
Abstract
Accumulating evidence supports the idea that cancer stem cells (CSCs) are those with the capacity to initiate tumors, generate phenotypical diversity, sustain growth, confer drug resistance, and orchestrate the spread of tumor cells. It is still controversial whether CSCs originate from normal stem cells residing in the tissue or cancer cells from the tumor bulk that have dedifferentiated to acquire stem-like characteristics. Although CSCs have been pointed out as key drivers in cancer, knowledge regarding their physiology is still blurry; thus, research focusing on CSCs is essential to designing novel and more effective therapeutics. The purinergic system has emerged as an important autocrine-paracrine messenger system with a prominent role at multiple levels of the tumor microenvironment, where it regulates cellular aspects of the tumors themselves and the stromal and immune systems. Recent findings have shown that purinergic signaling also participates in regulating the CSC phenotype. Here, we discuss updated information regarding CSCs in the purinergic system and present evidence supporting the idea that elements of the purinergic system expressed by this subpopulation of the tumor represent attractive pharmacological targets for proposing innovative anti-cancer therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The origin of cancer stem cells
Although cancer research has significantly progressed since the nineteenth century and several successful treatments have been developed since then, cancer is still a major cause of death worldwide. According to the World Health Organization, in 2020, cancer accounted for nearly 10 million deaths [1]. Currently, cancer is understood as a widely heterogeneous disease, and knowledge about intratumor complexity, which contributes to cancer progression and recurrence, therapy failure, and reduced overall survival, has been gained [2]. In 1855, Rudolf Virchow and Julius Cohnheim proposed that cancer results from the activation of dormant embryonic tissue remnants, giving rise to such a diverse population of cells. A modern interpretation of the observation made by those pathologists could be the cancer stem cell (CSC) model [3].
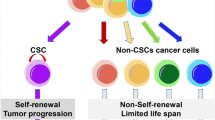
An important premise for the CSC model of cancer development is that the cells forming the tumor are phenotypically heterogeneous and hierarchically organized. This heterogeneity and hierarchy is supported by the differentiation grade of each cell with respect to an original ancestor [4]. In this hierarchy, notwithstanding the phenotypical diversity of the tumor tissue, only one type of cell has the competence to initiate cancer and is the source of each one of the phenotypes in a tumor; these are CSCs. CSCs display particular characteristics resembling normal stem cells (nSCs) (Fig. 1): (1) self-renewal potential to ensure the presence of tumor-initiating cells, a property that in nSCs is achieved by asymmetric cell division [5]; and (2) differentiation potential to give rise to the phenotypical diversity of a tumor [6]. In addition, the CSC subset of the tumor bulk also acquires resistance to conventional therapy, thus making them a possible cause of cancer recurrence [7].

Origin and fate of cancer stem cells. Cancer stem cells are highly plastic cells in the tumor bulk. The hypothesis of their origin transits between two different proposals: that cancer stem cells originate from a genetic mutation of normal stem cells or that tumor cells are capable of dedifferentiating from a differentiated state to an undifferentiated state. Like normal stem cells, cancer stem cells can self-renew and divide asymmetrically, giving rise to an unlimited number of cancer stem cells or cells with compromised cell fates, respectively. Thus, cancer stem cells have the capacity to initiate a new tumor and generate phenotypically different tumor cells that sustain tumor growth, are resistant to current therapies, and have the motility to spread throughout the body and form secondary tumors. Created with BioRender.com
CSCs were first identified in the experiments by Bonnet and Dick in 1997, where they proved that only a small subpopulation (0.1–1%) of acute myeloid leukemia (AML) could restore the disease after being transferred to immune-deficient mice, and subsequent propagation of the disease was also possible [8]. These cells (with the phenotype CD34+CD38neg) resembled those in the hematopoietic system. Similar observations were made for solid tumors in breast epithelial cancer. In a seminal work by Michael Clarke’s group, tumor cells were isolated according to their individual phenotypes and xenotransplanted into immunocompromised SCID mice. Clarke and colleagues found that only cells with a CD44+/CD24low/−ESA+ phenotype had the ability to induce a new tumor with full phenotypic diversity [9]; in turn, the tumor could be sequentially propagated. In parallel, human brain tumor-initiating cells were identified; in this case, it was demonstrated that only cells positive for the surface marker CD133 could originate cells with similar phenotypes to those observed in patients [10].
Thanks to these primordial studies, much effort has been focused on detecting CSCs in different tumor tissues, such as the ovary, prostate, lung, pancreas, neck and head, and colon [11,12,13,14,15,16,17], and many glycoproteins, including CD44, CD133, CD38, CD24, CD34, and CD73, have been linked to stem-like properties [18]. Different tumor markers have been identified depending on the type of cancer being studied. Hence, CSCs are not a single entity but rather a complex system with myriad phenotypes.
A question inherent to the CSC model is the origin of these tumor-initiating entities (Fig. 1). First, it was proposed that CSCs originate from nSCs through the accumulation of mutations in specific loci coding for tumor suppressors and/or oncogenes, resulting in malignant transformation [6, 19,20,21]. The CSC hypothesis has identified strategies for novel cancer cell treatment based on the stem cell theory, which involves a hierarchical organization from undifferentiated cells toward progenitors with restricted differentiation potential and finally terminal differentiation. Therefore, CSCs have greater oncological plasticity potential than more differentiated cancer cells. A comparison of CSCs with nSCs would provide common mechanisms and facilitate the treatment of recurrent tumor lapses, metastasis, and drug resistance. Nevertheless, untransformed nSCs and CSCs cannot be directly compared due to the complexity of their embryogenesis and stem cell development, as well as their epigenetic backgrounds. Furthermore, Teng and collaborators contest the CSC hypothesis and argue that tumor cells maximize their survival potential through a dedifferentiation process that occurs during microenvironmental stress (i.e., chemotherapy conditions) rather than through a hierarchical tumor development cascade [4]. In summary, interesting alternatives have been proposed, such as the acquisition of CSC properties by differentiated cells through different mechanisms, including gene transfer, environmental influence, and genomic instability [22, 23]. CSC phenotype acquisition is a fundamental question that may involve more than one mechanism and will therefore continue to generate controversy and require further investigation.
The microenvironment for CSCs
Physiologically “normal” stem cells, here named nSCs, reside within specific conditions that maintain the stem-cell state; these include cell-to-cell and cell-to-extracellular matrix (ECM) interactions and signals that repress and activate diverse cell fate programs in order to preserve self-renewal and keep a dormant state. The “stem cell niche” was first proposed by Schofield in 1978 based on the observation that the spleen is unable to support the hematopoietic stem cell state in the same way that the bone marrow can, concluding that there are specific conditions (i.e., a microenvironment) that support the stemness and differentiation capability of stem cells [24]. Since then, several nSC niches have been identified in adult tissues such as the hematopoietic system, skin, intestine, brain, and muscle [25]. A niche is thus defined by its functionality rather than by its location, as nSC proximity to certain components of the microenvironment does not indicate that the stem state is supported.
As mentioned previously in this review, the tumor is a complex and heterogeneous entity containing not only cancerous cells but also resident stromal host cells, soluble factors, and the ECM, all of which favor tumor growth and progression [26]. Thus, it is not surprising to realize that there are niches within the tumor that support a stem-like phenotype. In a similar manner to nSCs, CSCs reside in niches within the tumor microenvironment (TME), preserving their phenotypic plasticity and conferring immune suppression and protection against chemotherapy [27]. Many cell types have been described to comprise the CSC niche, such as fibroblasts, endothelial cells, adipocytes, myeloid-derived suppressor cells (MDSCs), macrophages, and CD4+ T-cells, among others [28]. Altogether, these diverse cell types orchestrate a malignant phenotype, allowing chemotherapy and radiotherapy resistance, cancer initiation and recurrence, and metastatic capability. Cancer-associated fibroblasts (CAFs) have been linked to a tumor-supportive environment in different models. For example, in a head and neck cancer model, cancer cell-secreted transforming growth factor beta (TGF-β) augmented periostin, an ECM component, leading to proliferation, migration, and metastasis [29]. Similarly, in a breast cancer model, normal fibroblasts that were irradiated and injected into the mouse mammary fat pad overexpressed TGF-β and hepatocyte growth factor (HGF), thereby supporting tumor initiation [30].
Endothelial cells have also been associated with favoring a malignant phenotype in a prostate cancer model, where human umbilical vein endothelial cells (HUVEC) co-cultured with prostate cancer cell lines favored tumor initiation and metastasis, thus activating autophagy, which has been related to a stem-like phenotype [31]. In addition to these stromal cells, immune cells are recruited and re-educated in order to favor malignancy and stem-likeness. In an ovarian cancer model, CSCs isolated from the OVCAR-3 (a metastatic ovarian carcinoma cell line) interacted with macrophages polarized to an M2 phenotype, which diminished chemotherapeutic sensitivity and increased invasion in a transwell assay and tumor formation in a mouse xenotransplant model. These processes were thought to be ignited by WNT pathway overactivation [32].
The TME is not just a silent spectator but rather an active component within the tumor bulk. In addition to the previously mentioned cell types, the TME also contains non-cellular elements, such as the ECM, which in healthy conditions helps to maintain tissue structure and homeostasis but in cancer has been related to supporting malignancy and metastasis. The ECM can perpetuate a malignant stem-like phenotype, given that modifications in the ECM can alter fibroblasts, endothelial cells, and immune cell functions, which can further reinforce ECM modifications and prolong the malignancy [33]. Moreover, the ECM can provide an anchoring site for CSCs through their matrix-interacting receptors, including integrins, CD47, and CD44. The latter has been used as a CSC marker in several in vitro models and patient-derived samples. Its binding to hyaluronic acid (present within the ECM) has been proven to activate NANOG, thereby eliciting the expression of pluripotent stem cell regulators (e.g., Rex1 and Sox2). CD44 has also been shown to interact with STAT-3, promoting multidrug-resistance transporter MDR1 expression, which results in chemotherapy resistance [34].
