
Abstract
Hybridization between plant species can have a number of biological consequences; interspecific hybridization has been tied to speciation events, biological invasions, and diversification at the level of genes, metabolites, and phenotypes. This study aims to provide evidence of transgressive segregation in the expression of primary and secondary metabolites in hybrids between Jacobaea vulgaris and J. aquaticus using an NMR-based metabolomic profiling approach. A number of F2 hybrid genotypes exhibited metabolomic profiles that were outside the range encompassed by parental species. Expression of a number of primary and secondary metabolites, including jacaronone analogues, chlorogenic acid, sucrose, glucose, malic acid, and two amino acids was extreme in some F2 hybrid genotypes compared to parental genotypes, and citric acid was expressed in highest concentrations in J. vulgaris. Metabolomic profiling based on NMR is a useful tool for quantifying genetically controlled differences between major primary and secondary metabolites among plant genotypes. Interspecific plant hybrids in general, and specifically hybrids between J. vulgaris and J. aquatica, will be useful for disentangling the ecological role of suites of primary and secondary metabolites in plants, because interspecific hybridization generates extreme metabolomic diversity compared to that normally observed between parental genotypes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Hybridization between plant species can have a host of both immediate and long-term consequences for plant metabolism. On one hand, interspecific hybridization can sometimes lead to the expression of novel secondary metabolites in hybrid lines (Orians 2000). For example, novel sesquiterpene lactones were generated in hybrids between two Helianthus species, resulting from novel combinations of basic structural skeletons from one species with side chains from the other (Buschmann and Spring 1995). On the other hand, primary and secondary metabolites can be expressed in extreme concentrations within interspecific hybrid genotypes (Orians 2000), and these extremes can translate into novel plant phenotypes. For instance when secondary metabolites involved in plant-herbivore defense are expressed in extremely high or low concentrations, interspecific hybrids can be more or less resistant than parents to attack by herbivores (Strauss 1994). Interspecific hybrids can often be clearly distinguished from parental species based on their biochemical phenotypes. For example, natural Jacobaea hybrids can be identified based on unique profiles of pyrrolizidine alkaloids (Kirk et al. 2004), interspecific eucalypt hybrids can be distinguished from parents based on intermediacy in concentrations of foliar metabolites (Nahrung et al. 2009), and interspecific Solanum hybrids can be distinguished from parents based on the presence of two novel glycoalkaloids (Savarese et al. 2009).
The additional variation in plant metabolism generated by interspecific hybridization is interesting for several reasons. First, new variation can provide the basis for new ecological interactions and novel evolutionary trajectories as a result of selection. In oaks for example, interspecific hybridization affects biological and ontogenic variation in the expression of secondary metabolites, and this variation correlates to the community structure of leaf-miners in the hybrid oak population (Yarnes et al. 2008). Second, variation in metabolism within natural interspecific hybrids can have consequences with regard to biodiversity of plant communities and the other species that they support. For instance, biodiversity of arthropods associated with hybrids of Eucalyptus spp. (Whitham et al. 1994; Dungey et al. 2000) and oak spp. (Nakamura et al. 2010) was found to be higher than that associated with parental species. Last, interspecific hybridization creates novel variation that biologists can use to study the effects of primary and secondary metabolites on plant growth and on interactions between plants and their environment (Hochwender et al. 2000; Orians et al. 2010).
In recent years, both natural and artificial interspecific hybrids have been used increasingly for the latter purpose. Because segregation occurs in F2 and later generation hybrids, these can be used to look for tradeoffs, correlations, and other genetic relationships between different traits. Segregation in F2 and later generation interspecific hybrids can lead to a decoupling of traits that are correlated within single species as a result of genetic linkage (Hochwender et al. 2000; Lexer et al. 2003; Orians et al. 2010). Also, interspecific hybrids often exhibit much greater variation than parental species as a result of transgressive segregation (Rieseberg et al. 1999), and this variation is useful for ecologists and evolutionary biologists. For example, Orians et al. (2010) were able to use high levels of variation in plant growth and expression of defense compounds within F2 families of interspecific hybrid willow seedlings to show transient tradeoffs between growth and the expression of defense compounds. These tradeoffs are often difficult to show within single species, where variation in growth and defense traits is lower (Orians et al. 2003).
