6), 335–346.
Davis, J., & Goadrich, M. (2006). The relationship between precision-recall and roc curves. New York: ACM.
Fawcett, T. (2004). ROC graphs: Notes and practical considerations for researchers. Machine Learning, 31, 1–38.
Fisher, R. A. (1937). The design of experiments. Edinburgh: Oliver and Boyd.
Golland, P., Liang, F., et al. (2005). Permutation tests for classification. Learning Theory, 501–515.
Günther, H., & Gleason, R. W. (1980). NMR spectroscopy: An introduction. New York: Wiley.
Hu, C., van Dommelen, J., et al. (2008). RPLC-ion-trap-FTMS method for lipid profiling of plasma: Method validation and application to p53 mutant mouse model. Journal of Proteome Research, 7(11), 4982–4991.
Kind, T., Tolstikov, V., et al. (2007). A comprehensive urinary metabolomic approach for identifying kidney cancer. Analytical Biochemistry, 363(2), 185–195.
Larsen, T. M., Dalskov, S., et al. (2009). The Diet, Obesity and Genes (Diogenes) Dietary Study in eight European countries—a comprehensive design for long-term intervention. Obesity Reviews, 11(1), 76–91.
Lindgren, F., Hansen, B., et al. (1996). Model validation by permutation tests: Applications to variable selection. Journal of Chemometrics, 10(5–6), 521–532.
Lino, C. M., & Silveira, M. (2006). Evaluation of organochlorine pesticides in serum from students in Coimbra, Portugal: 1997–2001. Environmental Research, 102(3), 339–351.
Lloyd, G. R., Ahmad, S., et al. (2009). Pattern recognition of inductively coupled plasma atomic emission spectroscopy of human scalp hair for discriminating between healthy and hepatitis C patients. Analytica Chimica Acta, 649(1), 33–42.
Martin, J. T. (1958). Agricultural spray chemicals. Occupational Medicine, 8(1), 11.
Mielke, P. W., & Berry, K. J. (2007). Permutation methods: a distance function approach. New York: Springer.
Pesarin, F., & Salmaso, L. (2010). Permutation tests for complex data: Theory, applications and software. New York: Wiley.
Smit, S., van Breemen, M. J., et al. (2007). Assessing the statistical validity of proteomics based biomarkers. Analytica Chimica Acta, 592(2), 210–217.
Stevens, S. S. (1946). On the theory of scales of measurement. Science, 103(2684), 677–680.
Sun, W., & Wright, F. A. (2010). A geometric interpretation of the permutation p-value and its application in eQTL studies. Annals, 4(2), 1014–1033.
Trygg, J., & Wold, S. (2002). Orthogonal projections to latent structures (O-PLS). Journal of Chemometrics, 16(3), 119–128.
van Velzen, E. J. J., Westerhuis, J. A., et al. (2008). Multilevel data analysis of a crossover designed human nutritional intervention study. Journal of Proteome Research, 7(10), 4483–4491.
Weckwerth, W., Loureiro, M. E., et al. (2004). Differential metabolic networks unravel the effects of silent plant phenotypes. Proceedings of the National Academy of Sciences of the United States of America, 101(20), 7809–7814.
Westerhuis, J. A., Hoefsloot, H. C. J., et al. (2008a). Assessment of PLSDA cross validation. Metabolomics, 4(1), 81–89.
Westerhuis, J. A., van Velzen, E. J. J., et al. (2008b). Discriminant Q 2 (DQ 2) for improved discrimination in PLSDA models. Metabolomics, 4(4), 293–296.
Xia, J., Psychogios, N., et al. (2009). MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Research, 37(Suppl 2), W652.
Yang, J., Xu, G., et al. (2004). Diagnosis of liver cancer using HPLC-based metabonomics avoiding false-positive result from hepatitis and hepatocirrhosis diseases. Journal of Chromatography B, 813(1–2), 59–65.
Younos, T. M., & Weigmann, D. L. (1988). Pesticides: a continuing dilemma. Journal (Water Pollution Control Federation), 60(7), 1199–1205.
Acknowledgments
This project was financed by the Netherlands Metabolomics Centre (NMC) which is a part of the Netherlands Genomics Initiative/Netherlands Organisation for Scientific Research. Authors gratefully acknowledge Ferdi van Dorsten (NMC, Unilever), Rob Vreeken, Jorne Troost and Iryna Paliukhovich (NMC, LACDR/Leiden University) for providing UPLC-MS data set and Jacques Vervoort (NMC, Wageningen University) for providing NMR data set. John Donners and Willem Vermin (SARA, the Dutch National High Performance Computing and e-Science Support Center, www.sara.nl) are also acknowledged for the help in implementing multi-thread calculations.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Szymańska, E., Saccenti, E., Smilde, A.K. et al. Double-check: validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 8 (Suppl 1), 3–16 (2012). https://doi.org/10.1007/s11306-011-0330-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-011-0330-3