Early in tumor development, there is reciprocal communication between the components of the TME and cancerous cells, promoting angiogenesis, nutrient supply, and waste removal. Many soluble factors constitute the TME, such as growth factors, hormones, and signaling molecules. TGF-β has been one of the most studied and well-characterized of these factors. TGF-β can have a dichotomous effect as it limits or preserves CSC populations. In gastric carcinoma, it was shown to decrease the side population by down-regulating ABCG2, an important chemotherapy efflux mediator [35]. On the other hand, several studies have proved that TFG-β facilitates the stem-like phenotype in solid tumors (e.g., breast cancer, glioblastoma, and leukemia) [36] by increasing CD133 and CD44 expression, thereby potentiating the stem-like populations. Other growth factors have been linked to promoting stemness in CSCs, such as epidermal growth factor (EGF) and fibroblast growth factor (FGF). In colon cancer, EGF proved to be essential for CSC selection and maintenance, and its inhibition promoted CSC apoptosis due to a decreased basal activation of Akt and ERK [37]. Conversely, FGF was demonstrated to induce a malignant phenotype in “healthy” induced pluripotent stem cells, where chronic exposure to this growth factor led to a CSC population that no longer required FGF in order to sustain its survival [38].
As previously stated, the TME includes soluble signaling molecules regulating the cancerous phenotype, and given that the purinergic system is ubiquitous and modulates many cell processes (e.g., proliferation, differentiation, and migration), ATP concentrations within the intra-tumor milieu can reach the millimolar range. In healthy tissues, they remain below the micromolar range, as Pellegatti and Di Virgilio elegantly demonstrated [39]. Extracellular ATP (eATP) is actively secreted into the extracellular space in a basal manner. Once there, ATP is metabolized to ADP, then AMP, and finally to adenosine (ADO) by the ectonucleotidases CD39 and CD73. Several stressors, such as hypoxic conditions (highly observed in necrotic tumors), acute and chronic inflammation (in the TME), and anti-cancer therapy-induced cell death, promote the accumulation of eATP and, thus, ADO. Within the TME, eATP and ADO seem to play contradictory roles since ATP is a “find me” signal, recruiting the immune system, and ADO is an immune suppressor [40]. Moreover, there is evidence suggesting that ATP can also induce immune suppressive regulatory T cell (Treg) expression through dendritic cell activation by chemotherapy-induced cell death [41]. Altogether, this information suggests that the primary function of the purinergic system, namely ATP and ADO, is purely related to the immune system; however, this could not be further from the truth, as we will argue in the next sections by explaining in detail the role of ATP in the TME.
Nucleotide and nucleoside signaling
Chemistry
Purines are an extended family of aromatic and heterocyclic molecules. 9H-purine, the simplest of these compounds, is formed by two rings: a pyrimidine fused with an imidazole. The alternating presence of single and double bonds among N and C atoms allows the formation of conjugated systems characterized by the coincidence of an overall low molecular energy and, at the same time, high molecular stability. The chemical underpinning of these properties is the existence of interatomic conjugated bonds connected by π-orbitals with delocalized electrons [42]. Different nucleobases, including purines, have been detected in carbonaceous meteorites [43]. In addition, it has been widely reported that adenine can be formed from simpler molecules, such as NH4+/NH3, CN−, CO, CH4, H2, and formamide, under abiotic primordial conditions [44]. These facts strongly suggest that purines, as well as many other “organic” molecules, were already present in early terrestrial times and were selected during the prebiotic evolution that occurred prior to the emergence of the first living entities on our planet [45].
The two principal purines with biological activities are adenine (6-amino-purine) and guanine (2-amino-6-hydroxypurine). In this review, we only address the importance and characteristics of adenine and its relationship with nucleoside and nucleotides in purinergic signaling through their actions on different membrane receptors. ADO contains a molecule of adenine attached to a ribofuranose moiety via a β-N (9)-glycosidic bond. ADO shows keto-enol tautomerism, being the keto form predominant at pH 7 [46]. In addition, the rotation of the glycosidic bond allows the presence of two ADO conformations: syn and anti. The syn conformation is favored in physiological conditions [47]. ADO is a polar molecule with a partition coefficient XlogP3 value of − 1.1, where negative coefficients are indicative of hydrophilic compounds. Its dipole moment has been calculated to be 3.93 Debye, showing an orientation toward the region between C6 and N3, and being located in the middle of the purine and sugar rings [48].
In contrast to ADO, ATP is a nucleotide with a negatively charged triphosphate moiety and a net electrical charge of − 4 at physiological pH. ATP and other nucleotide triphosphates form organic complexes with divalent cations, most commonly Mg2+. Mg2+ can coordinate with ATP4− by either all three phosphate groups (C3 configuration) or by the terminal β and γ phosphate groups only (C2 configuration) [49]. Because of its ionic characteristics, the XlogP3 partition coefficient of ATP is clearly more negative (− 5.7) than the one calculated for ADO (− 1.1). Molecular dynamics simulation of ATP has suggested that the conformations of this nucleotide in water change drastically when ATP is bound to diverse proteins, especially in the C2 of the ribose ring, the adenine ring, as well as the torsion angles of the glycosyl bond and the bond between phosphate and ribose [50].
In addition to signaling through purine nucleotides and nucleosides, the UTP and UDP pyrimidines, pyrimidine dinucleotides (i.e., Ap4A), and sugar nucleotides (UDP-glucose and UDP-galactose) activate specific receptor subtypes (reviewed in [51]).
Intracellular metabolism
ATP and ADO are part of a large set of interconnected purine intermediates that play diverse metabolic and signaling roles as extracellular and intracellular molecules by means of catabolic and anabolic transformations. These metabolic conversions are particularly relevant since only the liver and kidney are fully capable of synthesizing purines de novo from simpler units such as glycine, glutamine, aspartate, formate, and HCO3− [52]. Hence, some tissues (e.g., brain and muscle) express the salvage pathway that allows the formation of purine nucleotides from metabolic intermediates like hypoxanthine. Therefore, purine transit is established between these tissues (receptors) and the liver (emitting source), with ADO and hypoxanthine in plasma and erythrocytes being the metabolic intermediates. This metabolic communication is subjected to circadian regulation [53].
ATP is a key factor in energy metabolism that controls intracellular electron fluxes from reducing nutrients to mitochondrial oxygen. In the sixties, Atkinson and Walton postulated that the proportion of adenine nucleotides, known as the adenylate energy charge ([ATP] + ½[ADP]/[ATP] + [ADP] + [AMP]), regulates the balance between anabolic and catabolic reactions [54]. This concept was further tested and ratified with the identification of a set of allosteric enzymes sensitive to the AMP/ATP ratio that modulates the biosynthetic and degradative metabolic pathways, as well as with the existence of energy sensors such as AMP-kinase [55]. ADO treatment is capable of increasing the hepatic energy charge in vivo by enhancing mitochondrial activity [56].
ADO plays various intracellular roles. ADO catabolic transformation results in uric acid in mammals without uricase, a free radical scavenger [57]. The redox enzyme that produces uric acid, xanthine dehydrogenase, can also act as an oxidase in Ca2+-promoted protein alteration. Xanthine oxidase has been proposed as a source of the free radical anion superoxide (·O2−) in pathological conditions such as inflammation and ischemia [58]. ADO can also combine with the sulfur-containing molecule homocysteine to form S-adenosyl-L-homocysteine (SAH). SAH is an inhibitor of methylation reactions that depend on S-adenosyl methionine (SAM). Therefore, ADO can indirectly modulate the formation of methylated intermediates and influence processes including neurotransmission, epigenetics, and membrane fluidity [59].
Signal transduction
In addition to its well-known role in energy exchange reactions, ATP is an important signaling molecule with characterized release mechanisms and specific receptors. In 1929, researchers showed that adenine influences cardiac rhythm, thus indicating that ATP plays a role in extracellular signaling [60]. In 1972, Burnstock proposed ATP as a non-adrenergic and non-cholinergic neurotransmitter [61], giving rise to a new field of study: the purinergic system. At first, his proposals were not accepted by the scientific community but were gradually earning a place in the field thanks to the cloning of purinergic receptors that mediate the signal in response to extracellular purines. These receptors have been classified into two major types based on agonist selectivity: P1 adenosine receptors and P2 nucleotide receptors. The former are conformed by four receptors (A1, A2A, A2B, and A3), and the latter are divided into two main subtypes: P2X and P2Y receptors, which are ligand-gated ion channels and G-protein-coupled receptors, respectively [62].
To date, seven P2X subunits have been cloned in mammals (P2X1-P2X7), and these subunits can form either a homotrimer or heterotrimer that acts as a ligand-gated ion channel exclusively responsive to ATP [63], allowing Ca2+ and Na+ influx and K+ efflux. Each subunit has two membrane-spanning domains (TM1 and TM2) with an intracellular N-terminus and C-terminus, and most of the protein is extracellular with the ligand-binding region at the intersection of two subunits [63]. Regarding P2Y G-protein-coupled receptors, eight genes have been cloned in mammals (P2RY1, P2RY2, P2RY4, P2RY6, P2RY11-14). The preferred natural agonists are ATP (P2Y11), ADP (P2Y1, P2Y12, and P2Y13), UTP (P2Y2 and P2Y4), UDP (P2Y6), and UDP-sugars (P2Y14). P2Y1, 2, 4, and 6 act through Gq-phospholipase C β (PLCβ), producing inositol-triphosphate (IP3), which causes Ca2+ release from the endoplasmic reticulum, and diacylglycerol (DAG), which activates PKC. P2Y12-14 receptors act through Gi protein, causing adenylyl cyclase inhibition and a reduction in cyclic adenosine monophosphate (cAMP) levels; and P2Y11 receptor activates Gs protein, activating adenylyl cyclase and augmenting cAMP levels [64].
As molecular messengers, ATP and ADO can act as counterparts of each other, recognizing their specific receptors. For example, ATP is mostly a pro-inflammatory molecule, whereas ADO plays an important anti-inflammatory role. The fine-tuned regulation of these antagonist actions indeed depends on the set of ATP and ADO receptors expressed by the cellular system; they are also contingent on the activity of a set of extracellular hydrolytic enzymes that turn ATP into ADP, AMP, and ADO. For example, ATP can be converted into ADO in a two-step enzymatic process involving the CD39 (with apyrase-like action) and CD73 [65].