In this study, we used a metabolomics approach to investigate the expression of a suite of primary and secondary metabolites in a family of F2 hybrids resulting from an interspecific cross between Jacobaea vulgaris (syn. Senecio jacobaea) and J. aquatica (syn. Senecio aquaticus). These two species are differentiated in terms of both habitat preferences and the suite of natural enemies that attacks them (Kirk et al. 2005a). Natural interspecific hybrids between J. vulgaris and J. aquatica occur in The Netherlands, and substantial backcrossing to J. vulgaris provides potential for introgression of novel patterns of secondary metabolite expression (Kirk et al. 2004). A previous study found that F1 interspecific hybrids were generally intermediate to both parents or equal to one parent with regard to the expression of the most variable compounds in the metabolome (Kirk et al. 2005b). In this study, we aimed to determine (i) whether F2 interspecific hybrid genotypes exhibit high levels of metabolomic variation, (ii) which metabolites are responsible for most of this variation, and (iii) whether there is evidence for transgressive segregation in the expression of primary and secondary metabolites responsible for most of the metabolomic variation. This is one of the first studies of interspecific hybrid phytochemistry that incorporates a wide array of both primary and secondary metabolites (but see Leiss et al. 2009a).
2 Materials and methods
2.1 Study system
Jacobaea vulgaris (Tansy ragwort or Common ragwort) and J. aquaticus (marsh ragwort) are both native to Europe, and are closely related but not sister species (Pelser et al. 2003). Jacobaea vulgaris mainly grows in disturbed dry, sandy soils while J. aquatica grows in seasonally flooded habitats with high soil organic matter (Kirk et al. 2005a). Natural interspecific hybrids between J. vulgaris and J. aquatica occur at The Zwanenwater nature reserve in The Netherlands (Kirk et al. 2004). For this study, J. vulgaris seeds were collected at Meijendel, a coastal dune area in the North of The Hague (The Netherlands), and J. aquatica seeds were collected at the Zwanenwater Reserve (The Netherlands). Seeds of the two species were germinated in petri dishes and transplanted to pots (humidity 70%, temperature 20°C, light 16 h with sensitivity of 11.3 ± 1.6 SD μmol photons/m2/s, dark 8 h). One J. vulgaris genotype was crossed with one J. aquatica genotype by rubbing flower heads together (Kirk et al. 2005c). Parental genotypes were randomly selected from natural populations of J. vulgaris and J. aquatica that represent ecotypes found to hybridize in nature (Kirk et al. 2004). Two F1 genotypes from the initial parental (P) cross were crossed reciprocally which each other to produce an F2 generation (a number of F1 crosses were made, and we selected the family that produced the greatest number of viable F2 genotypes). 102 F2 genotypes were used in this experiment, including 56 genotypes from one F1 mother, and 46 F2 genotypes from the second F1 mother. The P, F1 and F2 genotypes were maintained in tissue culture according to Kirk et al. (2010) and could be cloned to perform experiments with replicate genotypes. We used ca. 2 cloned replicates per F2 genotype and 4 cloned replicates per P and F1 genotype.
2.2 Plant growth and harvesting
Genotypes in tissue culture were maintained on multiplying medium, and were transplanted to rooting medium two weeks prior to potting (Kirk et al. 2010). Once roots had developed, plants were potted in 1.3 liter pots filled with 95% sand collected from Meijendel, and 5% potting soil (Slingerland Potgrond Company, Zoeterwoude, The Netherlands) mixed with 1.5 g l−1 Osmocote slow release fertilizer (Scotts®, Marysville, USA; N:P:K = 15:9:11). The plants were grown in a climate chamber for 6 weeks (humidity 70%, light 16 h at 20°C, dark 8 h at 20°C).
After 6 weeks the plants were harvested; whole plants were removed from the soil and shoots (all aerial parts) were removed by cutting with scissors immediately above the root crown. Shoots were immediately wrapped separately in aluminum foil and were stored in a Styrofoam box with liquid nitrogen until harvesting (performed between 10:00 am and 11:00 am) was complete, after which they were stored for approximately one month at −80°C. Before extractions were carried out, shoots were freeze-dried for four days in a freeze drier with a collector temperature of −55°C (Labconco Free Zone® 12 liter Freeze Dry System, Kansas City, USA). Shoots were then ground to a fine powder and were stored at −20°C for one week until extraction.
2.3 Extraction of plant material
Dry material (30 mg) was a 2 ml-microtube to which 2 ml of 50% CH3OH-d 4 in buffer (90 mM KH2PO4 in D2O) containing 0.05% TMSP (trimethyl silyl propionic acid sodium salt-d 4, w/v) were added. The mixture was vortexed at room temperature for 30 s, ultrasonicated for 1 min, and centrifuged at 10,968×g at 4°C for 20 min. 700 μl of the supernatant was taken for NMR analysis, which was carried out within 30–60 min. from extraction.
2.4 Nuclear magnetic resonance measurements
NMR simultaneously detects most groups of organic compounds in a mixed sample, including carbohydrates, organic and fatty acids, amino acids, and some secondary metabolites (Schripsema 2010). Proton (1H) NMR functions by detecting protons attached to compounds in a sample, such that one compound is typically represented by multiple signals (protons) in the NMR spectrum. 1H NMR displays proton signals based on the bonding of each proton to other atoms in compound. The details of our NMR protocol are described in Appendix.
It is not possible to report the number of compounds appearing in the NMR spectra because multiple peaks represent each compound and identification of all compounds is impractical. However, it is usually estimated that NMR detects a range of 30–50 of the most highly accumulated plant compounds (Kirk et al. 2005b; Leiss et al. 2009a). In our experience, metabolites occurring in concentrations less than ≈1 μmol mg−1 are generally not detectable using the methods described here. Also, the sensitivity of the analysis depends on the nature of target compounds. Some areas of the spectra, such as the region where many carbohydrates are found, contain many signals, and metabolites occurring in this region may be difficult to detect if present in low concentrations. Conversely, relatively few signals occur in the phenolic region of the spectra, and even weak signals may be detected here.
The identification of compounds was based on comparison to compounds previously reported to occur in NMR-spectra of J. aquatica, J. vulgaris, and their hybrids (Kirk et al. 2005b; Leiss et al., 2009a). Also, two-dimensional NMR spectra were applied for identification purposes (Appendix), as well as our in-house database (700 individual metabolites). J-resolved spectra were used for deconvolution of multiplicity, Proton–Proton couplings were confirmed by 1H–1H-correlated spectroscopy (COSY). Proton–13C couplings were obtained by Heteronuclear Single Quantum Coherence (HSQC) for direct coupling and Heteronuclear Multiple Bonds Correlation (HMBC) for long-range coupling. Table 1 reports the chemical shifts and coupling constants of compounds reported in the results. No compounds were identified in this study that were not identified in previous studies of these species and their interspecific hybrids. In this study, we do not discuss the expression of pyrrolizidine alkaloids (PAs), which are a well known class of secondary metabolites in Jacobaea species, because it is difficult to detect structural variations in PAs from a bulk extract using the methods described here. We carried out a separate study of PA expression using LC-MS, the results of which will be published separately (Chen et al. in review).
2.5 Data analysis
2.5.1 Initial data handling
The 1H-NMR spectra were automatically exported as a spreadsheet using AMIX software (ver. 3.7, Bruker Biospin, Karlsruhe, Germany). The spectra, which were measured in chemical shift (ppm), were binned (bucketed) into bins of equal width (0.04 ppm; 243 bins total), and the peaks represented within each bin were integrated (the area under the spectra was calculated). Integrated signals were scaled (standardized) to the internal standard TMSP, so that peak intensities represent intensity relative to the internal standard. Integrated and standardized signals were used for all further quantitative analysis of the data. The region from δ − 0.4 to 10.0 was included in the analysis, and the region from δ 4.7 to 5.0 was excluded from the analysis because the water signal masked all other signals in this region.