For a long time, it has been reported that purinergic signaling plays important roles in many physiological and pathological events. Haulica et al. reported in 1973 that, in addition to neurotransmission and blood coagulation, ADO exerts a hypnogenic action that was later confirmed [66]. Both ATP and ADO can act as coronary vasodilator agents [67]. Additionally, in the context of the orchestrated immune response, the alternative actions of ATP and ADO have been described [65]. Purinergic system malfunction contributes to the mechanisms of various illnesses, including cancer, diabetes, gout, osteoporosis, and cardiovascular, neurological, and psychiatric diseases [62, 68, 69].
Purinergic signaling in cancer
Recent reviews highlight the diversity and plasticity of purinergic signaling in the context of cancer [70,71,72,73]. The purinergic system in the TME has a particular configuration that gives it a prominent role in cancer progression, with some notable characteristics: (1) cancer cells have a high capacity to produce ATP as a result of metabolic adaptations such as the Warburg effect, making cancerous cells energetically sustainable [74]; (2) ATP efflux to the interstitium is increased due to the boost in ATP synthesis, resulting in a high concentration of nucleotides (hundreds of µM) in the TME, which is enough to activate any purinergic receptor subtypes [39]; (3) purinergic receptors are widely expressed in tumor cells [75], and some subtypes, such as P2X7 receptor, are overexpressed in specific cancers [76,77,78,79]; and (4) the expression of ecto-nucleotidases (mainly CD73) contributes to regulating purinergic ligand concentrations in the TME [80]. CD39 and CD73 promote an immunosuppressive environment in the TME [81]. CD73, which is rate-limiting in the degradation of AMP into ADO, regulates tumor proliferation and progression and has therefore been defined as a prognostic marker for tumor survival [82].
The aforementioned characteristics suggest that the purinergic system is a fundamental element of the TME with a dual role. At the cellular social level, it mainly induces immunosuppression by mediating interactions with host immune cells. It also exerts autocrine-paracrine actions by directly regulating processes such as metabolism, cell proliferation, and cell migration and inducing epithelial-to-mesenchymal transition (EMT).
The purinergic system at the cellular social level: from “find me” signaling to evasion of antitumor immune response
ATP has been categorized as a “find me” signal that triggers the immune response. In the cancer context, ADO—a direct product of ATP hydrolysis catalyzed by the sequential actions of CD39 and CD73 ecto-nucleotidases—elicits an immunosuppressive response by modulating the phenotype of tumor-infiltrated immune cells [83, 84]. Thus, the identity and proportion of purinergic ligands directly contribute to the balance of antitumor immune attack.
The find me role of ATP has been described in the context of tissue damage generated by conditions such as hypoxia, inflammation or necrosis. Under these conditions, ATP acts as a damage-associated molecular pattern (DAMP) [85], attracting dendritic cells, macrophages, and neutrophils to prompt damage resolution [86, 87]. In cancer conditions, especially when anti-cancer therapies induce cell death, extracellular ATP increments notably in the TME, and nucleotides can activate receptors in resident non-cancerous cells. For instance, in dendritic cells, ATP activates P2X7 receptors, inducing NLRP3 inflammasome assembly and the release of IL-1β, a proinflammatory cytokine with the capacity to induce an immunogenic response through the regulation of CD8+ T cells [88, 89]. On the other hand, actions through P2X and P2Y receptors can determine macrophage subpopulations that promote tumor development (tumor-associated macrophages), thereby establishing a protective TME for cancer cells and inducing CSC development and dissemination [90, 91].
Moreover, a consequence of the increase in extracellular ATP concentration is ADO accumulation in the TME as a result of ecto-nucleotidase activity [92]. ADO, acting through ADO receptor-dependent mechanisms, inhibits the antitumor immune response. Thus, after CL8-1 tumor melanoma cell xenotransplantation in mice, pharmacological inhibition or genetic deletion of the A2A receptor enhanced the inhibition of tumor growth, vascularization, and the destruction of metastases by incoming antitumor T lymphocytes [92], demonstrating that the ADO/A2A receptor pathway is essential for the modulation of immune host-tumor interactions. Furthermore, phenotypic modulation to inhibit the antitumor immune response via the ADO/A2A receptor pathway was documented for Tregs [93,94,95], T effector cells [95,96,97], natural killer cells [98, 99], and myeloid cells [100,101,102]. On the other hand, in medulloblastoma cells, CD79 overexpression reduced tumor proliferation, possibly by inducing differentiation and apoptosis through A1 receptor activation [82].
Autocrine-paracrine actions of purinergic signaling in TME
As mentioned above, purinergic receptors are widely expressed in cancerous tissues [75], and ATP is available in the TME [39]; consequently, purinergic-mediated autocrine-paracrine communication regulates diverse physiological aspects in cancer cells, such as cell proliferation and cell migration.
P2X7 and P2Y2 receptors are two of the most studied purinergic receptors. Although P2X7 receptor was originally described as a cytotoxic receptor promoting cell death, it has been demonstrated to activate proliferative pathways in the TME, such as Ca2+-dependent and independent ERK phosphorylation [103,104,105], mTOR-HIF1-α-VEGF [106], and PI3K-AKT [107, 108], as well as tumor metastasis [109]. P2X7 can induce cell death and promote tumor proliferation and survival. These divergent actions have been related to two splice variants of the P2RX7 receptor gene: the P2X7A and B isoforms. While the P2X7A variant comprises the full-length receptor, capable of pore formation with cytotoxic activity, the truncated P2X7B variant lacks the C-terminal tail and thus cannot form pores. However, ion channel opening and consequent downstream cellular signaling functions in this receptor contribute to the TME and promote tumor progression, metastasis, and chemotherapy resistance (reviewed by [110]). Tumor-promoting actions for P2X7 receptor have been described for mesothelioma [78], pancreatic carcinoma [105, 111], ovarian carcinoma [79], osteosarcoma cells [106], and neuroblastoma cells [109].
On the other hand, P2Y2 receptor is coupled to Gq heterotrimeric G-proteins, and its activation induces Ca2+ release from intracellular storage by the PLC/IP3 pathway [64]. P2Y2 activity has also been related to the activation of PI3K/Akt [112, 113], mitogen-activated protein kinases (MAPK) ERK [114,115,116] and JNK [116, 117], and mTOR kinase [118]. P2Y2 receptor-induced proliferation has been described for cancers in a broad group of tissues, such as the lung [119], breast [115, 120], ovary [121], cervix [122], liver [123], and stomach [124]. P2X7 receptor has been associated with the induction of cell migration and/or the triggering of EMT in the lung [125], breast [126, 127], prostate [128], and colorectal cancer cells [108]. P2Y2 receptor modulates these processes in the breast [115, 120, 129], prostate [128, 130], ovary [131], liver [123], stomach [132], and pancreas [133]. Thus, purinergic signaling constitutes a set of autocrine-paracrine signals that contribute to the regulation of multiple processes to support cancer progression.
Purinergic signaling in CSCs
The implication of ATP and the purinergic system in nSCs is gradually becoming clearer. And while only a few studies have explored the role of the purinergic system exclusively in CSCs, extensive research has assessed the importance of purinergic receptors in stem-like traits that confer a malignant phenotype. Within tumor heterogeneity, CSCs are central to cancer phenotype regulation given their chemoresistance [134], enhanced DNA repair mechanisms [135], higher reactive oxygen species scavenging capacity [136], and metastatic competence [137]. In the next sections, we present some of the most recent literature concerning purinergic system components and their role in stem-like traits (summarized in Table 1).
ATP
One of the best-characterized systems in stem cell biology is the hematopoietic system, as well as the role of purinergic system in stem cell differentiation and pool regulation in hematopoiesis. As Paredes-Gamero and coworkers have revised [159], P2 receptors are differentially expressed in the different stages of hematopoiesis, and ATP is released by different cells in the hematopoietic niche (endothelial cells and osteoblasts). ATP may act as a regulator for hematopoietic stem cells (HSCs). One study reported that ATP increased a CD34+ cell population from adult healthy donors [160], while another study showed how ATP reduced the percentage of HSCs and myeloid progenitors [161]. Although the effect of ATP on stem cell physiology might seem contradictory, its influence on HSCs is evident.
As shown previously with nSCs, ATP might regulate CSC populations. In a primordial study published in 2012, the addition of exogenous ATP to glioblastoma cell lines (U87, C343, and C6) reduced spheroid size and numbers in the spheroid-formation assay and lessened the expression of the stem cell markers CD133 and OCT-4 [162]. In a similar way, ATP decreased the viability of patient-derived CSCs and increased their sensitivity to chemotherapy in glioblastoma [158] and acute myeloid lymphoma [157], thus implying that ATP can impact CSC survival. On the other hand, purinergic signaling could promote chemoresistance as ATP hydrolysis by CD73 enhanced temozolomide cytotoxicity in the glioblastoma cell lines M059J and U251, thereby supporting cell cycle arrest and cell death [163].
CD73
Ecto-5′-nucleotidase (NT5E/CD73) is one of the most extensively studied components of the purinergic system and a highly important regulator of extracellular ATP concentration. Thus, it is not surprising that there is information about its role in CSCs. High expression of CD73 has been correlated with stemness markers in different models. For example, in cell lines derived from patients with stage 4 neuroblastoma, CD73 protein levels were correlated with the presence of stemness markers (SOX2, CD44, and ALDH) and transcripts of EMT markers (N-cadherin, vimentin, Notch) [151] (Fig. 2). In another study, hepatocellular carcinoma cell lines were sorted based on CD73 expression levels. According to its findings, CD73High had a high expression of the stemness marker SOX9 and a high capacity for spheroid formation. However, when CD73 was knocked down, SOX9 was downregulated, and spheroid formation was abolished [164]. In addition, a renal carcinoma cell line (786-O) was sorted in a similar way, and the CD73High population upregulated Oct-3/4 expression and was enriched in mytomycin C-resistant cells [165], which was not observed in the CD73low population. In this same study, CD73 was evaluated in spheroids derived from 786-O cell lines, and higher levels of the ecto-nucleotidase were found in contrast with the monolayer culture.