2.6 Principal component analysis
Principal component analysis (PCA) is a visual method that facilitates separation of plant groups according to overall metabolic fingerprints, and is based on unsupervised clustering requiring no knowledge of the data set; PCA acts to reduce the dimensionality of multivariate data while preserving most of the variance within it (Eriksson et al. 2001). The principal components can be displayed graphically as a ‘scores’ plot (Fig. 1). This plot is useful for observing any groupings in the data set. PCA models are constructed using all the samples in the study. Coefficients by which the original variables must be multiplied to obtain the PC are called loadings. The numerical value of a loading of a given variable on a PC shows how much the variable has in common with that component (Massart et al. 1988). Thus for NMR data, loading plots can be used to detect the spectral regions and their corresponding metabolites responsible for the separation in the data. Principal component analysis (PCA) was performed with SIMCA-P (ver. 12.0, Umetrics, Umeå, Sweden).

Quantitative variation in the 1H NMR spectra from an interspecific hybrid Jacobaea family. Spectra are derived from ethanol–water extracts of individual samples of one F1 hybrid genotype (F1), three F2 hybrid genotypes (genotypes 70146, 60202, 60179), one Jacobaea aquatica genotype (A), and one J. vulgaris (J) genotype. TMSP: internal standard. F2 genotypes shown are extreme compared to A and J with regard to one or more PC axis (illustrated in Fig. 2)
2.7 Quantification of individual metabolites
We quantified a number of individual metabolites based on one individual signal from each metabolite that was selected from a non-crowded region of the 1H NMR spectra (indicated in Table 1). Only a subset of metabolites could be individually quantified because for some metabolites, all signals overlapped with those from other metabolites. Quantities of metabolites are expressed in relative intensity (based on integration under spectral peaks, and relative to the internal standard TMSP), because NMR intensities are proportional to the absolute molar concentration.
3 Results and discussion
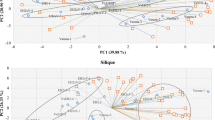
In this study, we characterized metabolomic variation among genotypes from a family of interspecific hybrids of J. vulgaris and J. aquatica. Two parental individuals (one genotype of each species, number of replicates of each genotype (n) = 3 and 4 respectively for J. vulgaris and J. aquatica), two F1 genotypes (n = 4), and 102 F2 genotypes (n = 2) were analysed. Obvious quantitative differences were observed among the 1H NMR spectra of different genotypes (Fig. 1). PCA provided a global overview of the differences between genotypes, and facilitated the identification of spectral regions and associated metabolites that discriminate between genotypes. The first four principal components (PCs) explained 64.1% of the variance in the metabolomic dataset. This is lower than the amount of variance explained by the first three axes in a previous study that incorporated only the parental species and the F1 hybrids (Kirk et al. 2005b). The two F1 genotypes appeared to be intermediate to J. vulgaris and J. aquatica based on all PC axes. Score plots (Fig. 2) showed that many F2 genotypes exhibited scores outside of those observed for both the P genotypes and the F1 genotypes based on PC 2 (Fig. 2a) and 3 (Fig. 2b). Compounds that explained variation in these axes included glutamine and glutamic acid, chlorogenic acid and jacaronone analogues, sucrose, glucose and aspartic acid (Fig. 3). Based on PCs 1 (Fig. 2a) and 4 (Fig. 2b), most F2 genotypes were intermediate to both parents or similar to one parent. Compounds that explained variation in these axes include fructose, mannitol, stachyose, citric acid, malic acid, and succinic acid (Fig. 3). There was substantial variation among F2 genotypes, and many of these exhibited variation in metabolomic profiles outside the range found in their parents and grandparents (Figs. 1, 2).

PCA analysis of 1H NMR spectra of extract from the shoots of one Jacobaea vulgaris genotype (n = 3; large blue dots), one J. aquaticus genotype (n = 4; large red dots), two interspecific F1 hybrid genotypes (n = 4 for each genotype; large black dots), and 102 F2 hybrid genotypes (n = 2 for each genotype; small grey dots). Score plots of PC1 against PC2 (a), and PC3 against PC4 (b) are shown. The large central ellipse represents Hotelling T2 with 95% confidence in score plots. The 1H NMR spectra of the three extreme (relative to parents) F2 genotypes indicated in this figure are shown in Fig. 1 (Color figure online)

Scatter plots of loadings of PC1 plotted against PC2 (a), and of PC3 plotted against PC4 (b). Dots on the scatter plot represent the magnitude of the correlation between each PC and the signal in individual bins (n = 243) relative to the internal standard TMSP. Each compound is generally represented by multiple signals, and signals from the same compound tend to co-vary within the PCA space. Signals were assigned to their associated metabolites, and these are circled and labeled in the scatter plot
It was not possible to quantify all compounds identified in this study due to some overlap in the signals of compounds within the spectra. However, we quantified a number of primary and secondary metabolites (Fig. 4) that showed distinct and non-overlapping signals in non-crowded regions of the spectra, including phenylpropanoids (chlorogenic acid), quinones (jacaronone analogues), sugars (sucrose and glucose), organic acids (malic acid and citric acid), and amino acids (alanine and threonine). We found evidence that transgressive segregation occurred in both directions with regard to all compounds except for citric acid. In other words, a considerable number of F2 genotypes exhibited extremely high and extremely low expression of all quantified compounds except for citric acid. In the latter case, J. vulgaris genotypes expressed the highest relative concentration of citric acid, and relative concentration of citric acid in all other genotypes was lower. Some F2 genotypes expressed extremely low concentrations of citric acid relative to the P and F1 generations. We also found that one F1 genotype expressed a relatively low concentration of glucose compared to parental species (Fig. 4).

Histograms representing the distributions of genotypes according to relative concentration of chlorogenic acid (a), jacaronone analogues (b), citric acid (c), malic acid (d), sucrose (e), glucose (f), alanine (g), and threonine (h). Arrows above figures indicate relative concentrations of given compounds in F1 hybrids (f), Jacobaea aquaticus (a), and J. vulgaris (J)
We set out to test whether extreme expression of both primary and secondary metabolites occurs in the interspecific F2 hybrids of J. vulgaris and J. aquaticus. Of the eight metabolites that we quantified, seven showed extreme concentrations in some F2 hybrid genotypes relative to both F1 hybrid genotypes and to parental species. Extreme concentrations were expressed in regard to both primary metabolites, such as amino acids (alanine and threonine), malic acid, glucose, and sucrose, and to secondary metabolites including chlorogenic acid and jacaronone analogues. It is sometimes argued that variation in primary metabolism should be constrained by the physiological demands of the plant, and variation in secondary metabolism should be greater due to varying degrees and direction of selection pressures through time and space. Citric acid was found to be an exception in that no hybrid genotypes contained higher concentrations of citric acid than the J. vulgaris genotype. Citric acid is an important intermediary in the Krebs cycle, and it is possible that it has already reached a physiological maximum in J. vulgaris. In any case, this finding suggests that all genes that have a positive effect on the concentration of citric acid are present in J. vulgaris.
With regard to the other seven metabolites that were quantified, the continuous distribution of F2 phenotypes and the presence of extreme F2 phenotypes suggests that their expression is controlled by several genes. Also, it is likely that both J. vulgaris and J. aquaticus possess genes of opposite effect on the expression of these metabolites (in other words, distinct genes that up- and down-regulate their production and/or accumulation), which is the basis for transgressive segregation (Rieseberg et al. 1999). High variation in F2 hybrid provides a promising future avenue for further ecological and evolutionary studies. For example, phenylpropanoids including chlorogenic acid and quinones (jacaronone analogues) are thought to play a variety of roles in interactions between plants and their environments. Leiss et al. (2009b) recently used a metabolomics approach to show that chlorogenic acid concentrations are correlated with thrips resistance in chrysanthemum. Jacaronone analogues have been shown to have insecticidal activity against houseflies (Xu et al. 2003) and can inhibit growth of the generalist herbivore Spodoptera litturalis (Lajide et al. 1996). Variation in concentrations of primary metabolites such as sugars may also play an important role in interactions with herbivores and pathogens (Zangerl and Berenbaum 2004), since high concentrations of energy-rich compounds are attractive to natural enemies.