Purinergic signaling in the process of the epithelial-mesenchymal transition in cancer cells. Epithelial-mesenchymal transition (EMT) involves the transformation of epithelial cells into mesenchymal-like cells, characterized by the loss of epithelial characteristics and the acquisition of mesenchymal traits. This transition is associated with increased migratory capacity, invasiveness, and the development of stem-like properties, leading to the formation of cancer stem cells (CSCs). CSCs are resistant to standard cancer treatments and contribute to tumor recurrence and metastasis. Purinergic signaling encompasses signaling pathways mediated by purine nucleotides, such as adenosine triphosphate (ATP), as well as its breakdown products, such as adenosine, facilitated by the ectoenzyme CD73. These signaling molecules interact with specific purinergic receptors expressed on the cell surface, including ADORA, P2X, and P2Y receptors. Extensive research has implicated purinergic signaling in the regulation of EMT and the maintenance of CSCs. Notably, the P2X7 receptor has emerged as a key player in this process. Activation of P2X7 isoform B has been associated with the induction of EMT and the expression of EMT-related transcription factors, such as TWIST and SNAIL, as well as mesenchymal markers like vimentin and N-cadherin. Additionally, P2X7 receptor activation has been linked to the acquisition of stem-like properties by cancer cells. Another important component of the purinergic signaling pathway involved in EMT and presence of stemness markers (SOX2, CD44 and ALDH) is CD73, an ectoenzyme responsible for generating extracellular adenosine. Adenosine can activate specific adenosine receptors, including ADORA A2A and A3 receptors, which have been shown to promote EMT and the expression of EMT markers. While the role of P2Y receptors in EMT is not well-studied, the activation of P2X7 isoform A and ADORA A2B receptors appears to counterbalance the EMT process. Overall, modulating purinergic signaling pathways, particularly by targeting P2X7 isoform B, CD73, and ADORA A2A and A3 receptors, shows potential as a therapeutic strategy to inhibit EMT, disrupt the stem-like properties of cancer cells, and enhance the effectiveness of cancer treatments. Created with BioRender.com
Other studies have shown that spheroids derived from cancer cell lines have a higher CD73 expression than their parental cell line. In a cervical cancer cell line (CaSki), CD73 was increased in spheroids in contrast to their monolayer cultures, thus promoting a higher extracellular concentration of ADO. The authors of the study mention the plausible positive feedback loop between ADO and CD73, given that ADO promotes TGF-\(\beta\) expression, which further enhances CD73 expression [145, 146]. To strengthen these findings, Bertolini and coworkers identified CD73 overexpression in spheroids from NSCLC cell lines and a higher production of ADO [147].
CD73 knockdown has been evaluated as well, and similar results have been found in different models. In a gall bladder cancer cell line NOZ, spheroids presented a high expression of CD73 and knockdown using siRNA impeded spheroid formation [148]. Furthermore, in ovarian cancer cell lines and fresh tumor tissue, CD73 silencing using siRNA downregulated EMT markers and spheroid formation capacity [166]. Other studies have evaluated chemotherapy resistance, as it is a well-established stem-like trait. Chemotherapy resistance was shown to be impeded after CD73 knockdown using siRNAs in both glioblastoma and breast cancer cell lines [149, 150]. Moreover, CD73 silencing using siRNA-containing nanoparticles in murine cancer cell lines from colon, breast, and melanoma diminished cell migration, proliferation, and resistance to doxorubicin [167].
Adenosine receptors
ADO is present in high concentrations in the TME, and its relevance as an immune modulator can be reviewed elsewhere. Here, we will focus on the role of adenosine receptors (ADORA) in stem-like properties. In glioblastoma cell lines, there is homogenous information showing that adenosine receptors A1, A2A, A2B, and A3 are overexpressed in spheroids and are correlated with stemness markers (CD133), EMT markers (vimentin, SNAIL, TWIST, and CDH1), and pharmacological antagonism. Knockdown of this receptors using siRNAs abolishes spheroid formation capacity, increases the sensitivity to chemotherapy, and impedes tumorigenesis [138, 140, 142, 168, 169]. In breast cancer, there is clear evidence that A2B receptor is correlated with a stem-like phenotype, since A2B is overexpressed in spheroids from different cell lines (MCF7, MDA-MB-231, SUM149, and SUM169) and its pharmacological activation in such CSCs enhances ALDH activity. On the other hand, A2B knockdown diminishes spheroid formation capacity and stemness markers (NANOG, SOX2, KLF4, and ALDH activity) [141, 143, 144]. To reinforce the importance of ADO receptors in CSCs, one study evaluated A2A in a gastric cancer model (MKN-45 cell line) and showed that receptor activation induced transcription factors that are essential for stemness (Nanog, OCT-4, SOX2, and CD44) and favored radioresistance [139] (Fig. 2).
P2Y receptors
In comparison to the other elements of the purinergic system, P2Y receptors have been poorly assessed in CSCs. In a neuroblastoma model (SH-SY5Y cell line), P2Y4 receptor pharmacological activation induced differentiation to neurites, as did transient overexpression [156]. Although P2Y2 receptor is one of the most interesting targets for cancer research, there is only one paper evaluating, albeit tangentially, its role in stem-like properties. In patient-derived breast cancer cells, P2Y2 receptor levels were correlated with the stemness marker Notch-4 [155]. The lack of information on the topic of P2Y receptors, CSCs, and stem-like traits in cancer is evident.
P2X7 receptor
P2X7 receptor, which induces tumor proliferation, contributes to the maintenance of embryonic stem cell pluripotency [170, 171]. And similar functions are expected in maintaining a pool of CSCs capable of causing tumor relapse and chemoresistance [172]. A piece of evidence points to P2X7 receptor as a key player in metabostemness, the metabolic reprogramming of cancer cells toward an undifferentiated phenotype (i.e., CSCs). P2X7 receptor modulation is associated with metabolic targets closely related to cellular events for stemness phenotype acquisition [173]. Importantly, Lameu and coworkers have focused on the functions of P2X7 receptor and its isoforms in promoting the phenotypic transition of the tumor into a stemness stage and EMT. They have observed that P2X7A is important for triggering CSC differentiation, since knockdown cells for this isoform remain in an undifferentiated state. In contrast, P2X7B activation is implicated in EMT [110, 172], promoting treatment resistance and metastatic formation.
Similar to P2Y receptors, P2X7 receptor involvement in CSCs has been studied solely in a tangential way. In this context, using glioblastoma models, P2X7 activation has been related to the acquisition of EMT markers in mRNA (CDH2, SNAIL, Zeb1) and proteins (N-cadherin, ZEB1, vimentin, and TWIST) [152], as well as the promotion of spheroid formation [154]. Other cancer models, such as osteosarcoma and colorectal cancer cell lines, have demonstrated similar effects, where P2X7 activation increased EMT markers both at mRNA and protein levels [106, 112]. Yet, there is a study evaluating the presence of P2X7 receptor in different leukemia-initiating cells. This study used an induced murine model of acute myeloid leukemia, and P2X7 receptor was detected at higher levels in leukemia-initiating cells and granulocyte-monocyte progenitors [153]. Although the latter findings are remarkable, to our knowledge, there are no other papers characterizing P2X7 or their function in CSCs.
Purinergic system and EMT
Thus far, we have described several purinergic elements and their implication in CSC biology. To elaborate on the purinergic system’s involvement in the stem-like phenotype, we will discuss how purinergic signaling modulates one important transdifferentiation program that has been shown to be continuously active in stem cells: EMT.
EMT is a transdifferentiation program in which epithelial cells gain mesenchymal characteristics, and it is involved in embryonic development, tissue repair, and cancer cell migration. EMT has been related to stem-like properties. Mammary epithelial cells undergoing EMT exhibited an amplified stem-like phenotype in terms of spheroid formation, soft agar colonies, and tumorigenic properties [174].
Considering that EMT confers stem-like properties, we focused on papers concerning the importance of the purinergic system in EMT. First, a study revealed that, in glioblastoma cell lines, hypoxia favored CD73 and ADO A3 receptor expression, which is notable considering the hypoxic conditions within the tumor. The study also showed that A3 receptor pharmacological inhibition decreased EMT markers such as TWIST, SNAIL, vimentin, and N-cadherin in such cell lines [168] (Fig. 2). With respect to P2 receptors, in colorectal cancer cell lines, pharmacological activation of P2X7 receptor led to EMT activation, as demonstrated by augmented vimentin, Snail, and fibronectin expression and decreased E-cadherin expression [108]. In neuroblastoma, a counterbalance of P2X7 isoforms was observed to promote EMT, pointing to an epithelial-prone P2X7A-related effect and P2X7B as an EMT-favoring isoform [172] (Fig. 2).
CSCs are highly resistant to chemotherapy and radiotherapy. Therefore, the paper published by Nguyen and coworkers is quite fascinating. According to the authors, a radioresistant pancreatic cancer cell line favored a mesenchymal state, which showed downregulation in E-cadherin and upregulation in vimentin and, surprisingly, CD73. In this same study, CD73 expression interference with shRNA led to radiosensitivity and an epithelial phenotype, evidencing the importance of CD73 in acquiring a mesenchymal phenotype [175].
Concluding remarks
ATP and ADO in the purinergic system are important for the regulation of tumor cell proliferation and malignant progression, as well as for modulating the immune response and TME biology. There is abundant information highlighting the importance of the purinergic system in nSCs but not in CSCs, even when it has been proven that the system occurs in healthy conditions and is relevant in cancer pathologies. ATP depletion by CD73 activity might be involved in stem-like properties, but it raises the question of whether ATP depletion or ADO generation creates such a phenotype. Furthermore, conclusions should be drawn with caution, given that the phenotype produced by purinergic receptor activity depends on the tissue in question, the receptor being studied, and the experimental conditions. Diverse elements of the purinergic system have been shown to play essential roles in maintaining stemness, especially CD73, A2B, and P2X7 receptor, yet there is an obvious lack of understanding about P2Y receptors. The purinergic system is undoubtedly a promising field of knowledge for comprehending CSC biology.
Data availability
Not applicable.
References
Cancer. https://www.who.int/news-room/fact-sheets/detail/cancer. Accessed 13 Mar 2023
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. https://doi.org/10.1016/J.CELL.2011.02.013
Huntly BJP, Gilliland DG (2005) Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer 5:311–321. https://doi.org/10.1038/NRC1592
Teng YD, Wang L, Kabatas S et al (2018) Cancer stem cells or tumor survival cells? Stem Cells Dev 27:1466–1478. https://doi.org/10.1089/scd.2018.0129
Yoo YD, Kwon YT (2015) Molecular mechanisms controlling asymmetric and symmetric self-renewal of cancer stem cells. J Anal Sci Technol 6. https://doi.org/10.1186/S40543-015-0071-4
Vermeulen L, Sprick MR, Kemper K et al (2008) Cancer stem cells - old concepts, new insights. Cell Death Differ 15:947–958. https://doi.org/10.1038/cdd.2008.20
Kuşoğlu A, Biray Avcı Ç (2019) Cancer stem cells: a brief review of the current status. Gene 681:80–85. https://doi.org/10.1016/J.GENE.2018.09.052
Bonnet D, Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3:730–737. https://doi.org/10.1038/NM0797-730
Al-Hajj M, Wicha MS, Benito-Hernandez A et al (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100:3983–3988. https://doi.org/10.1073/PNAS.0530291100
Singh SK, Clarke ID, Hide T, Dirks PB (2004) Cancer stem cells in nervous system tumors. Oncogene 23:7267–7273. https://doi.org/10.1038/sj.onc.1207946
Szotek PP, Pieretti-Vanmarcke R, Masiakos PT et al (2006) Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian inhibiting substance responsiveness. Proc Natl Acad Sci U S A 103:11154–11159. https://doi.org/10.1073/PNAS.0603672103
Collins AT, Berry PA, Hyde C et al (2005) Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 65:10946–10951. https://doi.org/10.1158/0008-5472.CAN-05-2018
Li C, Heidt DG, Dalerba P et al (2007) Identification of pancreatic cancer stem cells. Cancer Res 67:1030–1037. https://doi.org/10.1158/0008-5472.CAN-06-2030
Eramo A, Lotti F, Sette G et al (2008) Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ 15:504–514. https://doi.org/10.1038/SJ.CDD.4402283
Hermann PC, Huber SL, Herrler T et al (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1:313–323. https://doi.org/10.1016/J.STEM.2007.06.002
Prince ME, Sivanandan R, Kaczorowski A et al (2007) Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A 104:973–978. https://doi.org/10.1073/PNAS.0610117104
O’Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445:106–110. https://doi.org/10.1038/NATURE05372
Ailles LE, Weissman IL (2007) Cancer stem cells in solid tumors. Curr Opin Biotechnol 18:460–466. https://doi.org/10.1016/J.COPBIO.2007.10.007
Leedham S, Schier S, Thliveris AT et al (2005) From gene mutations to tumours–stem cells in gastrointestinal carcinogenesis. Cell Prolif 38:387–405. https://doi.org/10.1111/J.1365-2184.2005.00359.X
White AC, Lowry WE (2015) Refining the role for adult stem cells as cancer cells of origin. Trends Cell Biol 25:11–20. https://doi.org/10.1016/J.TCB.2014.08.008
Ayob AZ, Ramasamy TS (2018) Cancer stem cells as key drivers of tumour progression. J Biomed Sci 25. https://doi.org/10.1186/S12929-018-0426-4
Bergsmedh A, Szeles A, Henriksson M et al (2001) Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc Natl Acad Sci U S A 98:6407–6411. https://doi.org/10.1073/PNAS.101129998
Atashzar MR, Baharlou R, Karami J et al (2020) Cancer stem cells: a review from origin to therapeutic implications. J Cell Physiol 235:790–803. https://doi.org/10.1002/JCP.29044
Schofield R (1978) The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 4:7–25
Ferraro F, Lo Celso C, Scadden D (2010) Adult stem cels and their niches. Adv Exp Med Biol 695:155–168. https://doi.org/10.1007/978-1-4419-7037-4_11
Anderson NM, Simon MC (2020) The tumor microenvironment. Curr Biol 30:R921–R925. https://doi.org/10.1016/J.CUB.2020.06.081
Plaks V, Kong N, Werb Z (2015) The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16:225–238. https://doi.org/10.1016/J.STEM.2015.02.015
Motohara T, Yoshida GJ, Katabuchi H (2021) The hallmarks of ovarian cancer stem cells and niches: exploring their harmonious interplay in therapy resistance. Semin Cancer Biol 77:182–193. https://doi.org/10.1016/J.SEMCANCER.2021.03.038
Qin X, Yan M, Zhang J et al (2016) TGFβ3-mediated induction of periostin facilitates head and neck cancer growth and is associated with metastasis. Sci Rep 6. https://doi.org/10.1038/SREP20587
Kuperwasser C, Chavarria T, Wu M et al (2004) Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A 101:4966–4971. https://doi.org/10.1073/PNAS.0401064101
Zhao R, Bei X, Yang B et al (2018) Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J Exp Clin Cancer Res 37. https://doi.org/10.1186/S13046-018-0884-2
Raghavan S, Mehta P, Xie Y et al (2019) Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J Immunother Cancer 7. https://doi.org/10.1186/S40425-019-0666-1
Lu P, Weaver VM, Werb Z (2012) The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196:395–406. https://doi.org/10.1083/JCB.201102147
Bourguignon LYW, Peyrollier K, Xia W, Gilad E (2008) Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem 283:17635–17651. https://doi.org/10.1074/JBC.M800109200
Ehata S, Johansson E, Katayama R et al (2011) Transforming growth factor-β decreases the cancer-initiating cell population within diffuse-type gastric carcinoma cells. Oncogene 30:1693–1705. https://doi.org/10.1038/ONC.2010.546
Bellomo C, Caja L, Moustakas A (2016) Transforming growth factor β as regulator of cancer stemness and metastasis. Br J Cancer 115:761–769. https://doi.org/10.1038/BJC.2016.255
Feng Y, Dai X, Li X et al (2012) EGF signalling pathway regulates colon cancer stem cell proliferation and apoptosis. Cell Prolif 45:413–419. https://doi.org/10.1111/J.1365-2184.2012.00837.X
Sheta M, Hassan G, Afify SM et al (2021) Chronic exposure to FGF2 converts iPSCs into cancer stem cells with an enhanced integrin/focal adhesion/PI3K/AKT axis. Cancer Lett 521:142–154. https://doi.org/10.1016/J.CANLET.2021.08.026
Pellegatti P, Raffaghello L, Bianchi G et al (2008) Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One 3. https://doi.org/10.1371/JOURNAL.PONE.0002599
Feng LL, Cai YQ, Zhu MC et al (2020) The yin and yang functions of extracellular ATP and adenosine in tumor immunity. Cancer Cell Int 20. https://doi.org/10.1186/S12935-020-01195-X
Lecciso M, Ocadlikova D, Sangaletti S et al (2017) ATP release from chemotherapy-treated dying leukemia cells elicits an immune suppressive effect by increasing regulatory T cells and tolerogenic dendritic cells. Front Immunol 8. https://doi.org/10.3389/FIMMU.2017.01918
Raczyńska ED, Makowski M (2014) Geometric consequences of electron delocalization for adenine tautomers in aqueous solution. J Mol Model 20. https://doi.org/10.1007/S00894-014-2234-4
Martins Z (2018) The nitrogen heterocycle content of meteorites and their significance for the origin of life. Life 8. https://doi.org/10.3390/LIFE8030028. (Basel, Switzerland)
Kitadai N, Maruyama S (2018) Origins of building blocks of life: a review. Geosci Front 9:1117–1153. https://doi.org/10.1016/J.GSF.2017.07.007
Enchev V, Angelov I, Dincheva I et al (2021) Chemical evolution: from formamide to nucleobases and amino acids without the presence of catalyst. J Biomol Struct Dyn 39:1–16. https://doi.org/10.1080/07391102.2020.1792986
Cysewski P (2005) An ab initio DFT characteristics of tautomeric properties of hydroxyl radical modified nucleosides in polar and non-polar environments. Zeitschrift Phys Chemie 219:213–234. https://doi.org/10.1524/ZPCH.219.2.213.57306/MACHINEREADABLECITATION/RIS
Butanda-Ochoa A, Höjer G, Díaz-Muñoz M (2003) Modulation of the skeletal muscle Ca2+ release channel/ryanodine receptor by adenosine and its metabolites: a structure-activity approach. Bioorganic Med Chem 11:3029–3037. https://doi.org/10.1016/S0968-0896(03)00155-X
Butanda-Ochoa A, Hojer G, Morales-Tlalpan V, Diaz-Munoz M (2006) Recognition and activation of ryanodine receptors by purines. Curr Med Chem 13:647–657. https://doi.org/10.2174/092986706776055715
Buelens FP, Leonov H, De Groot BL, Grubmüller H (2021) ATP-magnesium coordination: protein structure-based force field evaluation and corrections. J Chem Theory Comput 17:1922–1930. https://doi.org/10.1021/ACS.JCTC.0C01205
Kobayashi E, Yura K, Nagai Y (2013) Distinct conformation of ATP molecule in solution and on protein. Biophys 9:1–12. https://doi.org/10.2142/BIOPHYSICS.9.1. (Nagoya-shi, Japan)
Jacobson K, Costanzi S, Ohno M et al (2004) Molecular recognition at purine and pyrimidine nucleotide (P2) receptors. Curr Top Med Chem 4:805–819. https://doi.org/10.2174/1568026043450961
Pareek V, Pedley AM, Benkovic SJ (2021) Human de novo purine biosynthesis. Crit Rev Biochem Mol Biol 56:1–16. https://doi.org/10.1080/10409238.2020.1832438