4 Concluding remarks
Based on this study, we can identify interspecific hybrid ragwort genotypes that express high and low concentration of compounds including phenylpropanoids and sugars, and we can use them for future experiments incorporating herbivores and pathogens, in order to elucidate that role of these compounds on susceptibility of plants to attack by natural enemies. Also, using metabolomic analysis of interspecific hybrids combined with genetic analysis is a promising avenue for elucidating the genetic architecture of metabolomic expression. Such analyses may provide a more detailed understanding of the physiological and evolutionary constraints associated with the production and accumulation of primary and secondary metabolites in plants. NMR-based metabolomic profiling is a useful tool for identifying genotypic differences in major plant metabolites, and can yield major advances in chemo-ecological studies (Macel et al. 2010; Prince and Pohnert 2010).
References
Buschmann, H., & Spring, O. (1995). Sesquiterpene lactones as a result of interspecific hybridization in Helianthus species. Phytochemistry, 39, 367–371.
Dungey, H. S., Potts, B. M., Whitham, T. G., & Li, H. F. (2000). Plant genetics affects arthropod community richness and composition: Evidence from a synthetic eucalypt hybrid population. Evolution, 54, 1938–1946.
Eriksson, L., Johansson, E., Kettaneh-Wold, N., & Wold, S. (2001). Multi-and megavariate data analysis. Umeå: Umetrics Academy.
Hochwender, C. G., Fritz, R. S., & Orians, C. M. (2000). Using hybrid systems to explore the evolution of tolerance to damage. Evolutionary Ecology, 14, 509–521.
Kim, H. K., Choi, Y. H., & Verpoorte, R. (2010a). NMR-based metabolomic analysis of plants. Nature Protocols, 3, 536–549.
Kim, H. K., Saifullah, K., Wilson, E. G., Kricun, S. D. P., Meissner, A., Göraler, S., et al. (2010b). Metabolic classification of south American Ilex Species by NMR-based metabolomics. Phytochemistry, 71, 773–784.
Kirk, H., Choi, Y. H., Kim, H. K., Verpoorte, R., & van der Meijden, E. (2005a). Comparing metabolomes: The chemical consequences of hybridization in plants. New Phytologist, 167, 613–622.
Kirk, H., Macel, M., Klinkhamer, P. G. L., & Vrieling, K. (2004). Natural hybridization between Senecio jacobaea and Senecio aquaticus: Molecular and chemical evidence. Molecular Ecology, 13, 2267–2274.
Kirk, H., Vrieling, K., & Klinkhamer, P. G. L. (2005b). Maternal effects and heterosis influence the fitness of plant hybrids. New Phytologist, 166, 685–694.
Kirk, H., Vrieling, K., & Klinkhamer, P. G. L. (2005c). Reproductive fitness of hybrids between Senecio jacobaea and S aquaticus (Asteraceae). American Journal of Botany, 92, 1467–1473.
Kirk, H., Vrieling, K., van der Meijden, E., & Klinkhamer, P. G. L. (2010). Species by environment interactions affect pyrrolizidine alkaloid expression in Senecio jacobaea, Senecio aquaticus, and their hybrids. Journal of Chemical Ecology, 36, 378–387.
Lajide, L., Escoubas, P., & Mizutani, J. (1996). Cyclohexadienones-insect growth inhibitors from the foliar surface and tissue extracts of Senecio cannabifolius. Experientia, 52, 259–263. doi:10.1007/BF01920719.
Leiss, K. A., Choi, Y. H., Abdel-Farid, I. B., Verpoorte, R., & Klinkhamer, P. G. L. (2009a). NMR metabolomics of thrips (Frankliniella occidentalis) resistance in Senecio hybrids. Journal of Chemical Ecology, 35, 219–229.