de Sánchez VC, Hernández-Muñoz R, Díaz-Muñoz M et al (1983) Circadian variations of adenosine level in blood and liver and its possible physiological significance. Life Sci 33:1057–1064. https://doi.org/10.1016/0024-3205(83)90661-6
Atkinson DE, Walton GM (1967) Adenosine triphosphate conservation in metabolic regulation. J Biol Chem 242:3239–3241. https://doi.org/10.1016/s0021-9258(18)95956-9
Lin SC, Hardie DG (2018) AMPK: sensing glucose as well as cellular energy status. Cell Metab 27:299–313. https://doi.org/10.1016/J.CMET.2017.10.009
Chagoya de Sánchez V, Brunner A, Piña E (1972) In vivo modification of the energy charge in the liver cell. Biochem Biophys Res Commun 46:1441–1445. https://doi.org/10.1016/S0006-291X(72)80138-4
Pasalic D, Marinkovic N, Feher-Turkovic L (2012) Uric acid as one of the important factors in multifactorial disorders--facts and controversies. Biochem Med 22:63–75. https://doi.org/10.11613/BM.2012.007
Chung HY, Baek BS, Song SH et al (1997) Xanthine dehydrogenase/xanthine oxidase and oxidative stress. Age 20:127–140. https://doi.org/10.1007/S11357-997-0012-2. (Omaha)
Su X, Wellen KE, Rabinowitz JD (2016) Metabolic control of methylation and acetylation. Curr Opin Chem Biol 30:52–60. https://doi.org/10.1016/J.CBPA.2015.10.030
Drury AN, Szent-Györgyi A (1929) The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol 68:213–237. https://doi.org/10.1113/JPHYSIOL.1929.SP002608
Burnstock G (1972) Purinergic nerves. Pharmacol Rev 24:509–581
Huang Z, Xie N, Illes P et al (2021) From purines to purinergic signalling: molecular functions and human diseases. Signal Transduct Target Ther 6. https://doi.org/10.1038/S41392-021-00553-Z
Coddou C, Yan Z, Obsil T et al (2011) Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev 63:641–683. https://doi.org/10.1124/PR.110.003129
Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50:413–492. https://doi.org/10.1007/978-3-642-28863-0_5
Faas MM, Sáez T, de Vos P (2017) Extracellular ATP and adenosine: the Yin and Yang in immune responses? Mol Aspects Med 55:9–19. https://doi.org/10.1016/j.mam.2017.01.002
Haulicǎ I, Ababei L, Brǎnişlteanu D, Topoliceanu F (1973) Letter: preliminary data on the possible hypnogenic role of adenosine. J Neurochem 21:1019–1020. https://doi.org/10.1111/J.1471-4159.1973.TB07549.X
García-Baizán A, Millor M, Bartolomé P et al (2019) Adenosine triphosphate (ATP) and adenosine cause similar vasodilator effect in patients undergoing stress perfusion cardiac magnetic resonance imaging. Int J Cardiovasc Imaging 35:675–682. https://doi.org/10.1007/S10554-018-1494-Y
Andrejew R, Glaser T, Oliveira-Giacomelli Á et al (2019) Targeting purinergic signaling and cell therapy in cardiovascular and neurodegenerative diseases. Adv Exp Med Biol 1201:275–353. https://doi.org/10.1007/978-3-030-31206-0_14
Ribeiro DE, Petiz LL, Glaser T et al (2023) Purinergic signaling in cognitive impairment and neuropsychiatric symptoms of Alzheimer’s disease. Neuropharmacology 226. https://doi.org/10.1016/J.NEUROPHARM.2022.109371
Campos-Contreras ADR, Díaz-Muñoz M, Vázquez-Cuevas FG (2020) Purinergic signaling in the hallmarks of cancer. Cells 9. https://doi.org/10.3390/CELLS9071612
Alvarez CL, Troncoso MF, Espelt MV (2022) Extracellular ATP and adenosine in tumor microenvironment: roles in epithelial-mesenchymal transition, cell migration, and invasion. J Cell Physiol 237:389–400. https://doi.org/10.1002/JCP.30580
Vultaggio-Poma V, Sarti AC, Di Virgilio F (2020) Extracellular ATP: a feasible target for cancer therapy. Cells 9:1–22. https://doi.org/10.3390/CELLS9112496
Kepp O, Bezu L, Yamazaki T et al (2021) ATP and cancer immunosurveillance. EMBO J 40. https://doi.org/10.15252/EMBJ.2021108130
Nakajima EC, Van Houten B (2013) Metabolic symbiosis in cancer: refocusing the Warburg lens. Mol Carcinog 52:329–337. https://doi.org/10.1002/MC.21863
Reyna-Jeldes M, Díaz-Muñoz M, Madariaga JA et al (2021) Autocrine and paracrine purinergic signaling in the most lethal types of cancer. Purinergic Signal 17:345–370. https://doi.org/10.1007/S11302-021-09785-8
Solini A, Cuccato S, Ferrari D et al (2008) Increased P2X7 receptor expression and function in thyroid papillary cancer: a new potential marker of the disease? Endocrinology 149:389–396. https://doi.org/10.1210/EN.2007-1223
Adinolfi E, Raffaghello L, Giuliani AL et al (2012) Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res 72:2957–2969. https://doi.org/10.1158/0008-5472.CAN-11-1947
Amoroso F, Capece M, Rotondo A et al (2015) The P2X7 receptor is a key modulator of the PI3K/GSK3β/VEGF signaling network: evidence in experimental neuroblastoma. Oncogene 34:5240–5251. https://doi.org/10.1038/ONC.2014.444
Vázquez-Cuevas FG, Martínez-Ramírez AS, Robles-Martínez L et al (2014) Paracrine stimulation of P2X7 receptor by ATP activates a proliferative pathway in ovarian carcinoma cells. J Cell Biochem 115:1955–1966. https://doi.org/10.1002/JCB.24867
Haas CB, Lovászi M, Braganhol E et al (2021) Ectonucleotidases in inflammation, immunity, and cancer. J Immunol 206:1983–1990. https://doi.org/10.4049/JIMMUNOL.2001342
Ruiz-Fernández de Córdoba B, Martínez-Monge R, Lecanda F (2023) ENPP1 immunobiology as a therapeutic target. Clin Cancer Res OF1–OF10. https://doi.org/10.1158/1078-0432.CCR-22-1681
Cappellari AR, Pillat MM, Souza HDN et al (2015) Ecto-5’-nucleotidase overexpression reduces tumor growth in a xenograph medulloblastoma model. PLoS One 10. https://doi.org/10.1371/JOURNAL.PONE.0140996
Allard B, Beavis PA, Darcy PK, Stagg J (2016) Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol 29:7–16. https://doi.org/10.1016/J.COPH.2016.04.001
Mello P de A, Coutinho-Silva R, Savio LEB (2017) Multifaceted effects of extracellular adenosine triphosphate and adenosine in the tumor-host interaction and therapeutic perspectives. Front Immunol 8. https://doi.org/10.3389/FIMMU.2017.01526
Di Virgilio F, Vultaggio-Poma V, Falzoni S, Giuliani AL (2023) Extracellular ATP: a powerful inflammatory mediator in the central nervous system. Neuropharmacology 224. https://doi.org/10.1016/J.NEUROPHARM.2022.109333
Elliott MR, Chekeni FB, Trampont PC et al (2009) Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461:282–286. https://doi.org/10.1038/NATURE08296
Trautmann A (2009) Extracellular ATP in the immune system: more than just a “danger signal.” Sci Signal 2. https://doi.org/10.1126/SCISIGNAL.256PE6
Ghiringhelli F, Apetoh L, Tesniere A et al (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1Β-dependent adaptive immunity against tumors. Nat Med 15:1170–1178. https://doi.org/10.1038/nm.2028
Aymeric L, Apetoh L, Ghiringhelli F et al (2010) Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res 70:855–858. https://doi.org/10.1158/0008-5472.CAN-09-3566
Aramini B, Masciale V, Grisendi G et al (2021) Cancer stem cells and macrophages: molecular connections and future perspectives against cancer. Oncotarget 12:230–250. https://doi.org/10.18632/ONCOTARGET.27870
Arnaud-Sampaio VF, Rabelo ILA, Bento CA et al (2020) Using cytometry for investigation of purinergic signaling in tumor-associated macrophages. Cytometry A 97:1109–1126. https://doi.org/10.1002/CYTO.A.24035
Ohta A, Gorelik E, Prasad SJ et al (2006) A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A 103:13132–13137. https://doi.org/10.1073/PNAS.0605251103
Deaglio S, Dwyer KM, Gao W et al (2007) Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204:1257–1265. https://doi.org/10.1084/JEM.20062512
Ma SR, Deng WW, Liu JF et al (2017) Blockade of adenosine A2A receptor enhances CD8+ T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol Cancer 16. https://doi.org/10.1186/S12943-017-0665-0
Zarek PE, Huang CT, Lutz ER et al (2008) A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 111:251–259. https://doi.org/10.1182/BLOOD-2007-03-081646
Erdmann AA, Gao ZG, Jung U et al (2005) Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood 105:4707–4714. https://doi.org/10.1182/BLOOD-2004-04-1407
Csóka B, Himer L, Selmeczy Z et al (2008) Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. FASEB J 22:3491–3499. https://doi.org/10.1096/FJ.08-107458
Young A, Ngiow SF, Gao Y et al (2018) A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res 78:1003–1016. https://doi.org/10.1158/0008-5472.CAN-17-2826
Neo SY, Yang Y, Record J et al (2020) CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J Clin Invest 130:1185–1198. https://doi.org/10.1172/JCI128895
Montalbán del Barrio I, Penski C, Schlahsa L et al (2016) Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages - a self-amplifying, CD39- and CD73-dependent mechanism for tumor immune escape. J Immunother Cancer 4. https://doi.org/10.1186/S40425-016-0154-9