Leiss, K. A., Maltese, F., Choi, Y. H., Verpoorte, R., & Klinkhamer, P. G. L. (2009b). Identification of chlorogenic acid as a resistance factor for thrips in chrysanthemum. Plant Physiology, 150, 1567–1575.
Lexer, C., Randell, R. A., & Rieseberg, L. H. (2003). Experimental hybridization as a tool for studying selection in the wild. Ecology, 84, 1688–1699.
Macel, M., van Dam, N. M., & Keurentjes, J. J. B. (2010). Metabolomics: The chemistry between ecology and genetics. Molecular Ecology Resources, 10, 583–593.
Massart, D. L., Vandeginste, B. G. M., Deming, S. N., Michotte, Y., & Kauffman, L. (1988). Chemometrics: A textbook. New York, USA: Elsevier.
Nahrung, H. F., Waugh, R., & Hayes, R. A. (2009). Corymbia species and hybrids: Chemical and physical foliar attributes and implications for herbivory. Journal of Chemical Ecology, 35, 1043–1053.
Nakamura, M., Asanuma, M., & Hiura, T. (2010). Differential effects of host plant hybridization on herbivore community structure and grazing pressure on forest canopies. Oikos, 119, 1445–1452.
Orians, C. M. (2000). The effects of hybridization in plants on secondary chemistry: Implications for the ecology and evolution of plant-herbivore interactions. American Journal of Botany, 87, 1749–1756.
Orians, C. M., Hochwender, C. G., Fritz, R. S., & Snall, T. (2010). Growth and chemical defense in willow seedlings: Trade-offs are transient. Oecologia, 163, 283–290.
Orians, C. M., Lower, S., Fritz, R. S., & Roche, B. M. (2003). The effects of plant genetic variation and soil nutrients on secondary chemistry and growth in a shrubby willow, Salix sericea: Patterns and constraints on the evolution of resistance traits. Biochemical Systematics and Ecology, 31, 233–247.
Pelser, P. B., Gravendeel, B., & van der Meijden, R. (2003). Phylogeny reconstruction in the gap between too little and too much divergence: The closest relatives of Senecio jacobaea (asteraceae) according to DNA sequences and aflps. Molecular Phylogenetics and Evolution, 29, 613–628.
Prince, E. K., & Pohnert, G. (2010). Searching for signals in the noise: Metabolomics in chemical ecology. Analytical and Bioanalytical Chemistry, 396, 193–197.
Rieseberg, L. H., Archer, M. A., & Wayne, R. K. (1999). Transgressive segregation, adaptation and speciation. Heredity, 83, 363–372.
Savarese, S., Andolfi, A., Cimmino, A., Carputo, D., Frusciante, L., & Evidente, A. (2009). Glycoalkaloids as biomarkers for recognition of cultivated, wild, and somatic hybrids of potato. Chemistry & Biodiversity, 6, 437–446.
Schripsema, J. (2010). Application of NMR in plant metabolomics: Techniques, problems and prospects. Phytochemical Analysis, 21, 14–21.
Strauss, S. Y. (1994). Levels of herbivory and parasitism in host hybrid zones. Trends in Ecology & Evolution, 9, 209–214.
Whitham, T. G., Morrow, P. A., & Potts, B. M. (1994). Plant hybrid zones as centers of biodiversity—the herbivore community of 2 endemic Tasmanian eucalypts. Oecologia, 97, 481–490.
Xu, H. H., Zhang, N. J., & Casida, J. E. (2003). Insecticides in Chinese medicinal plants: Survey leading to jacaranone, a neurotoxicant and glutathione-reactive quinol. Journal of Agriculture and Food Chemistry, 51, 2544–2547. doi:10.1021/jf021164x.
Yarnes, C. T., Boecklen, W. J., Tuominen, K., & Salminen, J. P. (2008). Hybridization affects seasonal variation of phytochemical phenotypes in an oak hybrid complex (Quercus gambelii × Quercus grisea). International Journal of Plant Sciences, 169, 567–578.
Zangerl, A. R., & Berenbaum, M. R. (2004). Genetic variation in primary metabolites of Pastinaca sativa; can herbivores act as selective agents? Journal of Chemical Ecology, 30, 1985–2002.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Appendix: Technical description of NMR measurements
Appendix: Technical description of NMR measurements
NMR experiments were carried out according to previously reported protocols from out lab (Kim et al. 2010a, b). 1D-1H NMR spectra, 2D J-resolved spectra as well as 1H–1H homonuclear and inverse detected 1H–13C correlation experiments were recorded at 25°C on a Bruker 600 MHz AVANCE II NMR spectrometer (600.13 MHz proton frequency) equipped with TCI cryoprobe and Z-gradient system. CD3OD was used for internal lock purposes. For 1D-1H NMR spectra a total of 32768 data points were recorded covering a spectral window of 9615 Hz. 128 scans of a standard one-pulse sequence with 30° flip angle for excitation and presaturation during 2.0 s relaxation delay with an effective field of γB1 = 50 Hz for suppression of the residual H2O signal was employed. An exponential window function with a line broadening factor of 0.3 Hz was applied prior to Fourier transformation. The resulting spectra were manually phased and baseline corrected, and referenced to internal TMSP at 0.0 ppm. For 2D-J-resolved NMR spectra a data matrix of 62 × 16384 data points covering 50 × 7739.4 Hz were acquired using 16 scans for each increment in F1. Presaturation was applied during a relaxation delay of 1.5 s with an effective field of γB1 = 50 Hz. Data were zero-filled to 512 × 32768 points prior to magnitude mode Fourier transformation with a sine shape window functions in both dimensions. The resulting frequency domain data were tilted by 45°, and then symmetrized along the F2 dimension (F1 = 0 Hz) and referenced according to internal TMSP. From the resulting 2D J-resolved spectra 1D-projection along the F2 dimension were extracted using the build-in positive projection routine in Topspin (version 2.1, Bruker Biospin). 1H–1H double-quantum filter correlation spectroscopy (DQF-COSY) spectra were acquired with presaturation (γB1 = 50 Hz) during a relaxation delay of 1.5 s. A data matrix of 1024 × 2048 points covering 7739.4 × 7739.4 Hz was recorded with 8 scans for each increment. Data was zero filled to 2048 × 2048 points prior to States-TPPI type 2D Fourier transformation and a sine bell shaped window function was applied in both dimensions. Coherence order selective gradient heteronuclear single quantum coherence (HSQC) spectra were recorded for a data matrix of 256 × 2048 points covering 30182.7 × 7812.5 Hz with 64 scans for each increment. INEPT transfer delays were optimized for a heteronuclear coupling of 145 Hz and a relaxation delay of 1.5 s. was applied. Data was linear predicted to 512 × 2048 using 32 coefficients and then zero filled to 2048 × 2048 points prior to echo-anti echo type 2D Fourier transformation and a sine bell shaped window function shifted by π/2 in both dimensions was applied. 1D projection along the F1 axis was extracted using the build-in positive projection tool of Topspin (version 2.1, Bruker Biospin). For heteronuclear multiple bond correlation (HMBC) spectra a data matrix of 300 × 2048 points covering 33201.9 × 6265.6 Hz was recorded with 256 scans for each increment. A relaxation delay of 1.5 s and a coherence transfer delay optimized for a long range coupling of 8 Hz were applied. Data was linear predicted to 600 × 2048 points using 32 coefficients prior to echo-anti echo type 2D Fourier transformation and a sine bell shaped window function shifted by π/2 in the F1 dimension and π/6 in the F2 dimension was applied. The final spectrum was obtained by magnitude calculation along the F2 dimension.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kirk, H., Cheng, D., Choi, Y.H. et al. Transgressive segregation of primary and secondary metabolites in F2 hybrids between Jacobaea aquatica and J. vulgaris . Metabolomics 8, 211–219 (2012). https://doi.org/10.1007/s11306-011-0301-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-011-0301-8