Kreckler LM, Wan TC, Ge ZD, Auchampach JA (2006) Adenosine inhibits tumor necrosis factor-alpha release from mouse peritoneal macrophages via A2A and A2B but not the A3 adenosine receptor. J Pharmacol Exp Ther 317:172–180. https://doi.org/10.1124/JPET.105.096016
d’Almeida SM, Kauffenstein G, Roy C et al (2016) The ecto-ATPDase CD39 is involved in the acquisition of the immunoregulatory phenotype by M-CSF-macrophages and ovarian cancer tumor-associated macrophages: regulatory role of IL-27. Oncoimmunology 5. https://doi.org/10.1080/2162402X.2016.1178025
Bradford MD, Soltoff SP (2002) P2X7 receptors activate protein kinase D and p42/p44 mitogen-activated protein kinase (MAPK) downstream of protein kinase C. Biochem J 366:745–755. https://doi.org/10.1042/BJ20020358
Stefano L, Rössler OG, Griesemer D et al (2007) P2X(7) receptor stimulation upregulates Egr-1 biosynthesis involving a cytosolic Ca(2+) rise, transactivation of the EGF receptor and phosphorylation of ERK and Elk-1. J Cell Physiol 213:36–44. https://doi.org/10.1002/JCP.21085
Choi JH, Ji YG, Ko JJ et al (2018) Activating P2X7 receptors increases proliferation of human pancreatic cancer cells via ERK1/2 and JNK. Pancreas 47:643–651. https://doi.org/10.1097/MPA.0000000000001055
Zhang Y, Cheng H, Li W et al (2019) Highly-expressed P2X7 receptor promotes growth and metastasis of human HOS/MNNG osteosarcoma cells via PI3K/Akt/GSK3β/β-catenin and mTOR/HIF1α/VEGF signaling. Int J Cancer 145:1068–1082. https://doi.org/10.1002/IJC.32207
Vázquez-Cuevas FG, Cruz-Rico A, Garay E et al (2013) Differential expression of the P2X7 receptor in ovarian surface epithelium during the oestrous cycle in the mouse. Reprod Fertil Dev 25:971–984. https://doi.org/10.1071/RD12196
Zhang W jun, Luo C, Huang C et al (2021) PI3K/Akt/GSK-3β signal pathway is involved in P2X7 receptor-induced proliferation and EMT of colorectal cancer cells. Eur J Pharmacol 899. https://doi.org/10.1016/J.EJPHAR.2021.174041
Ulrich H, Ratajczak MZ, Schneider G et al (2018) Kinin and purine signaling contributes to neuroblastoma metastasis. Front Pharmacol 9. https://doi.org/10.3389/FPHAR.2018.00500
Arnaud-Sampaio VF, Rabelo ILA, Ulrich H, Lameu C (2020) The P2X7 receptor in the maintenance of cancer stem cells, chemoresistance and metastasis. Stem Cell Rev Rep 16:288–300. https://doi.org/10.1007/s12015-019-09936-w
Giannuzzo A, Pedersen SF, Novak I (2015) The P2X7 receptor regulates cell survival, migration and invasion of pancreatic ductal adenocarcinoma cells. Mol Cancer 14. https://doi.org/10.1186/S12943-015-0472-4
Dong C, Hu D, Liu S et al (2022) AKT/GSK-3beta/VEGF signaling is involved in P2RY2 activation-induced the proliferation and metastasis of gastric cancer. Carcinogenesis. https://doi.org/10.1093/CARCIN/BGAC095
Zhou Q, Liu S, Kou Y et al (2022) ATP promotes oral squamous cell carcinoma cell invasion and migration by activating the PI3K/AKT pathway via the P2Y2-Src-EGFR axis. ACS Omega 7:39760–39771. https://doi.org/10.1021/ACSOMEGA.2C03727
Zaparte A, Cappellari AR, Brandão CA et al (2021) P2Y2 receptor activation promotes esophageal cancer cells proliferation via ERK1/2 pathway. Eur J Pharmacol 891. https://doi.org/10.1016/J.EJPHAR.2020.173687
Eun SY, Ko YS, Park SW et al (2015) P2Y2 nucleotide receptor-mediated extracellular signal-regulated kinases and protein kinase C activation induces the invasion of highly metastatic breast cancer cells. Oncol Rep 34:195–202. https://doi.org/10.3892/OR.2015.3972
Buzzi N, Bilbao PS, Boland R, de Boland AR (2009) Extracellular ATP activates MAP kinase cascades through a P2Y purinergic receptor in the human intestinal Caco-2 cell line. Biochim Biophys Acta 1790:1651–1659. https://doi.org/10.1016/J.BBAGEN.2009.10.005
Wang T, Takikawa Y, Watanabe A et al (2014) Proliferation of mouse liver stem/progenitor cells induced by plasma from patients with acute liver failure is modulated by P2Y2 receptor-mediated JNK activation. J Gastroenterol 49:1557–1566. https://doi.org/10.1007/S00535-013-0927-6
Ito N, Ruegg UT, Takeda S (2018) ATP-induced increase in intracellular calcium levels and subsequent activation of mTOR as regulators of skeletal muscle hypertrophy. Int J Mol Sci 19. https://doi.org/10.3390/IJMS19092804
Schäfer R, Sedehizade F, Welte T, Reiser G (2003) ATP- and UTP-activated P2Y receptors differently regulate proliferation of human lung epithelial tumor cells. Am J Physiol Lung Cell Mol Physiol 285. https://doi.org/10.1152/AJPLUNG.00447.2002
Chadet S, Jelassi B, Wannous R et al (2014) The activation of P2Y2 receptors increases MCF-7 breast cancer cells migration through the MEK-ERK1/2 signalling pathway. Carcinogenesis 35:1238–1247. https://doi.org/10.1093/carcin/bgt493
Choi KC, Tai CJ, Tzeng CR et al (2003) Adenosine triphosphate activates mitogen-activated protein kinase in pre-neoplastic and neoplastic ovarian surface epithelial cells. Biol Reprod 68:309–315. https://doi.org/10.1095/BIOLREPROD.102.006551
Muscella A, Elia MG, Greco S et al (2003) Activation of P2Y2 receptor induces c-FOS protein through a pathway involving mitogen-activated protein kinases and phosphoinositide 3-kinases in HeLa cells. J Cell Physiol 195:234–240. https://doi.org/10.1002/JCP.10242
Xie R, Xu J, Wen G et al (2014) The P2Y2 nucleotide receptor mediates the proliferation and migration of human hepatocellular carcinoma cells induced by ATP. J Biol Chem 289:19137–19149. https://doi.org/10.1074/JBC.M113.540047
Hevia MJ, Castro P, Pinto K et al (2019) Differential effects of purinergic signaling in gastric cancer-derived cells through P2Y and P2X receptors. Front Pharmacol 10. https://doi.org/10.3389/FPHAR.2019.00612
Cao Y, Wang X, Li Y et al (2019) Extracellular and macropinocytosis internalized ATP work together to induce epithelial-mesenchymal transition and other early metastatic activities in lung cancer. Cancer Cell Int 19. https://doi.org/10.1186/S12935-019-0973-0
Tafani M, Schito L, Pellegrini L et al (2011) Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-κB. Carcinogenesis 32:1167–1175. https://doi.org/10.1093/carcin/bgr101
Xia J, Yu X, Tang L et al (2015) P2X7 receptor stimulates breast cancer cell invasion and migration via the AKT pathway. Oncol Rep 34:103–110. https://doi.org/10.3892/OR.2015.3979
Li WH, Qiu Y, Zhang HQ, et al (2015) P2Y2 Receptor and EGFR cooperate to promote prostate cancer cell invasion via ERK1/2 pathway. PLoS One 10:. https://doi.org/10.1371/JOURNAL.PONE.0133165
Zhang JL, Liu Y, Yang H et al (2017) ATP-P2Y2-β-catenin axis promotes cell invasion in breast cancer cells. Cancer Sci 108:1318–1327. https://doi.org/10.1111/CAS.13273
Li WH, Qiu Y, Zhang HQ et al (2013) P2Y2 receptor promotes cell invasion and metastasis in prostate cancer cells. Br J Cancer 109:1666–1675. https://doi.org/10.1038/BJC.2013.484
Martínez-Ramírez AS, Garay E, García-Carrancá A, Vázquez-Cuevas FG (2016) The P2RY2 receptor induces carcinoma cell migration and EMT through cross-talk with epidermal growth factor receptor. J Cell Biochem 117:1016–1026. https://doi.org/10.1002/JCB.25390
Reyna-Jeldes M, De la Fuente-Ortega E, Cerda D et al (2021) Purinergic P2Y2 and P2X4 receptors are involved in the epithelial-mesenchymal transition and metastatic potential of gastric cancer derived cell lines. Pharmaceutics 13. https://doi.org/10.3390/PHARMACEUTICS13081234
Choi JH, Ji YG, Lee DH (2013) Uridine triphosphate increases proliferation of human cancerous pancreatic duct epithelial cells by activating P2Y2 receptor. Pancreas 42:680–686. https://doi.org/10.1097/MPA.0B013E318271BB4B
Gaggianesi M, Di Franco S, Pantina VD et al (2021) Messing up the cancer stem cell chemoresistance mechanisms supported by tumor microenvironment. Front Oncol 11. https://doi.org/10.3389/FONC.2021.702642
Bao S, Wu Q, McLendon RE et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760. https://doi.org/10.1038/NATURE05236
Diehn M, Cho RW, Lobo NA et al (2009) Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458:780–783. https://doi.org/10.1038/NATURE07733
Shiozawa Y, Nie B, Pienta KJ et al (2013) Cancer stem cells and their role in metastasis. Pharmacol Ther 138:285–293. https://doi.org/10.1016/J.PHARMTHERA.2013.01.014
Daniele S, Zappelli E, Natali L et al (2014) Modulation of A1 and A2B adenosine receptor activity: a new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell Death Dis 5. https://doi.org/10.1038/CDDIS.2014.487
Liu G, Yang S, Liu Y et al (2022) The adenosine-A2a receptor regulates the radioresistance of gastric cancer via PI3K-AKT-mTOR pathway. Int J Clin Oncol 27:911–920. https://doi.org/10.1007/S10147-022-02123-X
Rocha JD, Uribe D, Delgado J et al (2022) A2B adenosine receptor enhances chemoresistance of glioblastoma stem-like cells under hypoxia: new insights into MRP3 transporter Function. Int J Mol Sci 23. https://doi.org/10.3390/IJMS23169022
Jafari SM, Joshaghani HR, Panjehpour M, Aghaei M (2018) A2B adenosine receptor agonist induces cell cycle arrest and apoptosis in breast cancer stem cells via ERK1/2 phosphorylation. Cell Oncol 41:61–72. https://doi.org/10.1007/s13402-017-0359-z
Liu TZ, Wang X, Bai YF et al (2014) The HIF-2alpha dependent induction of PAP and adenosine synthesis regulates glioblastoma stem cell function through the A2B adenosine receptor. Int J Biochem Cell Biol 49:8–16. https://doi.org/10.1016/J.BIOCEL.2014.01.007
Lan J, Lu H, Samanta D et al (2018) Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment. Proc Natl Acad Sci U S A 115:E9640–E9648. https://doi.org/10.1073/pnas.1809695115
Lan J, Wei G, Liu J et al (2022) Chemotherapy-induced adenosine A2B receptor expression mediates epigenetic regulation of pluripotency factors and promotes breast cancer stemness. Theranostics 12:2598–2612. https://doi.org/10.7150/THNO.70581
García-Rocha R, Monroy-García A, Hernández-Montes J et al (2019) Cervical cancer cells produce TGF-β1 through the CD73-adenosine pathway and maintain CD73 expression through the autocrine activity of TGF-β1. Cytokine 118:71–79. https://doi.org/10.1016/J.CYTO.2018.09.018
García-Rocha R, Monroy-García A, Carrera-Martínez M et al (2022) Evidence that cervical cancer cells cultured as tumorspheres maintain high CD73 expression and increase their protumor characteristics through TGF-β production. Cell Biochem Funct 40:760–772. https://doi.org/10.1002/CBF.3742
Bertolini G, Compagno M, Belisario DC et al (2022) CD73/adenosine pathway involvement in the interaction of non-small cell lung cancer stem cells and bone cells in the pre-metastatic niche. Int J Mol Sci 23. https://doi.org/10.3390/IJMS23095126
Cao L, Bridle KR, Shrestha R et al (2022) CD73 and PD-L1 as potential therapeutic targets in gallbladder cancer. Int J Mol Sci 23. https://doi.org/10.3390/IJMS23031565
Hallaj S, Heydarzadeh Asl S, Alian F et al (2020) Inhibition of CD73 using folate targeted nanoparticles carrying anti-CD73 siRNA potentiates anticancer efficacy of Dinaciclib. Life Sci 259. https://doi.org/10.1016/J.LFS.2020.118150
Azambuja JH, Schuh RS, Michels LR et al (2020) CD73 as a target to improve temozolomide chemotherapy effect in glioblastoma preclinical model. Cancer Chemother Pharmacol 85:1177–1182. https://doi.org/10.1007/S00280-020-04077-1
Jain D, Somasundaram DB, Aravindan S et al (2021) Prognostic significance of NT5E/CD73 in neuroblastoma and its function in CSC stemness maintenance. Cell Biol Toxicol. https://doi.org/10.1007/S10565-021-09658-1
Ziberi S, Zuccarini M, Carluccio M et al (2019) Upregulation of epithelial-to-mesenchymal transition markers and P2X7 receptors is associated to increased invasiveness caused by P2X7 receptor stimulation in human glioblastoma stem cells. Cells 9. https://doi.org/10.3390/CELLS9010085
He X, Wan J, Yang X et al (2021) Bone marrow niche ATP levels determine leukemia-initiating cell activity via P2X7 in leukemic models. J Clin Invest 131. https://doi.org/10.1172/JCI140242
Kwak SH, Shin S, Lee JH et al (2018) Synthesis and structure-activity relationships of quinolinone and quinoline-based P2X7 receptor antagonists and their anti-sphere formation activities in glioblastoma cells. Eur J Med Chem 151:462–481. https://doi.org/10.1016/J.EJMECH.2018.03.023
Kim DC, Jin H, Lee JS et al (2020) P2Y2R has a significant correlation with Notch-4 in patients with breast cancer. Oncol Lett 20:647–654. https://doi.org/10.3892/OL.2020.11630
Cavaliere F, Nestola V, Amadio S et al (2005) The metabotropic P2Y4 receptor participates in the commitment to differentiation and cell death of human neuroblastoma SH-SY5Y cells. Neurobiol Dis 18:100–109. https://doi.org/10.1016/J.NBD.2004.09.001
Salvestrini V, Orecchioni S, Talarico G et al (2017) Extracellular ATP induces apoptosis through P2X7R activation in acute myeloid leukemia cells but not in normal hematopoietic stem cells. Oncotarget 8:5895–5908. https://doi.org/10.18632/ONCOTARGET.13927
D’Alimonte I, Nargi E, Zuccarini M et al (2015) Potentiation of temozolomide antitumor effect by purine receptor ligands able to restrain the in vitro growth of human glioblastoma stem cells. Purinergic Signal 11:331–346. https://doi.org/10.1007/S11302-015-9454-7
Filippin KJ, de Souza KFS, de Araujo Júnior RT et al (2020) Involvement of P2 receptors in hematopoiesis and hematopoietic disorders, and as pharmacological targets. Purinergic Signal 16. https://doi.org/10.1007/S11302-019-09684-Z
Lemoli RM, Ferrari D, Fogli M et al (2004) Extracellular nucleotides are potent stimulators of human hematopoietic stem cells in vitro and in vivo. Blood 104:1662–1670. https://doi.org/10.1182/BLOOD-2004-03-0834
Barbosa CMV, Leon CMMP, Nogueira-Pedro A et al (2011) Differentiation of hematopoietic stem cell and myeloid populations by ATP is modulated by cytokines. Cell Death Dis 2. https://doi.org/10.1038/CDDIS.2011.49
Ledur PF, Villodre ES, Paulus R et al (2012) Extracellular ATP reduces tumor sphere growth and cancer stem cell population in glioblastoma cells. Purinergic Signal 8:39–48. https://doi.org/10.1007/s11302-011-9252-9
Scheffel TB, Rockenbach L, Cruz FF et al (2022) Inhibition of ATP hydrolysis as a key regulator of temozolomide resistance and migratory phenotype of glioblastoma cells. Biochem Biophys Res Commun 601:24–30. https://doi.org/10.1016/J.BBRC.2022.02.062
Ma XL, Hu B, Tang WG et al (2020) CD73 sustained cancer-stem-cell traits by promoting SOX9 expression and stability in hepatocellular carcinoma. J Hematol Oncol 13. https://doi.org/10.1186/S13045-020-0845-Z
Song L, Ye W, Cui Y et al (2017) Ecto-5’-nucleotidase (CD73) is a biomarker for clear cell renal carcinoma stem-like cells. Oncotarget 8:31977–31992. https://doi.org/10.18632/ONCOTARGET.16667
Lupia M, Angiolini F, Bertalot G et al (2018) CD73 Regulates stemness and epithelial-mesenchymal transition in ovarian cancer-initiating cells. Stem Cell Rep 10:1412–1425. https://doi.org/10.1016/j.stemcr.2018.02.009
Hajizadeh F, Moghadaszadeh Ardebili S, Baghi Moornani M et al (2020) Silencing of HIF-1α/CD73 axis by siRNA-loaded TAT-chitosan-spion nanoparticles robustly blocks cancer cell progression. Eur J Pharmacol 882. https://doi.org/10.1016/J.EJPHAR.2020.173235
Torres Á, Erices JI, Sanchez F et al (2019) Extracellular adenosine promotes cell migration/invasion of glioblastoma stem-like cells through A3 adenosine receptor activation under hypoxia. Cancer Lett 446:112–122. https://doi.org/10.1016/J.CANLET.2019.01.004
Erices JI, Niechi I, Uribe-Ojeda A et al (2022) The low affinity A2B adenosine receptor enhances migratory and invasive capacity in vitro and angiogenesis in vivo of glioblastoma stem-like cells. Front Oncol 12. https://doi.org/10.3389/FONC.2022.969993
Glaser T, Cappellari AR, Pillat MM et al (2012) Perspectives of purinergic signaling in stem cell differentiation and tissue regeneration. Purinergic Signal 8:523–537. https://doi.org/10.1007/S11302-011-9282-3
Glaser T, De Oliveira SLB, Cheffer A et al (2014) Modulation of mouse embryonic stem cell proliferation and neural differentiation by the P2X7 receptor. PLoS One 9. https://doi.org/10.1371/JOURNAL.PONE.0096281
Arnaud-Sampaio VF, Bento CA, Glaser T et al (2022) P2X7 receptor isoform B is a key drug resistance mediator for neuroblastoma. Front Oncol 12. https://doi.org/10.3389/FONC.2022.966404
Rabelo ILA, Arnaud-Sampaio VF, Adinolfi E et al (2021) Cancer metabostemness and metabolic reprogramming via P2X7 receptor. Cells 10. https://doi.org/10.3390/CELLS10071782
Mani SA, Guo W, Liao MJ et al (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715. https://doi.org/10.1016/J.CELL.2008.03.027
Nguyen AM, Zhou J, Sicairos B et al (2020) Upregulation of CD73 confers acquired radioresistance and is required for maintaining irradiation-selected pancreatic cancer cells in a mesenchymal state. Mol Cell Proteomics 19:375–389. https://doi.org/10.1074/MCP.RA119.001779
Acknowledgements
We are grateful to Jessica Gonzalez Norris for proofreading. Jose David Nuñez Ríos is a doctoral student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and received a fellowship (CVU 1044618) from CONACyT-Mexico.
Funding
This work was funded by PAPIIT-UNAM-México (IN205223 and IN202121) and FAPESP (2022/11093–9).
Author information
Authors and Affiliations
Contributions
J.D.N.-R. and F.G.V.-C. conceived the article; C.L. prepared figures; all authors wrote the main manuscript text; all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nuñez-Rios, J.D., Ulrich, H., Díaz-Muñoz, M. et al. Purinergic system in cancer stem cells. Purinergic Signalling (2023). https://doi.org/10.1007/s11302-023-09976-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11302-023-09976-5